

Abstract
Metabolic syndrome (MetS) consists of several medical conditions that collectively predict the risk for cardiovascular disease better than the sum of individual conditions. The risk of developing MetS in human depends on synergy of both genetic and environmental factors. Being a multifactorial condition with alarming rate of prevalence nowadays, establishment of appropriate experimental animal models mimicking the disease state in humans is crucial in order to solve the difficulties in evaluating the pathophysiology of MetS in human. This review aims to summarize the underlying mechanisms involved in the pathophysiology of dietary, genetic, and pharmacological models of MetS. Furthermore, we will discuss the usefulness, suitability, pros and cons of these animal models. Even though numerous animal models of MetS have been established, further investigations on the invention of new animal model and clarification of plausible mechanisms are still necessary to confer a better understanding to researchers on the selection of animal models for their studies.
Keywords: Antipsychotic drugs, Carbohydrate, Fat, Fructose, Glucocorticoid, Leptin, Sucrose
Background
Metabolic Syndrome (MetS) is characterized by the simultaneous occurrence of at least three of the following medical conditions, obesity, hyperglycemia, hypertension or dyslipidemia [1]. Metabolic syndrome poses a public healthcare problem worldwide owing to its increasing prevalence. Worldwide prevalence of MetS ranges from 10 to 84 % depending on age, gender, race, ethnicity and definition of MetS [2]. Approximately 20–25 % of world’s adult population is estimated to have MetS [3]. The prevalence of MetS in Malaysia was 22.9, 16.5 and 6.4 % based on the definitions by International Diabetes Federation (IDF), National Cholesterol Education Programme Adult Treatment Panel III (NCEP ATP III) and modified World Health Organization (WHO) respectively; whereby men have a higher prevalence compared to women [4].
Metabolic syndrome is a collection of various conditions, thus it does not have a single cause. Contributing factors for the features of MetS can be hereditary or environmental. Family history of type II diabetes, hypertension and insulin resistance and ethnic background are inevitable genetic factors that greatly increase the risk for developing MetS [5–8]. Furthermore, senescence is another important unalterable risk factor for MetS [9–11]. On the other hand, environmental risk factors for MetS are controllable. These include sedentary lifestyle, physical inactivity and eating habits [12]. Metabolic syndrome ultimately predisposes an individual to other medical complications. For instance, MetS causes increased risk of cardiovascular disease (CVD) [13], type II diabetes [14], non-alcoholic fatty liver disease [15], cancer (liver, pancreas, breast and bladder) [16–19], kidney and pancreatic dysfunction [20].
The deleterious effects of MetS draw research efforts in developing new interventions to reduce its burden on the healthcare system. Due to its multifactorial nature, selecting an adequate experimental model that best represents the pathophysiology of MetS in humans can be rather challenging. Rats and mice are the most common animal models used in investigating MetS. Some of the various approaches used to induce MetS in rodents include dietary manipulation, genetic modification and drugs. Previously, a review was produced by Panchal and Brown, which primarily suggested the rat model that displayed closest criteria to human MetS was induced by high-carbohydrate high-fat diet [20]. In this review, we collate and discuss the various animal models of MetS. The caveats and suitability of MetS animal models for research will also be discussed to provide the readers a comprehensive overview on the selection of the best animal models to meet their research purpose.
Diet-induced models of MetS
Numerous dietary approaches capable to induce MetS in animals have been reported. They included the use of a single type of diet or a combination of diets, such as high-fructose, high-sucrose (Table 1), high-fat (Table 2), high-fructose/high-fat, or high-sucrose/high-fat diets (Table 3). A number of dietary studies have become the cornerstone for the investigation of MetS because diet affects whole-body metabolism and regulation through effects on hormones, glucose metabolism, and lipid metabolism pathways. The most commonly used rodent strains in diet-induced models of MetS include Sprague-Dawley rats, Wistar rats, C57BL/6 J mice and Golden Syrian Hamster [21–24]. Here, we look into how these different diets give rise to various illnesses of MetS.
Table 1.
Effects of fructose- and sucrose-enriched diets on the development of MetS
| Researchers (Year) | Types of diet | Treatment length | Strains of animal | Components of metabolic syndrome | |||
|---|---|---|---|---|---|---|---|
| Obesity | Hyperglycemia | Hypertension | Dyslipidemia | ||||
| Thirunavukkarasu et al. [41] | High-fructose diet | 3 weeks | Male Wistar rats | - | ✓ | ✓ | - |
| Sanchez-Lozada et al. [42] | High-fructose diet (60 %) | 8 weeks | Male Sprague-Dawley rats | - | - | ✓ | ✓ |
| Fructose drinking water (10 %) | - | - | ✓ | ✓ | |||
| Shahraki et al. [105] | High-fructose diet | 8 weeks | Male Wistar rats | ✗ | ✓ | - | ✓ |
| Mahmoud and Elshazly [106] | Fructose drinking water (10 %) | 12 weeks | Male Wistar rats | ✓ | ✓ | ✓ | ✓ |
| Mansour et al. [107] | High-fructose diet | 16 weeks | Male Wistar albino rats | ✓ | ✓ | - | ✓ |
| Mamikutty et al. [108] | Fructose drinking water | 8 weeks | Male Wistar rats | ✓ | ✓ | ✓ | ✓ |
| Di Luccia et al. [109] | High-fructose diet | 8 weeks | Male Sprague-Dawley rats | ✓ | ✓ | - | ✓ |
| Jurgens et al. [35] | Fructose drinking water (15 %) | 10 weeks | Male NMRI mice | ✓ | - | - | - |
| Sucrose soft drink (10 %) | ✗ | - | - | - | |||
| Non-caloric soft drink | ✗ | - | - | - | |||
| Oron-Herman et al. [51] | High-sucrose diet | 7 weeks | Male spontaneously hypertensive rats | ✗ | ✓ | ✓ | ✗ |
| High-fructose diet | Male Sprague-Dawley rats | ✗ | ✓ | ✓ | ✓ | ||
| Aguilera et al. [45] | Sucrose drinking water (30 %) | 21 weeks | Male Wistar rats | ✓ | - | ✓ | ✓ |
| Vasanji et al. [49] | Sucrose drinking water (32 %) | 10 weeks | Male Sprague-Dawley rats | - | ✓ | - | ✓ |
| Pang et al. [50] | High-sucrose diet | 6 weeks | Male Sprague-Dawley rats | - | ✗ | ✓ | ✓ |
Table represents the effects of fructose- and sucrose-enriched diets on each component of MetS. The symbol ‘✓’ and ‘✗’ indicate the presence and absence of significant effect of the sign of MetS respectively, while ‘-’ indicates the effects on the component not being evaluated in the study
Table 2.
Effects of fat-enriched diet on the development of MetS
| Researchers (Year) | Types of diet | Treatment length | Strains of animal | Components of metabolic syndrome | |||
|---|---|---|---|---|---|---|---|
| Obesity | Hyperglycemia | Hypertension | Dyslipidemia | ||||
| Dobrian et al. [110] | High-fat diet | 10 weeks | Male Sprague-Dawley rats | ✓ | - | ✓ | ✓ |
| Ghibaudi et al. [59] | High-fat diet | 24 weeks | Male Sprague-Dawley weanling rats | ✓ | ✓ | - | ✓ |
| Rossmeisl et al. [111] | High-fat diet | 8 weeks | Male C57BL/6 J mice | ✓ | ✓ | - | ✓ |
| Male AKR/J (AKR) mice | ✓ | ✓ | - | ✓ | |||
| Gallou-Kabani et al. [112] | High-fat diet (60 %) | 20 weeks | Male & female C57Bl/6 J mice | ✓ | ✓ | - | ✓ |
| Male & female A/J mice | ✓ | ✗ | - | ✓ | |||
| Fraulob et al. [60] | High-fat diet | 16 weeks | Male C57BL/6 mice | ✓ | ✓ | - | ✓ |
| Graham et al. [61] | High-fat diet | 40 weeks | Male C57BL/6 mice | - | - | - | ✓ |
| Halade et al. [56] | High-fat diet | 24 weeks | Female C57Bl/6 J mice | ✓ | - | - | - |
| Davidson et al. [113] | High-fat diet | 24 weeks | Male Sprague-Dawley rats | ✓ | ✓ | - | ✓ |
| Pirih et al. [114] | High-fat diet | 13 weeks | C57BL/6 mice (wild type) | - | ✗ | - | ✓ |
| Hyperlipidemic (Ldlr−/−) mice | - | ✓ | - | ✓ | |||
| Podrini et al. [115] | High-fat diet | 12 weeks | Female C57BL/6NTac mice | ✓ | ✓ | - | ✓ |
| Xu et al. [57] | High-fat diet | 12 weeks | Male C57BL/6 mice | ✓ | - | - | - |
| Fujita and Maki [24] | High-fat diet | 4 weeks | Male C57BL/6 J mice | ✓ | - | - | ✓ |
| Gancheva et al. [116] | High-fat diet | 8 weeks | Male Wistar rats | ✓ | ✓ | - | ✓ |
| Li et al. [62] | High-fat diet | 16 weeks | Male C57BL/6 mice | ✓ | ✓ | - | ✓ |
| Suman et al. [23] | High-fat diet | 10 weeks | Male Wistar rats | ✓ | ✓ | ✓ | ✓ |
Table represents the effects of fat-enriched diet on each component of MetS. The symbol ‘✓’ and ‘✗’ indicate the presence and absence of significant effect of the sign of MetS respectively, while ‘-’ indicates the effects on the component not being evaluated in the study
Table 3.
