

Abstract
The elucidation of cancer pathogenesis has been hindered by limited access to patient samples, tumor heterogeneity and the lack of reliable model organisms. Characterized by their ability to self-renew indefinitely and differentiate into all cell lineages of an organism, pluripotent stem cells (PSCs), including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), provide a powerful and unlimited source to generate differentiated cells that can be used to study disease biology, facilitate drug discovery and development, and provide key insights for developing personalized therapies. This article reviews the recent developments and technologies converting PSCs into clinically relevant model systems for cancer research.
Keywords: Pluripotent Stem Cells, Cancer Disease Modeling, Genome Editing, Differentiation
Modeling Disease with Pluripotent Stem Cells
In 1998, Thomson and colleagues isolated human embryonic stem cells (ESCs) from blastocysts and developed a defined culture system to maintain the cells in vitro [1], opening a new avenue for medical research. Later, in 2006–2007, a breakthrough by the laboratories of Yamanaka and Thomson heralded the development of a new kind of pluripotent cells – induced pluripotent stem cells (iPSCs) [2–4]. Both groups demonstrated that somatic cells (e.g., dermal fibroblasts and peripheral blood) could be reprogrammed to an ES-like cell state by using a defined transcriptional factor cocktail (Yamanaka’s OCT4, SOX2, KLF4, c-MYC; or Thompson’s OCT4, SOX2, NANOG, LIN28) [5]. Over the past decade, subsequent advances facilitated the generation of iPSCs with chemicals, microRNA and modified RNA, or other gene delivery systems (retroviruses, adenoviruses, Sendai virus, transposons and plasmids) [5]. Applications for iPSCs include regenerative medicine, disease modelling, drug screening, and personalized therapy.
The unique combination of pluripotency and self-renewal distinguishes pluripotent stem cells (PSCs), including both ESCs and iPSCs, from all other cells (Figure 1A). The unlimited proliferative potential of these undifferentiated cells provides an arbitrarily large source of experimental material, while their pluripotency allows them to be coaxed into forming all adult tissue types. Well-defined protocols, including directed differentiation and organoid cultures have been developed to derive many major target tissues and cell types from PSCs of endodermal (liver, small intestine, stomach, thyroid and lung), mesodermal (muscle, bone, cartilage, kidney and blood) or ectodermal (epidermis, retinal and cerebral tissue) lineages [6–8].
Figure 1. Application of Pluripotent Stem Cells to Study Cancer-Associated Genetic Alterations.
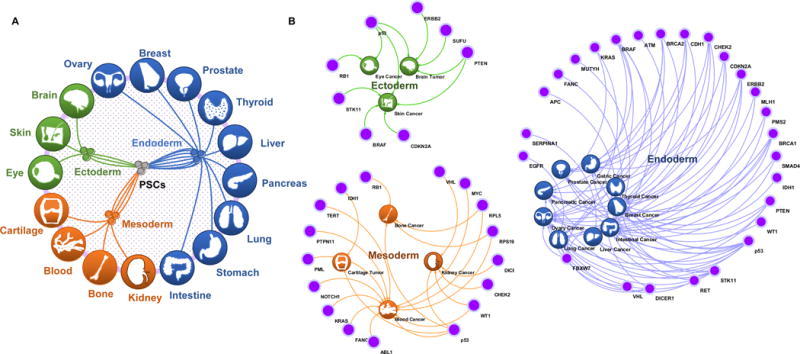
(A) PSCs are characterized by their capability to differentiate into all derivative cell types of the three germ layers. PSCs can form blood, kidney, bone and cartilage cells via the mesoderm; ovary, breast, prostate, thyroid, liver, pancreas, lung, stomach, and intestine cells via the endoderm; and brain, eye and skin cells via the ectoderm. (B) Loss of tumor suppressor genes, such as p53 mutation; or acquisition of oncogenes, such as ERBB2 amplification or ABL1 translocation, results in both hereditary and sporadic cancers in ectodermal, mesodermal, and endodermal tissues.
PSCs provide unparalleled advantages as a model system, allowing investigators to study a cell continuously from the moment it differentiates from a multipotent progenitor into a differentiated cell type of interest. The relevant genetic background for the model system can be introduced into PSCs using two primary strategies. In one approach, somatic cells from patients with genetic disorders are used to derive iPSC lines. These patient-derived iPSCs and their derivative differentiated tissues are then used to recapitulate a disease phenotype in vitro or shed light on disease-relevant mechanisms [9]. This approach has been applied successfully to study the genetic causes of neurodegeneration [10–12], mental disorder [13], heart disease [14–17], and metabolic disorders [18].