Effects of different diet combinations on the development of MetS
| Researchers (Year) | Types of diet | Treatment length | Strains of animal | Components of metabolic syndrome | |||
|---|---|---|---|---|---|---|---|
| Obesity | Hyperglycemia | Hypertension | Dyslipidemia | ||||
| Poudyal et al. [29] | High-carbohydrate high-fat diet | 16 weeks | Male Wistar rats | ✓ | ✓ | ✓ | ✓ |
| Panchal et al. [30] | High-carbohydrate high-fat diet | 16 weeks | Male Wistar rats | ✓ | ✓ | ✓ | ✓ |
| Hao et al. [31] | High-carbohydrate high-fat diet | 14 weeks | Male Wistar rats | ✓ | ✓ | ✓ | ✓ |
| Senaphan et al. [22] | High-carbohydrate high-fat diet | 16 weeks | Male Sprague-Dawley rats | ✗ | ✓ | ✓ | ✓ |
| Dissard et al. [117] | High-fat high-fructose diet | 32 weeks | Male C57BL/6 mice | ✓ | ✓ | ✗ | ✓ |
| Barrios-Ramos et al. [118] | Hypercholesterolemic diet & fructose drinking water | 4 weeks | Male Wistar rats | ✓ | ✓ | ✓ | ✓ |
| Gancheva et al. [116] | High-fat high-fructose diet | 8 weeks | Male Wistar rats | ✓ | ✓ | - | ✓ |
| Yang et al. [119] | High-fat high sucrose diet | 4 weeks | Male C57BL/6 J mice | ✓ | ✓ | - | ✓ |
| Zhou et al. [120] | High-sucrose high-fat diet | 48 weeks | Male Sprague-Dawley rats | ✓ | ✓ | - | ✓ |
Table represents the effects of different diet combinations on each component of MetS. The symbol ‘✓’ and ‘✗’ indicate the presence and absence of significant effect of the sign of MetS respectively, while ‘-’ indicates the effects on the component not being evaluated in the study
Carbohydrate-enriched diet
Carbohydrates can be divided into simple (e.g. monosaccharides and disaccharides) and complex (e.g. oligosaccharides and polysaccharides) forms. Carbohydrates are one of the essential nutrients acting as the main source of energy (short-term fuel) in the body because they are simpler to metabolize compared to fats. Adopting a sedentary lifestyle puts an individual into the conditions of high energy intake but low physical activity, thus increasing the tendency towards energy storage, overweight and finally obesity.
Carbohydrate metabolism begins from digestion in the small intestine to form glucose molecules, followed by absorption into the bloodstream and transportation into liver via the portal vein. When carbohydrate intake greatly exceeds daily energy requirements, blood glucose concentration will remain high and insulin is secreted by the pancreas to allow cells to uptake glucose. At this moment, the mechanisms involved in utilizing glucose are: (a) breakdown of glucose in the process of glycolysis, (b) glucose is converted to glycogen in the liver and muscles, and (c) insulin acts on adipose tissue to promote fatty acids synthesis and inhibit release of available fatty acids [25]. Prolonged excessive carbohydrate consumption causes sustained high glucose levels in the blood. Insulin is thus produced in proportion to lower the blood glucose. Therefore, high dietary carbohydrates are converted into fats for storage. Insulin sensitivity is also decreased. Substantial evidence has demonstrated a strong association between high carbohydrate intake and insulin resistance [26–28].
Information on the metabolic impact of carbohydrate on animal models of MetS is absent. Most of the diet regimens were designed with the combination of high-carbohydrate and high-fat. Two studies tracing metabolic changes in rats fed with high-carbohydrate high-fat diet are available. These studies adopted a high-carbohydrate high-fat diet (consist of 39.5 % sweetened condensed milk, 20 % beef tallow, 17.5 % fructose, 15.5 % powdered rat food, 2.5 % salt mixture, 5 % water) to induce MetS in an animal model. The researchers claimed it mimics more closely the human disease state compared to other methods of inducing MetS [29–31]. Test animals developed hypertension, impaired glucose tolerance, increased abdominal fat deposition, increased abdominal circumference, and altered lipid profile after 16 weeks on this diet. Another study by Senaphan and co-workers reported that high-carbohydrate high-fat diet with some modifications (35 % sweetened condensed milk, 20 % pork tallow, 17.5 % fructose, 20 % powdered rat food, 2.5 % salt mixture, 5 % water) provided similar outcomes as the previous study [22].
Ironically, the combination of high-carbohydrate with high-fiber was reported to confer hypolipidemic and hypoglycemic effects as evidenced in human studies. In a clinical study, high-carbohydrate high-fiber diet was suggested as dietary therapy in diabetic patients because this diet was capable of reducing postprandial plasma glucose, insulin response, cholesterol, and triglycerides levels [32]. Hence, the composition and combination of high-carbohydrate diet are important factors that must be taken into consideration for the induction of MetS.
Fructose-enriched diet
Fructose, commonly known as the fruit sugar, is one of the monosaccharides along with glucose and galactose. Nowadays, fructose is often used as a taste enhancer to make food more appetizing and tempting. There is no biological need for dietary fructose; it is only an intermediary molecule during glucose metabolism. The circulating concentration of fructose (~0.01 mmol/L) in peripheral blood is very low compared to glucose (~5.5 mmol/L) [33]. Interestingly, a small quantity of fructose produces a lower glycemic response to substitute sucrose and starch in the diet in diabetic patients [34]. Unfortunately, intake of fructose is excessive nowadays due to the consumption of artificially sweetened beverages and food.
Theoretically, a large influx of fructose into the liver causes accumulation of triglycerides and cholesterol because of its lipogenic (fat-producing) properties, subsequently leading to reduced insulin sensitivity, insulin resistance and glucose intolerance [35, 36]. Fructose consumption resulted in massive fructose uptake by the liver. Fructose is converted to fructose-1-phosphate, a reaction catalyzed by the enzyme phosphofructokinase in the presence of ATP. It is followed by the cleavage of fructose-1-phosphate into glyceraldehyde and dihydroxyacetone phosphate without the conversion of glucose to fructose-1,6-bisphosphate, an initial regulatory step of glycolysis [37]. Phosphofructokinase is a negative regulator for glucose metabolism, allowing fructose to enter into the glycolysis pathway continuously. Fructose-1,6-bisphosphate is then converted to pyruvate through the process of glycolysis. At this juncture, fructose is involved in several simultaneous processes: (a) a portion of the fructose is converted into lactate from pyruvate, (b) another portion produces triose-phosphate which readily converts to glucose or glycogen via gluconeogenesis, (c) carbons derived from the fructose can be converted into fatty acids, and (d) inhibition of hepatic lipid oxidation by fructose favours very low density lipoproteins (VLDL)-triglyceride synthesis and fatty acid re-esterification [38]. As a result, this refined carbohydrate is rapidly absorbed and readily metabolized by liver to produce glucose, glycogen, pyruvate, lactate, glycerol, and acyl-glycerol molecules.
Knowledge on fructose metabolism revealed the superiority of fructose-feeding for the induction of MetS in animal models in comparison with glucose or starch. Previous research indicated that glucose or starch-feeding is not as effective as fructose-feeding in inducing MetS [39]. In addition, mice fed with fructose gained more weight compared to mice fed with the same calories using starch [35]. The correlation between chronic high intake of dietary fructose with increased energy intake, body weight, adiposity, hypertriglyceridemia, hyperlipidemia, hypertension, glucose intolerance and decreased insulin sensitivity in laboratory animal, all leading to MetS, is indisputable [39, 40]. An animal study conducted by Thirunavukkarasu et al. [41] showed that increased blood pressure, glucose intolerance, and decreased insulin sensitivity were detected in rats fed with a fructose-enriched diet containing >60 % of total calories. Another study performed by Sanchez-Lozada et al. [42] reported that 10 % of fructose in drinking water resulted in the same effects as high dose of fructose (60 % in diet) in inducing hypertension and hyperlipidemia in male Sprague-Dawley rats, but they were less severe compared to high dose of fructose.
To sum up, fructose behaves more like a fat instead of a carbohydrate in both humans and animals. A low dose of fructose in drinking water (10 %) is sufficient to induce MetS in animals.
Sucrose-enriched diet
Sucrose, or table sugar, is a disaccharide found in cane or beet sugar. It consists of one fructose molecule and one glucose molecule. Sucrose has the same role as fructose to make food more palatable. When sucrose is consumed, it is cleaved into its constituents, i.e. glucose and fructose by the enzyme sucrase [43]. Both molecules are then taken up by their specific transport mechanisms. As outlined earlier, glucose uptake in glucose metabolism is negatively regulated by phosphofructokinase, leading to the continuous entry of fructose into the glycolytic pathway. Excess fructose will be converted into fat in the liver as fructose is a better substrate for fatty acid synthesis compared to glucose [44]. Thus, fructose is the main active ingredient contributing to the development of MetS in animals after sucrose consumption.
An animal study showed that administration of 30 % sucrose in drinking water led to the development of MetS in male Wistar rats with increased body weight, systolic blood pressure, insulin, triacylglycerol, total cholesterol, low density lipoproteins (LDL) cholesterol, and free fatty acids [45]. Besides, high sucrose supplementation is widely used for induction of whole body insulin resistance in rats, whereby high levels of plasma insulin was detected [45–48]. Concomitantly, animals treated with 32 % sucrose in drinking water exhibited hyperglycemia, hypertriglyceridemia, hypercholesterolemia, and increased body weight [49]. Another study by Pang et al. [50] reported that rats responded to sucrose supplementation (77 %) by a significant elevation in systolic blood pressure, plasma insulin, and triglycerides.
However, Kasim-Karakas et al. [21] revealed that only fructose-feeding increased fasting non-esterified fatty acids and triglycerides levels in the plasma and liver of Golden Syrian hamsters. However the increment was not found in sucrose-fed hamsters. Moreover, impaired glucose tolerance, significant increase of body weight and body fat were only detected in fructose-fed (15 %) rats, but not in other groups fed with a soft drink (10 % sucrose) and a diet soft drink (without calories) [35]. Fructose and sucrose supplementation also invoked distinct responses in two different animal models, i.e. Sprague-Dawley and spontaneous hypertensive rats, which represented environmentally and genetically acquired MetS respectively. Fructose enrichment in Sprague-Dawley rats caused hyperinsulinemia, hypertriglyceridemia, hypercholesterolemia, hypertension, and insulin resistance. Meanwhile, sucrose enrichment in spontaneous hypertensive rats only increased blood pressure and worsened insulin resistance [51].
These paradoxical outcomes accumulated from previous studies implied that high content of sucrose will ensure the success of MetS development in animal models. However, fructose appeared to be more superior than an equivalent amount of sucrose in inducing MetS because fructose exists as a free molecule while sucrose contains only 50 % fructose and 50 % glucose.
Fat-enriched diet
Fats are one of the three main macronutrients and are the most calorically dense macronutrient [52]. Fats, also known as triglycerides, are composed of esters of three fatty acid chains and glycerol. Lipid metabolism begins with the process of lipolysis. Plenty of glycerol and fatty acids diffuse freely into the bloodstream. Plasma free fatty acids are major substrates for hepatic VLDL-triglycerides production [53]. Approximately 70 % of released free fatty acids will be re-esterified (lipogenesis) to form triglycerides [54]. The rate of re-esterification is dependent on the rate of glycerol-3-phosphate production through glycolysis and the rate of fatty acid release from adipocytes [25]. The coupled actions of free and re-esterified fatty acids (triglycerides) form VLDL, which assists fats to circulate in the water-based solution of the bloodstream.
Many researchers have employed different types of high-fat diets that vary between 20 and 60 % of total energy. The source of the fat component may be either plant-derived oils (e.g. corn, safflower or olive oil) or animal-derived fats (e.g. beef tallow and lard) [55]. High-fat diets have been extensively used to induce MetS in experimental animals. More specifically, high-fat diets have been widely used to induce obesity in animals [56, 57]. Studies have also indicated that high-fat diet is effective in promoting hyperglycemia, insulin resistance, dyslipidemia and increased free fatty acids in the blood, either independently or concurrently [58].
A comprehensive study by Ghibaudi et al. [59] aimed to assess the chronic effect of dietary fats with different fat content (10, 32 and 45 %) on body adiposity and metabolism in rats. The findings demonstrated that energy intake, weight gain, fat mass, plasma glucose, cholesterol, triglycerides, free fatty acids, leptin, and insulin levels increased dose-dependently with increased dietary fat. Apart from that, mice fed with high-fat (60 %) diet exhibited greatly increased body mass, total fat pads, plasma triglyceride, high density lipoproteins (HDL) cholesterol, and LDL cholesterol levels [60]. Another animal model fed with high-fat diet displayed elevation of total cholesterol, LDL cholesterol, and unesterified cholesterol [61]. Later investigation has found that high-fat intake augmented body weight, total cholesterol, and leptin levels in male C57BL/6 J mice [24]. Another recent study indicated that mice fed with high-fat diet had increased body weight, plasma lipids, plasma insulin, and insulin resistance compared to mice fed with standard chow [62]. To conclude, the increased formation of VLDL helps to distribute assembled triglycerides synthesized by the liver resulting from overconsumption of high-fat diet. A high level of VLDL cholesterol can cause obesity, dyslipidemia and the build-up of cholesterol in arteries. The accumulation of triglycerides in the liver can cause insulin resistance.
A summary on the effects of different nutrients on whole body metabolism has been illustrated (Fig. 1). The lipotoxicity hypothesis (overproduction and accumulation of triglycerides in the non-adipose tissue such as liver, muscle, and pancreas) is the common criteria seen in the effect of different diets in the development of MetS. It is alarming that MetS can be contributed by these seemingly harmless essential nutrients.
Fig. 1.
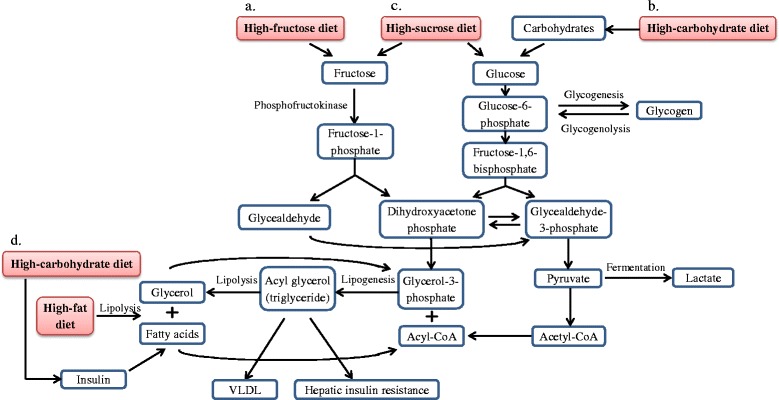
Summary of the effects of different diets on whole body metabolism. a High-fructose diet intake interferes glycolytic pathway by bypassing the rate-controlling step, the conversion of glucose-6-phosphate into fructose-1,6-bisphosphate. Phosphofructokinase acts as a negative regulator for glucose metabolism and allows fructose to enter the glycolytic pathway continuously to produce pyruvate, lactate, glycerol and acyl-glycerol. b When plenty of glucose is available during high dietary carbohydrate, glucose utilizing pathways are initiated: breakdown of glucose by glycolysis, conversion of glucose into glycogen via glycogenesis, and production of insulin which acts on adipose tissue to promote fatty acids synthesis. c Consumption of high-sucrose diet: sucrose separates into fructose and glucose molecules and enters their specific mechanisms as stated earlier. d Fats undergo lipolysis, glycerol and fatty acids are released into the blood. However, fatty acids released during lipolysis are re-esterified to form triglyceride. Overproduction of triglyceride through excessive intake of various nutrients is likely to cause accumulation of triglyceride in the liver, which will further lead to hepatic insulin resistance (reduced insulin sensitivity)
Genetic models of MetS
In addition to diet-induced MetS animal model, genetic animal models are imperative in order to investigate the pathogenesis of MetS caused by genetic factors. These genetic models of MetS are time-saving because the duration for the development of MetS is significantly shortened compared to diet-induced MetS.
Originally, leptin- or leptin receptor-deficient rodent models are used as genetically obese and diabetic experimental models. Numerous animal models are developed, such as leptin-deficient (ob/ob) mice, leptin receptor-deficient (db/db) mice, Zucker fatty (ZF) rats, Zucker diabetic fatty (ZDF) rats, DahlS.Z-Lepr fa /Lepr fa (DS/obese) rats, Goto-Kakizaki (GK) rats, obese spontaneous hypertensive rat (Koletsky rat), and the POUND mice™. Leptin, serving as an anti-obesity hormone by binding to leptin receptor, is secreted by mature adipocytes in proportion with the size of fat depots [63]. Circulating leptin is taken up into the hypothalamus to decrease food intake and eating appetite to increase energy expenditure via several signaling pathways. Thus, the occurrence of obesity in these models is basically owing to the abnormalities in leptin signaling, which result in hyperphagia (great desire on food), uncontrolled appetite, and reduced energy expenditure [64].
Leptin-deficient (ob/ob) and leptin receptor-deficient (db/db) mice are the models of single autosomal recessive mutation on leptin gene (chromosome 6) and leptin receptor gene (chromosome 4) respectively. Leptin-deficient (ob/ob) mice develop obesity, hyperinsulinemia and hyperglycemia with the absence of hypertension and dyslipidemia. Both hypertension and dyslipidemia did not develop even after 38 weeks of age [65]. Whereas leptin receptor-deficient (db/db) mice develop obesity, hyperglycemia, and dyslipidemia without hypertension [66, 67]. Hence, both of these animal models are excellent models for obesity and type II diabetes, but not for MetS. Zucker fatty rat, also known as leptin receptor-deficient obese rat, carries a missense mutation in the leptin receptor gene with homozygous fa allele, hallmarked by an increased circulating leptin level [68]. Obesity developed in these rats as early as between 3 to 5 weeks of life [69]. Instead of being genetically obese, ZF rats demonstrated hyperinsulinemia, insulin resistance, mild glucose intolerance, dyslipidemia, and hypertension [70, 71]. A variant of Zucker rat, known as Zucker Diabetic Fatty rat, is a selective inbred rat strain derived from ZF rat with high glucose levels [71]. Zucker Diabetic Fatty rats display hyperphagia caused by a non-functioning leptin receptor [72]. Moreover, ZDF rats recapitulate several phenotypes of type II diabetes (impaired glucose metabolism, hyperglycemia, and hyperinsulinemia) resulting from the defects of GLUT-2 and GLUT-4 transporter. Long-term severe diabetes leads to mild cardiac diastolic dysfunction in ZDF rats [73]. Hattori et al. [74] introduced a new animal model of MetS, namely DahlS.Z-Lepr fa /Lepr fa (DS/obese) rat strain, which was established from a cross between Dahl salt-sensitive rats and ZF rats. Higher systolic blood pressure, body weight, visceral fat mass, subcutaneous fat mass, and ratio of LDL cholesterol to HDL cholesterol levels were detected in DS/obese rats compared to DahlS.Z-Lepr + /Lepr + (DS/lean) rats fed on a normal diet, whereas fasting serum glucose concentration remained unchanged. After that, Murase et al. [75] further confirmed this strain of rat as a MetS animal model because female DS/obese rats developed elevated systolic blood pressure, body weight, insulin, triglycerides, LDL:HDL cholesterol ratio, visceral and subcutaneous fat mass.
Goto-Kakizaki (GK) rat, a leptin resistant animal model, is considered as one of the best non-obese inbred model of type II diabetes [76]. They spontaneously develop hyperleptinemia, hyperphagia, hyperglycemia, decreased β-cell function, increased gluconeogenesis, and accumulation of visceral fat [77, 78]. Goto-Kakizaki rat was established through repetitive selective breeding of Wistar rats with glucose intolerance over several generations [79, 80]. In light of the difficulties to access human pancreatic islet defect, this specific animal model representing human type II diabetes provide an opportunity to study the disease intensively. However, GK rats only act as genetic model representing certain aspects of MetS thus not a suitable animal model to represent MetS.