Alternatively, a genetic disease trait can be directly introduced into PSCs. This approach is aided greatly by recent major developments in gene delivery systems such as helper-dependent adenoviral vectors (HDAdVs) [19], adeno-associated viruses (AAVs) [20], gene manipulation approaches (RNAi [21, 22] and piggyBac transposases [23]), and genome editing tools (Zinc finger nuclease (ZFNs) [23–25], Transcription activator-like effector nucleases (TALENs) [26, 27], and clustered, regularly interspaced, short palindromic repeat/Cas9 (CRISPR/Cas9) [28, 29]). These technologies allow introducing alterations (deletions, amplifications, mutations or gene fusions) into ESCs or iPSCs of an arbitrary genetic background, allowing studying human monogenic and complex diseases as the pathology develops.
While the field of PSC-derived cancer research remains in its infancy, a number of PSC-derived cell lines have been generated to model disorders with a cancer predisposition (Table 1). Several groups have applied patient-derived iPSCs and/or engineered PSCs to phenocopy cancer features, explore disease mechanisms and screen potential therapeutic drugs [30–34]. Their experience highlights the potential of human PSCs in cancer studies by overcoming limitations related to availability of patient samples or translation of results from animal models or cell lines with inappropriate genetic backgrounds. Here, we outline the existing PSC cancer models and their potential applications to understanding cancer biology. We discuss how recent developments (e.g., genome-editing and cell differentiation technologies) in PSCs have transformed our understanding of cancer biology and paved the way for new therapeutic strategies. Finally we review some of the most promising model systems in which we anticipate this powerful technology will be applied.
Table 1.
Established PSCs models of cancer or diseases that predispose to cancer.
| Disease | Inheritance | Prevalence | Defected Gene | Cancer Type | Key Model Findings in Cancer | References |
|---|---|---|---|---|---|---|
| Alpha-1 antitrypsin deficiency | Autosomal codominant | 1 in 1,500 to 3,500 | SERPINA1 | Liver cancer | N/A | [18, 23, 77] |
| Ataxia-telangiectasia | Autosomal recessive | 1 in 40,000 to 100,000 | ATM | Leukemia and lymphoma | N/A | [78] |
| Diamond-Blackfan anemia | Autosomal dominant | 1 in 5,000,000 to 7,000,000 | RPL5, RPL11, RPL35A, RPS7, RPS10, RPS17, RPS19, RPS24, and RPS26 | Osteosarcoma | N/A | [79, 80] |
| Diffuse intrinsic pontine gliomas | Sporadic | 1 in 250,000 to 500,000 | H3.3 | Brain tumor | Undifferentiated epigenome structure and a primitive stem cell gene signature in NPCs with H3.3K27M expression, PDGFRA activation and p53 loss | [33] |
| Down syndrome | Sporadic | 1 in 800 | Chromosome 21 | Leukemia | N/A | [11, 81] |
| Dyskeratosis congenital | Autosomal dominant or recessive | 1 in 1,000,000 | DKC1, TERC, TERT, and TINF2 | Leukemia | N/A | [82, 83] |
| Dystrophic epidermolysis bullosa | Autosomal recessive | 1 in 150,000 to 1,000,000 | COL7A1 | Skin Cancer | N/A | [84, 85] |
| Fanconi anemia | Autosomal recessive | 1 in 160,000 | FANC genes, BRCA2, BRIP1, PALB2, and RAD51C | Leukemia | N/A | [86] |
| Glioblastoma multiforme | Sporadic | 1 in 33,000 to 50,000 | EGFR, PIK3CA, PTEN, and TP53 | Brain Tumor | Elevated PAX7 and GBM-associated gene signature in PTEN-deficient NSCs | [34] |
| Li-Fraumeni syndrome | Autosomal dominant | 1 in 20,000 | TP53 and CHEK2 | Osteosarcoma, breast cancer, brain tumor, and soft tissue sarcoma | Impaired expression of H19 and osteosarcoma signature in LFS osteoblasts | [30] |
| del(7q)-Myelodysplastic syndrome | Sporadic | 1 in 100,000 | Chromosome 7q | Leukemia | Impaired myeloid lineage differentiation in del(7q) iPSC cells dependent on HIPK2, ATP6V0E2, LUC7L2 and EZH2 | [32] |