Spontaneous hypertensive rat (SHR) was generated from outbreed between Wistar Kyoto male rats with noticeable elevated blood pressure and females with slight elevation of blood pressure, followed by selective inbreed of the offspring with highest blood pressure [81]. The SHR is used as an experimental model for genetically induced hypertension. A study by Potenza et al. [82] demonstrated that 12-week-old SHRs were hypertensive, hyperinsulinemic, and insulin resistant compared to Wistar-Kyoto rats. Spontaneously hypertensive rats generally do not develop hypercholesterolemia and hyperlipidemia unless they are put on a special diet regimen, such as high-cholesterol or high-fructose high-fat diet [83, 84]. Modification of SHR, known as obese SHR or Koletsky rat, was obtained by crossing a female SHR with a normotensive Sprague-Dawley male. Koletsky rats carry a nonsense mutation in the leptin receptor and possess interesting phenotypes, including obesity at 5 weeks of age, hypertriglyceridemia even with standard diet, hyperinsulinemia with normal blood glucose, and severe hypertension at 3 months of age [69]. Koletsky rats have been suggested as a more appropriate animal model for MetS compared to SHRs. The POUND mouse (C57BL/6NCrl-Lepr db-lb/Crl) was established in the last decade as another model fulfilling all the MetS criteria in a single animal. The animals were fed with Purina Diet ad libitum and showed obesity at 1 month of age, hyperinsulinemia and hyperglycemia at 18 weeks of age, increased leptin levels at 17–18 weeks of age, as well as increased cholesterol levels at 14 weeks of age [85].
Amongst all these leptin- and leptin receptor-related rodent models, ZF rats, ZDF rats, DS/obese rats, Koletsky rats and POUND mice are suitable models of MetS because these rats display all the conditions of MetS (Table 4). Genetic models are beneficial in elucidating the plausible molecular mechanisms involved in the development of certain disease states. However, there were only nine mutations have been identified in the leptin gene in 2014 and mutations were more prevalent in consanguineous marriages [86]. Thus mutations of leptin or leptin receptor rarely occur in humans, implying that they do not actually resemble the human disease state in real life.
Table 4.
Metabolic changes in genetic models of MetS
| Strain | Model | Mutation/deficiency | Abnormalities/metabolic changes | References |
|---|---|---|---|---|
| Leptin-deficient (ob/ob) mice | Obesity, type II diabetes | Autosomal recessive mutation on leptin gene (chromosome 6) | (a) Obese & increased body weight (Age: 4 weeks) (b) Hyperinsulinemia & hyperglycemia (Age: 4 weeks) (c) Impaired glucose tolerance (Age: 12 weeks) (d) Reduced blood pressure (e) Does not develop dyslipidemia |
[65, 121, 122] |
| Leptin receptor-deficient (db/db) mice | Obesity, type II diabetes | Autosomal recessive mutation on leptin receptor gene (chromosome 4) |
(a) Obese & increased body weight (Age: 6 weeks) (b) High fasting blood glucose (Age: 8 weeks) (c) Hyperinsulinemia & impaired glucose tolerance (Age: 12 weeks) (d) Unchanged blood pressure (e) Increased triglycerides, total cholesterol, LDL cholesterol, and free fatty acid (Age: 13 weeks) |
[66, 67] |
| Zucker fatty (ZF) rat | Metabolic syndrome | Missense mutation on leptin receptor gene | (a) Obese (Age: 3-5 weeks) (b) Hyperinsulinemia, insulin resistance (Age: 3 weeks) (c) Hypertension (Age: 12 weeks) (d) Hypercholesterolemia, hypertriglyceridemia |
[71] |
| Zucker Diabetic Fatty (ZDF) rat | Metabolic syndrome | Non-functional leptin receptor | (a) Obese (Age: 3–5 weeks) (b) Hyperinsulinemia, insulin resistance, hyperglycemia (Age: 13–15 weeks) (c) Mild hypertension (Age: 12–14 weeks) (d) Hypercholesterolemia, hypertriglyceridemia (Age: 20 weeks) |
[123] |
| DahlS.Z-Lepr fa /Lepr fa(DS/obese) rat | Metabolic syndrome | - | (a) Obese (Age: 18 weeks) (b) Hyperinsulinemia (Age: 18 weeks) (c) Unchanged serum glucose concentration (d) Hypertension (Age: 11–12 weeks) (e) Hypercholesterolemia & hypertriglyceridemia (Age: 18 weeks) |
[74, 75] |
| Goto-Kakizaki (GK) rat | Type II diabetes | Leptin resistance | (a) Non-obese (b) Hyperinsulinemia, insulin resistance & mild hyperglycemia (Age: 4 weeks) (c) Hyperlipidemia (Age: 8 weeks) (d) Unchanged blood pressure (Age: 14 months) |
[76, 78, 124, 125] |
| Spontaneous Hypertensive rat | Hypertension | - | (a) Hyperinsulinemia, insulin resistance (Age: 12 weeks) (b) Severe hypertension (Age: 4 weeks) |
[82, 126] |
| Obese Spontaneous Hypertensive rat (Koletsky rat) | Metabolic syndrome | Nonsense mutation of leptin receptor | (a) Obese, increased abdominal fat (Age: 5 weeks) (b) Hyperinsulinemia, insulin resistance (Age: 16–18 weeks) (c) Normal fasting blood glucose (d) Severe hypertension (Age: 12 weeks) (e) Hyperlipidemia (Age: 16–18 weeks) |
[69, 127] |
| The POUND mouse™ | Pre-diabetes/metabolic syndrome | Mutation in leptin receptor (deletion of axon 2 on chromosome 4) | (a) Obese (Age: 4 weeks) (b) Hyperinsulinemia, hyperglycemia (Age: 18 weeks) (c) Increased leptin levels (Age: 17–18 weeks), (d) Hypercholesterolemia (Age: 14 weeks) |
[85, 128] |
Drug/chemically-induced model of MetS
Glucocorticoid-induced MetS
Endogenous glucocorticoids are naturally occurring stress hormones secreted by the adrenal glands. Glucocorticoids bind to its receptors (glucocorticoid and mineralocorticoid receptors) to exert their effects on different tissues [87]. Apart from that, exogenous glucocorticoids are used as medicine to treat a wide range of human diseases, such as autoimmune disease and cancer. It is also used to prevent rejection in organ transplantation. However, glucocorticoid treatment brings about undesirable side effects such as body weight gain, glucose intolerance, impaired calcium homeostasis, osteoporosis, cataracts and central nervous system effects [88]. Both endogenous and exogenous glucocorticoids have been used to develop MetS in animal models [89].
Glucocorticoids cause MetS by acting directly on different tissues and organs (e.g. fat, liver, muscles, and kidneys) via several mechanisms: (1) glucocorticoids stimulate the differentiation of pre-adipocytes into mature adipocytes; (2) glucocorticoids increase lipolysis to release free fatty acids; (3) glucocorticoids increase proteolysis in muscle to increase free amino acids. Amino acid-induced mammalian target of rapamycin complex-1 (mTORC1) activation causes phosphorylation of insulin receptor substrate-1 (IRS-1), leading to the occurrence of insulin resistance; (4) glucocorticoids promote gluconeogenesis in liver and cause hyperglycemia; and (5) non-specific binding of glucocorticoids to its receptor in the kidneys causes an increase in sodium retention, potassium excretion, water retention, and plasma volume concomitantly with elevation of blood pressure [87, 88, 90].
Using laboratory animals, glucocorticoid-induced MetS has been done through various approaches, such as feeding [87, 91], daily intraperitoneal injections [92], or surgically implanted glucocorticoid pellets [93, 94]. All these different routes of administration of glucocorticoids resulted in almost similar outcomes. Mounting levels of corticosterone enhanced food intake, weight gain, abdominal fat accumulation, severe fasting hyperglycemia, insulin resistance, impaired glucose tolerance, hypertension, dyslipidemia, as well as deposition of lipids in visceral adipose, hepatic tissue and skeletal muscle in animals. Meanwhile, the removal of corticosterone reversed all these adverse conditions.
Antipsychotic-induced MetS
Antipsychotic drugs are medications used to treat neuropsychiatric disorders, for examples, schizophrenia, depression, and bipolar disorder [95]. Antipsychotic drugs have been associated with a high incidence of MetS, evidenced by body weight gain, increased visceral fat, impaired glucose tolerance, and insulin resistance in animal studies [96, 97]. However, the exact underlying mechanism involved in antipsychotic-induced MetS still remains an enigma. The proposed mechanism available currently is that the weight gain caused by antipsychotic treatment contributes to the development of diabetes and dyslipidemia [98]. Latest evidence demonstrated that administration of the second generation antipsychotic, olanzapine, via intraperitoneal injection or oral gavage interacted with gut microbiota and caused body weight gain, increased plasma free fatty acids, infiltration of macrophages in adipose tissue, and deposition of visceral fat in both rat and mouse models [97, 99]. Since antipsychotic drugs are important as treatment for psychiatric diseases, ongoing research is necessary to elucidate the plausible mechanisms involved in antipsychotic-induced MetS so that this side-effect can be avoided. The comparison between various types of MetS animal model has been summarized (Table 5).
Table 5.
Overall merits and caveats of various types of MetS animal model
| Diet-induced model of MetS Examples: Induction by fructose drinking water, high fat diet, and high-carbohydrate high-fat diet | |
| Pros | Cons |
| ➢ Suitable for the investigations of non-genetic lifestyle-dependent MetS in humans ➢ Inexpensive (dependent on the kind of diet) |
➢ Delayed onset of MetS ➢ A lengthy duration of diet regimen (usually takes up to 16 weeks) |
| Genetic model of MetS Examples: ZF rat, ZDF rat, DS/obese rat, Koletsky rat, POUND mouse | |
| Pros | Cons |
| ➢ Severe and spontaneous occurring MetS | ➢ Do not resemble the criteria of MetS in humans with intact leptin receptor gene ➢ Mutation in leptin or leptin receptor gene rarely occur in humans ➢ Costly ➢ Mutations/deficiencies in animals are not easily manipulated |
| Drug/chemical-induced model of MetS Example: Induction by glucocorticoids and antipsychotic drugs | |
| Pros | Cons |
| ➢ Suitable for the investigations of drug-related MetS in human ➢ Inexpensive |
➢ Delayed onset of MetS |
Other animal models of MetS
Other animal models of MetS are available despite those typical laboratory rodent models, such as the use of guinea pig, swine, Nile rat, and Sand rat. A male Hartley guinea pig model of MetS was successfully developed by exposure to high-fat, high-sucrose or high-fat high-fructose diet for 150 days [100, 101]. Additionally, Ossabaw swine model of MetS was developed after fed with high-fat, high-cholesterol atherogenic diet, evidenced by obesity, elevated arterial pressure, glucose intolerance, and hyperinsulinemia [102]. Nile rat (Arvicanthis niloticus) was introduced as a novel model of MetS that experiences onset of hyperglycemia, hypertension, dyslipidemia, and abdominal fat accumulation by age of one when rats were given laboratory chow diet [103]. Sand rat (Psammomys obesus), found mostly in North Africa, spontaneously develops obesity and diabetes under laboratory diets [104]. These MetS features have not been observed among the wild type of Nile and Sand rats.
Conclusions
In conclusion, the advantage of using animal models to study MetS is the ability to monitor histological, functional, biochemical, and morphological changes of MetS, which is difficult to conduct in humans. Subsequent studies are encouraged using combination or modification of existing established methods in order to successfully develop an animal model of MetS with the desired metabolic changes. Apart from pathophysiological similarity with human MetS, an excellent animal model should also be reproducible, simple, reliable, and affordable with minimal disadvantages.
Acknowledgements
The authors thank Universiti Kebangsaan Malaysia for supporting this work through research grant DIP-2014-040.
Funding
This work is fully supported by research grant (DIP-2014-040) from Universiti Kebangsaan Malaysia.
Availability of data and materials
Deposition of data and data sharing are not applicable to this review as no datasets were generated or analyzed.
Authors’ contributions
SKW performed literature search and drafted the manuscript; KYC, FHS, FA, and SIN provided critical review for the manuscript; SIN gave final approval for the publication of this manuscript.
Competing interests
The authors declare no competing interests.
Consent for publication
All contributing authors declare their consent for the final accepted version of manuscript to be considered for publication in Nutrition and Metabolism.
Ethics approval and consent to participate
Ethics approval and consent to participate are not applicable to this review.
Abbreviations
- CVD
Cardiovascular disease
- db/db mice
leptin receptor-deficient mice
- DS/lean rats
DahlS.Z-Lepr + /Lepr + rats
- DS/obese rats
DahlS.Z-Lepr fa /Lepr fa rats
- GLUT-2
glucose transporter-2
- GLUT-4
glucose transporter-4
- GK rats
Goto-Kakizaki rats
- HDL
High density lipoproteins
- IDF
International Diabetes Federation
- IRS-1
Insulin receptor substrate-1
- LDL
Low density lipoproteins
- MetS
Metabolic syndrome
- mTORC1
Mammalian target of rapamycin complex-1
- NCEP ATP III
National Cholesterol Education Programme Adult Treatment Panel III
- ob/ob mice
Leptin-deficient mice
- SHR
Spontaneous hypertensive rat
- VLDL
Very low density lipoproteins
- WHO
World Health Organization
- ZDF rats
Zucker diabetic fatty rats
- ZF rats
Zucker fatty rats
References
- 1.Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640–5. doi: 10.1161/CIRCULATIONAHA.109.192644. [DOI] [PubMed] [Google Scholar]
- 2.Kaur J. A comprehensive review on metabolic syndrome. Cardiol Res Pract. 2014;2014:21. doi: 10.1155/2014/943162. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3.Alberti G, Zimmet P, Shaw J. The IDF Consensus Worldwide Definition of the Metabolic Syndrome. Brussels: International Diabetes Federation; 2006. [Google Scholar]
- 4.Bee YT, Jr, Haresh KK, Rajibans S. Prevalence of Metabolic Syndrome among Malaysians using the International Diabetes Federation, National Cholesterol Education Program and Modified World Health Organization Definitions. Malays J Nutr. 2008;14:65–77. [PubMed] [Google Scholar]
- 5.Rampal S, Mahadeva S, Guallar E, Bulgiba A, Mohamed R, Rahmat R, et al. Ethnic differences in the prevalence of metabolic syndrome: results from a multi-ethnic population-based survey in Malaysia. PLoS One. 2012;7:e46365. doi: 10.1371/journal.pone.0046365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tan AK, Dunn RA, Yen ST. Ethnic disparities in metabolic syndrome in malaysia: an analysis by risk factors. Metab Syndr Relat Disord. 2011;9:441–51. doi: 10.1089/met.2011.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Das M, Pal S, Ghosh A. Family history of type 2 diabetes and prevalence of metabolic syndrome in adult Asian Indians. J Cardiovasc Dis Res. 2012;3:104–8. doi: 10.4103/0975-3583.95362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ranasinghe P, Cooray DN, Jayawardena R, Katulanda P. The influence of family history of hypertension on disease prevalence and associated metabolic risk factors among Sri Lankan adults. BMC Public Health. 2015;15:576. doi: 10.1186/s12889-015-1927-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonomini F, Rodella LF, Rezzani R. Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis. 2015;6:109–20. doi: 10.14336/AD.2014.0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Veronica G, Esther RR. Aging, metabolic syndrome and the heart. Aging Dis. 2012;3:269–79. [PMC free article] [PubMed] [Google Scholar]
- 11.Ghezzi AC, Cambri LT, Botezelli JD, Ribeiro C, Dalia RA, de Mello MA. Metabolic syndrome markers in wistar rats of different ages. Diabetol Metab Syndr. 2012;4:16. doi: 10.1186/1758-5996-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wagner A, Dallongeville J, Haas B, Ruidavets JB, Amouyel P, Ferrieres J, et al. Sedentary behaviour, physical activity and dietary patterns are independently associated with the metabolic syndrome. Diabetes Metab. 2012;38:428–35. doi: 10.1016/j.diabet.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 13.Lakka HM, Laaksonen DE, Lakka TA, Niskanen LK, Kumpusalo E, Tuomilehto J, et al. The metabolic syndrome and total and cardiovascular disease mortality in middle-aged men. JAMA. 2002;288:2709–16. doi: 10.1001/jama.288.21.2709. [DOI] [PubMed] [Google Scholar]
- 14.Aschner P. Metabolic syndrome as a risk factor for diabetes. Expert Rev Cardiovasc Ther. 2010;8:407–12. doi: 10.1586/erc.10.13. [DOI] [PubMed] [Google Scholar]
- 15.Vanni E, Bugianesi E, Kotronen A, De Minicis S, Yki-Jarvinen H, Svegliati-Baroni G. From the metabolic syndrome to NAFLD or vice versa? Dig Liver Dis. 2010;42:320–30. doi: 10.1016/j.dld.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 16.Johansen D, Stocks T, Jonsson H, Lindkvist B, Bjorge T, Concin H, et al. Metabolic factors and the risk of pancreatic cancer: a prospective analysis of almost 580,000 men and women in the Metabolic Syndrome and Cancer Project. Cancer Epidemiol Biomarkers Prev. 2010;19:2307–17. doi: 10.1158/1055-9965.EPI-10-0234. [DOI] [PubMed] [Google Scholar]
- 17.Haggstrom C, Stocks T, Rapp K, Bjorge T, Lindkvist B, Concin H, et al. Metabolic syndrome and risk of bladder cancer: prospective cohort study in the metabolic syndrome and cancer project (Me-Can) Int J Cancer. 2011;128:1890–8. doi: 10.1002/ijc.25521. [DOI] [PubMed] [Google Scholar]
- 18.Esposito K, Chiodini P, Colao A, Lenzi A, Giugliano D. Metabolic syndrome and risk of cancer: a systematic review and meta-analysis. Diabetes Care. 2012;35:2402–11. doi: 10.2337/dc12-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bjorge T, Lukanova A, Jonsson H, Tretli S, Ulmer H, Manjer J, et al. Metabolic syndrome and breast cancer in the me-can (metabolic syndrome and cancer) project. Cancer Epidemiol Biomarkers Prev. 2010;19:1737–45. doi: 10.1158/1055-9965.EPI-10-0230. [DOI] [PubMed] [Google Scholar]
- 20.Panchal SK, Brown L. Rodent models for metabolic syndrome research. J Biomed Biotechnol. 2011;2011:351982. doi: 10.1155/2011/351982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kasim-Karakas SE, Vriend H, Almario R, Chow LC, Goodman MN. Effects of dietary carbohydrates on glucose and lipid metabolism in golden Syrian hamsters. J Lab Clin Med. 1996;128:208–13. doi: 10.1016/S0022-2143(96)90013-X. [DOI] [PubMed] [Google Scholar]