| Noonan syndrome | Autosomal dominant | 1 in 1,000 to 2,500 | PTPN11, BRAF, KRAS, NRAS, RAF1, and SOS1 | Leukemia | Proliferation of CD33+ myeloid cells and elevated miR-15a and miR-223 in NS/JMML hematopoietic cells | [31] |
| Polycythaemia vera | Sporadic | 1 in 2,000 | JAK2 and TET2 | Leukemia | N/A | [87] |
| Shwachman-Diamond syndrome | Autosomal recessive | 1 in 100,000 to 1,000,000 | SBDS | Leukemia | N/A | [11, 88] |
| Werner syndrome | Autosomal recessive | 1 in 200,000 | WRN | Skin cancer, soft tissue sarcoma | N/A | [89, 90] |
| Wilms Tumor | Sporadic or Hereditary | 1 in 10,000 | WT1 | Kidney cancer | N/A | [91] |
Modeling Cancer with Pluripotent Stem Cells
Over the past forty years, researchers have used cancer cell lines, patient samples and small organism models (e.g., fruit fly, zebrafish and mouse) to study the molecular mechanisms of cancer initiation, progression and metastasis, but the complexity of the cancer genome and differences among species frequently limit clinical translation. Although there are iPSC models for a number of genetic diseases that predispose to cancer, to date, relatively few of these systems have been used to explore mechanisms of oncogenesis. We discuss several examples of these pioneer models below.
Li-Fraumeni syndrome (LFS)
LFS is an autosomal dominant inherited cancer syndrome that is characterized by early onset of a variety of tumor types, including soft tissue sarcoma and osteosarcoma, breast cancer, brain tumors, leukemia, and adrenocortical carcinoma [35]. Our group established a model of LFS using patient-derived iPSCs to delineate mechanisms of mutant p53 in osteosarcoma [30]. In this system, osteoblasts differentiated from LFS iPSC-derived mesenchymal stem cells recapitulate osteosarcoma features, including defective osteoblastic differentiation and tumorigenic ability. Gene expression in LFS osteoblasts is also similar to the expression profile in primary osteosarcomas, particularly the more aggressive phenotypes. LFS-derived osteoblasts are free of cytogenetic rearrangements, permitting study of early oncogenic mechanisms prior to accumulation of secondary genomic alterations. Expression of the long non-coding RNA H19 had been previously linked to p53 activity[36]; and transcriptome analysis suggested impaired expression of H19 in LFS osteoblasts and further functional studies showed that H19 is essential for normal osteogenesis and inhibition of tumorigenesis. The LFS iPSC disease model uncovered a previously unidentified role of p53 in osteogenic differentiation defects and tumorigenesis.
Noonan syndrome (NS)
NS is an autosomal dominant disorder characterized by a wide spectrum of congenital heart abnormalities, short stature, facial dimorphism and predisposition to hematological malignancies. A subset of NS patients develop juvenile myelomonocytic leukemia (JMML), an aggressive myelodysplastic and myeloproliferative neoplasm [37]. Mulero-Navarro et al. investigated the molecular mechanisms involved in NS-associated JMML harboring PTPN11 mutations using hematopoietic cells derived from NS/JMML patient specific-iPSCs [31]. These hematopoietic cells recapitulated several JMML characteristics including hypersensitivity to GM-CSF and increased myeloid population. Comparison of transcriptome profiles of controls and NS/JMML-derived CD33+ myeloid cells confirmed dysregulation of ERK and JAK-STAT signaling and proliferation of NS/JMML CD33+ myeloid cells. Expression levels of miR-15a and miR-223 were also elevated in these cells. Notably, dysregulation of miR-15a and miR-223 is commonly observed in mononuclear cells isolated from JMML patients harboring PTPN11 mutations. Using the NS/JMML iPSC model, Mulero-Navarro et al. demonstrated that inhibition of these miRNAs could restore normal myelopoiesis, providing a novel therapeutic target for PTPN11-mutated JMML.