- 22.Senaphan K, Kukongviriyapan U, Sangartit W, Pakdeechote P, Pannangpetch P, Prachaney P, et al. Ferulic acid alleviates changes in a rat model of metabolic syndrome induced by high-carbohydrate, high-fat diet. Nutrients. 2015;7:6446–64. doi: 10.3390/nu7085283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suman RK, Ray Mohanty I, Borde MK, Maheshwari U, Deshmukh YA. Development of an experimental model of diabetes co-existing with metabolic syndrome in rats. Adv Pharmacol Sci. 2016;2016:11. doi: 10.1155/2016/9463476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujita Y, Maki K. High-fat diet-induced obesity triggers alveolar bone loss and spontaneous periodontal disease in growing mice. BMC Obes. 2015;3:1. doi: 10.1186/s40608-016-0082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y. Metabolism: Carbohydrate. In: Mooren FC, editor. Encyclopedia of Exercise Medicine in Health and Disease. Berlin: Springer Berlin Heidelberg; 2012. pp. 570–3. [Google Scholar]
- 26.Bessesen DH. The role of carbohydrates in insulin resistance. J Nutr. 2001;131:2782s–6s. doi: 10.1093/jn/131.10.2782S. [DOI] [PubMed] [Google Scholar]
- 27.McClenaghan NH. Determining the relationship between dietary carbohydrate intake and insulin resistance. Nutr Res Rev. 2005;18:222–40. doi: 10.1079/NRR2005109. [DOI] [PubMed] [Google Scholar]
- 28.Gadgil MD, Appel LJ, Yeung E, Anderson CA, Sacks FM, Miller ER., 3rd The effects of carbohydrate, unsaturated fat, and protein intake on measures of insulin sensitivity: results from the OmniHeart trial. Diabetes Care. 2013;36:1132–7. doi: 10.2337/dc12-0869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poudyal H, Panchal S, Brown L. Comparison of purple carrot juice and beta-carotene in a high-carbohydrate, high-fat diet-fed rat model of the metabolic syndrome. Br J Nutr. 2010;104:1322–32. doi: 10.1017/S0007114510002308. [DOI] [PubMed] [Google Scholar]
- 30.Panchal SK, Poudyal H, Iyer A, Nazer R, Alam A, Diwan V, et al. High-carbohydrate high-fat diet-induced metabolic syndrome and cardiovascular remodeling in rats. J Cardiovasc Pharmacol. 2011;57:51–64. doi: 10.1097/FJC.0b013e3181feb90a. [DOI] [PubMed] [Google Scholar]
- 31.Hao L, Lu X, Sun M, Li K, Shen L, Wu T. Protective effects of L-arabinose in high-carbohydrate, high-fat diet-induced metabolic syndrome in rats. Food Nutr Res. 2015;59:28886. doi: 10.3402/fnr.v59.28886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Natale C, Annuzzi G, Bozzetto L, Mazzarella R, Costabile G, Ciano O, et al. Effects of a plant-based high-carbohydrate/high-fiber diet versus high-monounsaturated fat/low-carbohydrate diet on postprandial lipids in type 2 diabetic patients. Diabetes Care. 2009;32:2168–73. doi: 10.2337/dc09-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bray GA. How bad is fructose? Am J Clin Nutr. 2007;86:895–6. doi: 10.1093/ajcn/86.4.895. [DOI] [PubMed] [Google Scholar]
- 34.Bantle JP, Wylie-Rosett J, Albright AL, Apovian CM, Clark NG, Franz MJ, et al. Nutrition recommendations and interventions for diabetes: a position statement of the American Diabetes Association. Diabetes Care. 2008;31(Suppl 1):S61–78. doi: 10.2337/dc08-S061. [DOI] [PubMed] [Google Scholar]
- 35.Jurgens H, Haass W, Castaneda TR, Schurmann A, Koebnick C, Dombrowski F, et al. Consuming fructose-sweetened beverages increases body adiposity in mice. Obes Res. 2005;13:1146–56. doi: 10.1038/oby.2005.136. [DOI] [PubMed] [Google Scholar]
- 36.Basciano H, Federico L, Adeli K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr Metab. 2005;2:5. doi: 10.1186/1743-7075-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elliott SS, Keim NL, Stern JS, Teff K, Havel PJ. Fructose, weight gain, and the insulin resistance syndrome. Am J Clin Nutr. 2002;76:911–22. doi: 10.1093/ajcn/76.5.911. [DOI] [PubMed] [Google Scholar]
- 38.Rizkalla SW. Health implications of fructose consumption: A review of recent data. Nutr Metab (Lond) 2010;7:82. doi: 10.1186/1743-7075-7-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson RJ, Segal MS, Sautin Y, Nakagawa T, Feig DI, Kang DH, et al. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am J Clin Nutr. 2007;86:899–906. doi: 10.1093/ajcn/86.4.899. [DOI] [PubMed] [Google Scholar]
- 40.Schulze MB, Manson JE, Ludwig DS, Colditz GA, Stampfer MJ, Willett WC, et al. Sugar-sweetened beverages, weight gain, and incidence of type 2 diabetes in young and middle-aged women. JAMA. 2004;292:927–34. doi: 10.1001/jama.292.8.927. [DOI] [PubMed] [Google Scholar]
- 41.Thirunavukkarasu V, Anitha Nandhini AT, Anuradha CV. Lipoic acid attenuates hypertension and improves insulin sensitivity, kallikrein activity and nitrite levels in high fructose-fed rats. J Comp Physiol B. 2004;174:587–92. doi: 10.1007/s00360-004-0447-z. [DOI] [PubMed] [Google Scholar]
- 42.Sanchez-Lozada LG, Tapia E, Jimenez A, Bautista P, Cristobal M, Nepomuceno T, et al. Fructose-induced metabolic syndrome is associated with glomerular hypertension and renal microvascular damage in rats. Am J Physiol Renal Physiol. 2007;292:F423–9. doi: 10.1152/ajprenal.00124.2006. [DOI] [PubMed] [Google Scholar]
- 43.Elzouki AY, Stapleton F, Harfi H, Oh W, Whitley R, Nazer H. Textbook of clinical pediatrics: Springer Science & Business Media. 2011. [Google Scholar]
- 44.Williams G, Pickup J. The Handbook of Diabetes. 3. Oxford: Blackwell Science Limited; 2003. [Google Scholar]
- 45.Aguilera AA, Diaz GH, Barcelata ML, Guerrero OA, Ros RM. Effects of fish oil on hypertension, plasma lipids, and tumor necrosis factor-alpha in rats with sucrose-induced metabolic syndrome. J Nutr Biochem. 2004;15:350–7. doi: 10.1016/j.jnutbio.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 46.Davidoff AJ, Mason MM, Davidson MB, Carmody MW, Hintz KK, Wold LE, et al. Sucrose-induced cardiomyocyte dysfunction is both preventable and reversible with clinically relevant treatments. Am J Physiol Endocrinol Metab. 2004;286:E718–24. doi: 10.1152/ajpendo.00358.2003. [DOI] [PubMed] [Google Scholar]
- 47.Hintz KK, Aberle NS, Ren J. Insulin resistance induces hyperleptinemia, cardiac contractile dysfunction but not cardiac leptin resistance in ventricular myocytes. Int J Obes Relat Metab Disord. 2003;27:1196–203. doi: 10.1038/sj.ijo.0802389. [DOI] [PubMed] [Google Scholar]
- 48.Wold LE, Dutta K, Mason MM, Ren J, Cala SE, Schwanke ML, et al. Impaired SERCA function contributes to cardiomyocyte dysfunction in insulin resistant rats. J Mol Cell Cardiol. 2005;39:297–307. doi: 10.1016/j.yjmcc.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 49.Vasanji Z, Cantor EJ, Juric D, Moyen M, Netticadan T. Alterations in cardiac contractile performance and sarcoplasmic reticulum function in sucrose-fed rats is associated with insulin resistance. Am J Physiol Cell Physiol. 2006;291:C772–80. doi: 10.1152/ajpcell.00086.2005. [DOI] [PubMed] [Google Scholar]
- 50.Pang X, Zhao J, Zhang W, Zhuang X, Wang J, Xu R, et al. Antihypertensive effect of total flavones extracted from seed residues of Hippophae rhamnoides L. in sucrose-fed rats. J Ethnopharmacol. 2008;117:325–31. doi: 10.1016/j.jep.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 51.Oron-Herman M, Kamari Y, Grossman E, Yeger G, Peleg E, Shabtay Z, et al. Metabolic syndrome: comparison of the two commonly used animal models. Am J Hypertens. 2008;21:1018–22. doi: 10.1038/ajh.2008.218. [DOI] [PubMed] [Google Scholar]
- 52.Brown MA, Storlien LH, Huang X-F, Tapsell LC, Else PL, Higgins JA, et al. Dietary fat and carbohydrate composition: Metabolic disease. 2010. [PubMed] [Google Scholar]
- 53.Nielsen S, Karpe F. Determinants of VLDL-triglycerides production. Curr Opin Lipidol. 2012;23:321–6. doi: 10.1097/MOL.0b013e3283544956. [DOI] [PubMed] [Google Scholar]
- 54.Wolfe RR, Klein S, Carraro F, Weber JM. Role of triglyceride-fatty acid cycle in controlling fat metabolism in humans during and after exercise. Am J Physiol. 1990;258:E382–9. doi: 10.1152/ajpendo.1990.258.2.E382. [DOI] [PubMed] [Google Scholar]
- 55.Buettner R, Scholmerich J, Bollheimer LC. High-fat diets: modeling the metabolic disorders of human obesity in rodents. Obesity (Silver Spring) 2007;15:798–808. doi: 10.1038/oby.2007.608. [DOI] [PubMed] [Google Scholar]
- 56.Halade GV, Rahman MM, Williams PJ, Fernandes G. High fat diet-induced animal model of age-associated obesity and osteoporosis. J Nutr Biochem. 2010;21:1162–9. doi: 10.1016/j.jnutbio.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu F, Du Y, Hang S, Chen A, Guo F, Xu T. Adipocytes regulate the bone marrow microenvironment in a mouse model of obesity. Mol Med Rep. 2013;8:823–8. doi: 10.3892/mmr.2013.1572. [DOI] [PubMed] [Google Scholar]
- 58.Zivkovic AM, German JB, Sanyal AJ. Comparative review of diets for the metabolic syndrome: implications for nonalcoholic fatty liver disease. Am J Clin Nutr. 2007;86:285–300. doi: 10.1093/ajcn/86.2.285. [DOI] [PubMed] [Google Scholar]
- 59.Ghibaudi L, Cook J, Farley C, van Heek M, Hwa JJ. Fat intake affects adiposity, comorbidity factors, and energy metabolism of sprague-dawley rats. Obes Res. 2002;10:956–63. doi: 10.1038/oby.2002.130. [DOI] [PubMed] [Google Scholar]
- 60.Fraulob JC, Ogg-Diamantino R, Fernandes-Santos C, Aguila MB, Mandarim-de-Lacerda CA. A mouse model of metabolic syndrome: insulin resistance, fatty liver and non-alcoholic fatty pancreas disease (NAFPD) in C57BL/6 mice fed a high fat diet. J Clin Biochem Nutr. 2010;46:212–23. doi: 10.3164/jcbn.09-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Graham LS, Tintut Y, Parhami F, Kitchen CM, Ivanov Y, Tetradis S, et al. Bone density and hyperlipidemia: the T-lymphocyte connection. J Bone Miner Res. 2010;25:2460–9. doi: 10.1002/jbmr.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li Y, Lu Z, Zhang X, Yu H, Kirkwood KL, Lopes-Virella MF, et al. Metabolic syndrome exacerbates inflammation and bone loss in periodontitis. J Dent Res. 2015;94:362–70. doi: 10.1177/0022034514561658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ahima RS, Flier JS. Leptin. Annu Rev Physiol. 2000;62:413–37. doi: 10.1146/annurev.physiol.62.1.413. [DOI] [PubMed] [Google Scholar]
- 64.Wang B, Chandrasekera PC, Pippin JJ. Leptin- and leptin receptor-deficient rodent models: relevance for human type 2 diabetes. Curr Diabetes Rev. 2014;10:131–45. doi: 10.2174/1573399810666140508121012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Van den Bergh A, Vanderper A, Vangheluwe P, Desjardins F, Nevelsteen I, Verreth W, et al. Dyslipidaemia in type II diabetic mice does not aggravate contractile impairment but increases ventricular stiffness. Cardiovasc Res. 2008;77:371–9. doi: 10.1093/cvr/cvm001. [DOI] [PubMed] [Google Scholar]
- 66.Ae Park S, Choi MS, Cho SY, Seo JS, Jung UJ, Kim MJ, et al. Genistein and daidzein modulate hepatic glucose and lipid regulating enzyme activities in C57BL/KsJ-db/db mice. Life Sci. 2006;79:1207–13. doi: 10.1016/j.lfs.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 67.Dong YF, Liu L, Kataoka K, Nakamura T, Fukuda M, Tokutomi Y, et al. Aliskiren prevents cardiovascular complications and pancreatic injury in a mouse model of obesity and type 2 diabetes. Diabetologia. 2010;53:180–91. doi: 10.1007/s00125-009-1575-5. [DOI] [PubMed] [Google Scholar]
- 68.Pico C, Sanchez J, Oliver P, Palou A. Leptin production by the stomach is up-regulated in obese (fa/fa) Zucker rats. Obes Res. 2002;10:932–8. doi: 10.1038/oby.2002.127. [DOI] [PubMed] [Google Scholar]
- 69.Aleixandre de Artinano A, Miguel Castro M. Experimental rat models to study the metabolic syndrome. Br J Nutr. 2009;102:1246–53. doi: 10.1017/S0007114509990729. [DOI] [PubMed] [Google Scholar]
- 70.Augstein P, Salzsieder E. Morphology of pancreatic islets: a time course of pre-diabetes in Zucker fatty rats. Methods Mol Biol. 2009;560:159–89. doi: 10.1007/978-1-59745-448-3_12. [DOI] [PubMed] [Google Scholar]
- 71.Lehnen AM, Rodrigues B, Irigoyen MC, De Angelis K, Schaan BD. Cardiovascular changes in animal models of metabolic syndrome. J Diabetes Res. 2013;2013:761314. doi: 10.1155/2013/761314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marsh SA, Powell PC, Agarwal A, Dell’Italia LJ, Chatham JC. Cardiovascular dysfunction in Zucker obese and Zucker diabetic fatty rats: role of hydronephrosis. Am J Physiol Heart Circ Physiol. 2007;293:H292–8. doi: 10.1152/ajpheart.01362.2006. [DOI] [PubMed] [Google Scholar]
- 73.Daniels A, Linz D, van Bilsen M, Rutten H, Sadowski T, Ruf S, et al. Long-term severe diabetes only leads to mild cardiac diastolic dysfunction in Zucker diabetic fatty rats. Eur J Heart Fail. 2012;14:193–201. doi: 10.1093/eurjhf/hfr166. [DOI] [PubMed] [Google Scholar]
- 74.Hattori T, Murase T, Ohtake M, Inoue T, Tsukamoto H, Takatsu M, et al. Characterization of a new animal model of metabolic syndrome: the DahlS. Z-Leprfa/Leprfa rat. Nutr Diabetes. 2011;1:e1. doi: 10.1038/nutd.2010.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Murase T, Hattori T, Ohtake M, Abe M, Amakusa Y, Takatsu M, et al. Cardiac remodeling and diastolic dysfunction in DahlS.Z-Lepr(fa)/Lepr(fa) rats: a new animal model of metabolic syndrome. Hypertens Res. 2012;35:186–93. doi: 10.1038/hr.2011.157. [DOI] [PubMed] [Google Scholar]
- 76.Akash MS, Rehman K, Chen S. Goto-Kakizaki rats: its suitability as non-obese diabetic animal model for spontaneous type 2 diabetes mellitus. Curr Diabetes Rev. 2013;9:387–96. doi: 10.2174/15733998113099990069. [DOI] [PubMed] [Google Scholar]
- 77.Beddow SA, Samuel VT. Fasting hyperglycemia in the Goto-Kakizaki rat is dependent on corticosterone: a confounding variable in rodent models of type 2 diabetes. Dis Model Mech. 2012;5:681–5. doi: 10.1242/dmm.009035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maekawa F, Fujiwara K, Kohno D, Kuramochi M, Kurita H, Yada T. Young adult-specific hyperphagia in diabetic Goto-kakizaki rats is associated with leptin resistance and elevation of neuropeptide Y mRNA in the arcuate nucleus. J Neuroendocrinol. 2006;18:748–56. doi: 10.1111/j.1365-2826.2006.01470.x. [DOI] [PubMed] [Google Scholar]
- 79.Goto Y, Kakizaki M, Masaki N. Spontaneous diabetes produced by selective breeding of normal Wistar rats. Proc Jpn Acad. 1975;51:80–5. [Google Scholar]
- 80.Goto Y, Kakizaki M. The spontaneous-diabetes rat: a model of noninsulin dependent diabetes mellitus. Proc Jpn Acad Ser B: Phys Biol Sci. 1981;57:381–4. doi: 10.2183/pjab.57.381. [DOI] [Google Scholar]
- 81.Okamoto K, Aoki K. Development of a strain of spontaneously hypertensive rats. Jpn Circ J. 1963;27:282–93. doi: 10.1253/jcj.27.282. [DOI] [PubMed] [Google Scholar]
- 82.Potenza MA, Marasciulo FL, Chieppa DM, Brigiani GS, Formoso G, Quon MJ, et al. Insulin resistance in spontaneously hypertensive rats is associated with endothelial dysfunction characterized by imbalance between NO and ET-1 production. Am J Physiol Heart Circ Physiol. 2005;289:H813–22. doi: 10.1152/ajpheart.00092.2005. [DOI] [PubMed] [Google Scholar]
- 83.Girard A, Madani S, El Boustani ES, Belleville J, Prost J. Changes in lipid metabolism and antioxidant defense status in spontaneously hypertensive rats and Wistar rats fed a diet enriched with fructose and saturated fatty acids. Nutrition. 2005;21:240–8. doi: 10.1016/j.nut.2004.04.022. [DOI] [PubMed] [Google Scholar]
- 84.Lorkowska B, Bartus M, Franczyk M, Kostogrys RB, Jawien J, Pisulewski PM, et al. Hypercholesterolemia does not alter endothelial function in spontaneously hypertensive rats. J Pharmacol Exp Ther. 2006;317:1019–26. doi: 10.1124/jpet.105.098798. [DOI] [PubMed] [Google Scholar]
- 85.The C57BL/6NCrl-Leprdb-lb/Crl Mouse: A Model for Metabolic Syndrome/Pre-Diabetes [http://www.criver.com/files/pdfs/rms/pound/rm_rm_r_pound_mouse_fact_sheet.aspx]. Accessed Jun 2016.
- 86.Mazen I, Amr K, Tantawy S, Farooqi IS, El Gammal M. A novel mutation in the leptin gene (W121X) in an Egyptian family. Mol Genet Metab Rep. 2014;1:474–6. doi: 10.1016/j.ymgmr.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fransson L, Franzén S, Rosengren V, Wolbert P, Sjöholm Å, Ortsäter H. β-cell adaptation in a mouse model of glucocorticoid-induced metabolic syndrome. J Endocrinol. 2013;219:231–41. doi: 10.1530/JOE-13-0189. [DOI] [PubMed] [Google Scholar]
- 88.Ferris HA, Kahn CR. New mechanisms of glucocorticoid-induced insulin resistance: make no bones about it. J Clin Invest. 2012;122:3854–7. doi: 10.1172/JCI66180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wong SK, Chin KY, Suhaimi FH, Ahmad F, Ima-Nirwana S. The Relationship between Metabolic Syndrome and Osteoporosis: A Review. Nutrients. 2016;8:347. [DOI] [PMC free article] [PubMed]
- 90.Yoon MS. The Emerging Role of Branched-Chain Amino Acids in Insulin Resistance and Metabolism. Nutrients. 2016;8:405. [DOI] [PMC free article] [PubMed]
- 91.Fransson L, Dos Santos C, Wolbert P, Sjoholm A, Rafacho A, Ortsater H. Liraglutide counteracts obesity and glucose intolerance in a mouse model of glucocorticoid-induced metabolic syndrome. Diabetol Metab Syndr. 2014;6:3. doi: 10.1186/1758-5996-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rafacho A, Giozzet VA, Boschero AC, Bosqueiro JR. Functional alterations in endocrine pancreas of rats with different degrees of dexamethasone-induced insulin resistance. Pancreas. 2008;36:284–93. doi: 10.1097/MPA.0b013e31815ba826. [DOI] [PubMed] [Google Scholar]
- 93.Christ-Crain M, Kola B, Lolli F, Fekete C, Seboek D, Wittmann G, et al. AMP-activated protein kinase mediates glucocorticoid-induced metabolic changes: a novel mechanism in Cushing’s syndrome. FASEB J. 2008;22:1672–83. doi: 10.1096/fj.07-094144. [DOI] [PubMed] [Google Scholar]
- 94.Shpilberg Y, Beaudry JL, D’Souza A, Campbell JE, Peckett A, Riddell MC. A rodent model of rapid-onset diabetes induced by glucocorticoids and high-fat feeding. Dis Model Mech. 2012;5:671–80. doi: 10.1242/dmm.008912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wofford MR, King DS, Harrell TK. Drug-induced metabolic syndrome. J Clin Hypertens (Greenwich) 2006;8:114–9. doi: 10.1111/j.1524-6175.2006.04751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wu C, Yuen J, Boyda HN, Procyshyn RM, Wang CK, Asiri YI, et al. An evaluation of the effects of the novel antipsychotic drug lurasidone on glucose tolerance and insulin resistance: a comparison with olanzapine. PLoS One. 2014;9:e107116. doi: 10.1371/journal.pone.0107116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Davey KJ, Cotter PD, O’Sullivan O, Crispie F, Dinan TG, Cryan JF, et al. Antipsychotics and the gut microbiome: olanzapine-induced metabolic dysfunction is attenuated by antibiotic administration in the rat. Transl Psychiatry. 2013;3:e309. doi: 10.1038/tp.2013.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chase P, Neumiller JJ. Antipsychotic-induced diabetes mellitus. US Pharm. 2012;37:39–44. [Google Scholar]
- 99.Morgan AP, Crowley JJ, Nonneman RJ, Quackenbush CR, Miller CN, Ryan AK, et al. The antipsychotic olanzapine interacts with the gut microbiome to cause weight gain in mouse. PLoS One. 2014;9:e115225. doi: 10.1371/journal.pone.0115225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Caillier B, Pilote S, Patoine D, Levac X, Couture C, Daleau P, et al. Metabolic syndrome potentiates the cardiac action potential-prolonging action of drugs: a possible ‘anti-proarrhythmic’ role for amlodipine. Pharmacol Res. 2012;65:320–7. doi: 10.1016/j.phrs.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 101.Patoine D, Levac X, Pilote S, Drolet B, Simard C. Decreased CYP3A expression and activity in guinea pig models of diet-induced metabolic syndrome: is fatty liver infiltration involved? Drug Metab Dispos. 2013;41:952–7. doi: 10.1124/dmd.112.050641. [DOI] [PubMed] [Google Scholar]
- 102.Neeb ZP, Edwards JM, Alloosh M, Long X, Mokelke EA, Sturek M. Metabolic syndrome and coronary artery disease in Ossabaw compared with Yucatan swine. Comp Med. 2010;60:300–15. [PMC free article] [PubMed] [Google Scholar]
- 103.Noda K, Melhorn MI, Zandi S, Frimmel S, Tayyari F, Hisatomi T, et al. An animal model of spontaneous metabolic syndrome: Nile grass rat. FASEB J. 2010;24:2443–53. doi: 10.1096/fj.09-152678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chaabo F, Pronczuk A, Maslova E, Hayes K. Nutritional correlates and dynamics of diabetes in the Nile rat (Arvicanthis niloticus): a novel model for diet-induced type 2 diabetes and the metabolic syndrome. Nutr Metab (Lond) 2010;7:29. doi: 10.1186/1743-7075-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shahraki MR, Harati M, Shahraki AR. Prevention of high fructose-induced metabolic syndrome in male wistar rats by aqueous extract of Tamarindus indica seed. Acta Med Iran. 2011;49:277–83. [PubMed] [Google Scholar]
- 106.Mahmoud AA, Elshazly SM. Ursodeoxycholic acid ameliorates fructose-induced metabolic syndrome in rats. PLoS One. 2014;9:e106993. doi: 10.1371/journal.pone.0106993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mansour SM, Zaki HF, Ezz-El-Din S. Beneficial effects of co-enzyme Q 10 and rosiglitazone in fructose-induced metabolic syndrome in rats. Bull Fac Pharm Cairo Univ. 2013;51:13–21. doi: 10.1016/j.bfopcu.2012.10.001. [DOI] [Google Scholar]
- 108.Mamikutty N, Thent ZC, Sapri SR, Sahruddin NN, Mohd Yusof MR, Haji Suhaimi F. The establishment of metabolic syndrome model by induction of fructose drinking water in male Wistar rats. BioMed Res Int. 2014;2014:263897. doi: 10.1155/2014/263897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Di Luccia B, Crescenzo R, Mazzoli A, Cigliano L, Venditti P, Walser JC, et al. Rescue of fructose-induced metabolic syndrome by antibiotics or faecal transplantation in a rat model of obesity. PLoS One. 2015;10:e0134893. doi: 10.1371/journal.pone.0134893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dobrian AD, Davies MJ, Prewitt RL, Lauterio TJ. Development of hypertension in a rat model of diet-induced obesity. Hypertension. 2000;35:1009–15. doi: 10.1161/01.HYP.35.4.1009. [DOI] [PubMed] [Google Scholar]
- 111.Rossmeisl M, Rim JS, Koza RA, Kozak LP. Variation in type 2 diabetes--related traits in mouse strains susceptible to diet-induced obesity. Diabetes. 2003;52:1958–66. doi: 10.2337/diabetes.52.8.1958. [DOI] [PubMed] [Google Scholar]
- 112.Gallou-Kabani C, Vige A, Gross MS, Rabes JP, Boileau C, Larue-Achagiotis C, et al. C57BL/6J and A/J mice fed a high-fat diet delineate components of metabolic syndrome. Obesity (Silver Spring) 2007;15:1996–2005. doi: 10.1038/oby.2007.238. [DOI] [PubMed] [Google Scholar]
- 113.Davidson EP, Coppey LJ, Dake B, Yorek MA. Effect of treatment of sprague dawley rats with AVE7688, enalapril, or candoxatril on diet-induced obesity. J Obes. 2011;2011:9. doi: 10.1155/2011/686952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pirih F, Lu J, Ye F, Bezouglaia O, Atti E, Ascenzi MG, et al. Adverse effects of hyperlipidemia on bone regeneration and strength. J Bone Miner Res. 2012;27:309–18. doi: 10.1002/jbmr.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Podrini C, Cambridge EL, Lelliott CJ, Carragher DM, Estabel J, Gerdin AK, et al. High-fat feeding rapidly induces obesity and lipid derangements in C57BL/6 N mice. Mamm Genome. 2013;24:240–51. doi: 10.1007/s00335-013-9456-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gancheva S, Zhelyazkova-Savova M, Galunska B, Chervenkov T. Experimental models of metabolic syndrome in rats. Scr Sci Med. 2015;47:14–21. [Google Scholar]
- 117.Dissard R, Klein J, Caubet C, Breuil B, Siwy J, Hoffman J, et al. Long term metabolic syndrome induced by a high fat high fructose diet leads to minimal renal injury in C57BL/6 mice. PLoS One. 2013;8:e76703. doi: 10.1371/journal.pone.0076703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Barrios-Ramos J, Garduño-Siciliano L, Loredo-Mendoza M, Chamorro-Cevallos G, Jaramillo-Flores M. A quick model for the induction of metabolic syndrome markers in rats. Intern Med. 2014;4:2. [Google Scholar]
- 119.Yang ZH, Miyahara H, Takeo J, Katayama M. Diet high in fat and sucrose induces rapid onset of obesity-related metabolic syndrome partly through rapid response of genes involved in lipogenesis, insulin signalling and inflammation in mice. Diabetol Metab Syndr. 2012;4:32. doi: 10.1186/1758-5996-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhou X, Han D, Xu R, Li S, Wu H, Qu C, et al. A model of metabolic syndrome and related diseases with intestinal endotoxemia in rats fed a high fat and high sucrose diet. PLoS One. 2014;9:e115148. doi: 10.1371/journal.pone.0115148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dubuc PU. The development of obesity, hyperinsulinemia, and hyperglycemia in ob/ob mice. Metabolism. 1976;25:1567–74. doi: 10.1016/0026-0495(76)90109-8. [DOI] [PubMed] [Google Scholar]
- 122.Mark AL, Shaffer RA, Correia ML, Morgan DA, Sigmund CD, Haynes WG. Contrasting blood pressure effects of obesity in leptin-deficient ob/ob mice and agouti yellow obese mice. J Hypertens. 1999;17:1949–53. doi: 10.1097/00004872-199917121-00026. [DOI] [PubMed] [Google Scholar]
- 123.Leonard BL, Watson RN, Loomes KM, Phillips AR, Cooper GJ. Insulin resistance in the Zucker diabetic fatty rat: a metabolic characterisation of obese and lean phenotypes. Acta Diabetol. 2005;42:162–70. doi: 10.1007/s00592-005-0197-8. [DOI] [PubMed] [Google Scholar]
- 124.Goto Y, Kakizaki M, Masaki N. Production of spontaneous diabetic rats by repetition of selective breeding. Tohoku J Exp Med. 1976;119:85–90. doi: 10.1620/tjem.119.85. [DOI] [PubMed] [Google Scholar]
- 125.Portha B, Giroix MH, Tourrel-Cuzin C, Le-Stunff H, Movassat J. The GK rat: a prototype for the study of non-overweight type 2 diabetes. Methods Mol Biol. 2012;933:125–59. doi: 10.1007/978-1-62703-068-7_9. [DOI] [PubMed] [Google Scholar]
- 126.Dickhout JG, Lee RM. Blood pressure and heart rate development in young spontaneously hypertensive rats. Am J Physiol. 1998;274:H794–800. doi: 10.1152/ajpheart.1998.274.3.H794. [DOI] [PubMed] [Google Scholar]
- 127.Tofovic SP, Jackson EK. Rat models of the metabolic syndrome. Methods Mol Med. 2003;86:29–46. doi: 10.1385/1-59259-392-5:29. [DOI] [PubMed] [Google Scholar]
- 128.Baldwin W, McRae S, Marek G, Wymer D, Pannu V, Baylis C, et al. Hyperuricemia as a mediator of the proinflammatory endocrine imbalance in the adipose tissue in a murine model of the metabolic syndrome. Diabetes. 2011;60:1258–69. doi: 10.2337/db10-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Deposition of data and data sharing are not applicable to this review as no datasets were generated or analyzed.