Myelodysplastic syndrome (MDS)
MDS is a bone marrow disorder that leads to defective hematopoiesis and a disposition to develop anemia, cytopenia and leukemia. Sporadic loss of one copy of the long arm of chromosome 5 [del(5q)] and/or chromosome 7 [del(7q)] is a characteristic cytogenetic abnormality in MDS [38]. Kotini et al. established del(7q) MDS iPSCs from patient hematopoietic stem cells with loss of chromosome 7q and iPSCs derived from normal fibroblast isogenic controls [32]. iPSC cells with del(7q) recapitulated the phenotype of impaired myeloid lineage differentiation seen in MDS. This defective differentiation potential could be reproduced by engineering hemizygosity of definite 7q segments in normal iPSCs and could be rescued by spontaneous acquisition of an extra chromosome 7. Through phenotype-rescue screening, Kotini et al. identified HIPK2, ATP6V0E2, LUC7L2 and EZH2 as haploinsufficient genes involved in del(7q) MDS-associated hematopoietic defects.
Diffuse intrinsic pontine gliomas (DIPGs)
DIPGs are rare highly aggressive pediatric brain tumors that arise from glial tissue. Somatic p.Lys27Met substitution in histone 3.3 (H3.3K27M) is commonly detected in patients with DIPGs and is associated with poor survival [39, 40]. Funato at al. engineered human ESC-derived neural progenitor cells (NPCs) with heterozygous H3.3K27M mutations [33]. To mimic genetic alterations found in clinical samples of H3.3K27M-mutated DIPGs, ESC-derived NPCs were transduced with lentiviruses also carrying constitutively active PDGFRA(D842V) and p53 shRNA. In this NPC model, H3.3K27M expression synergized with PDGFRA activation and p53 loss, culminating in neoplastic transformation. Genome-wide analyses of H3.3K27M-transformed NPCs revealed that they maintain both an undifferentiated epigenome structure and a primitive stem cell gene signature, enabling their tumorigenic potential. Using a small-molecule chemical library screen of compounds targeting epigenetic regulators, they identified the MEN1 inhibitor MI-2 as a potential drug for the subset of DIPGs harboring the H3.3K27M mutation. This study demonstrates the potential of PSCs for drug screening.
Glioblastoma multiforme (GBM)
GBM, also known as grade IV astrocytoma, is a highly malignant brain tumor derived from glial cells. While GBMs are genetically very diverse, mutations in PTEN are common and correlate with increased invasion, drug resistance and tumor recurrence [41]. Duan et al. engineered PTEN-deficient ESCs using a TALEN-based genome editing methodology and derived neural stem cells (NSCs) to model GBM [34]. PTEN-deficient NSCs displayed the GBM-associated gene signature and formed intracranial tumors in vivo. Duan et al. found that elevated levels of PAX7 contributed to neoplastic transformation by producing more aggressive phenotypes. Elevated PAX7 expression can be explained directly by PTEN deficiency since PTEN interacts with CREB/CBP and co-occupies the PAX7 promoter. Screening for anti-cancer compounds in PTEN-deficient NSCs suggested mitomycin C as a potential drug.
Directed Differentiation and Organoids
The potential of PSCs in modeling cancer is critically dependent on availability of defined methods to differentiate PSCs into the tissues from which tumors arise. The generation of specific cell or tissue types by directed differentiation and organoids is a fast-growing field in stem cell research [6–8].
Directed differentiation
Directed differentiation is the application of a temporally defined set of external factors or culture conditions in order to produce a cell population enriched for a desired lineage. For example, we have employed a directed differentiation protocol to derive mesenchymal stem cells (MSCs) from p53-mutant iPSCs. While the MSCs exhibited no obvious deficits and were free of chromosomal abnormalities, key oncogenic features representative of osteosarcoma emerged after their subsequent differentiation into osteoblasts [30].
PSC differentiation protocols have been established for generation of hepatic cells [42–44], intestinal tissue [45], thyroid follicular cells [46], airway epithelial cells [47–49], renal progenitor cells [50, 51], retinal cells [52–55], epithelial stem cells [56], epidermal keratinocytes [57], myeloid cells [58, 59], neurons [34, 60], MSCs [61], and melanocytes [62], among many others. Application of these protocols to cancer patient-derived iPSCs and engineered ESCs should allow research of cancers of these organs.
Directed PSC differentiation protocols pose particular challenges to model cancer. PSCs can form teratomas, a rare tumor type from a normal genetic background. While differentiated cells derived from PSCs do not have this tumorigenic potential, even small contamination of desired differentiated cells with pluripotent cells can give the false appearance of tumorigenic properties. An effective differentiation protocol and purification scheme is therefore essential for any cancer study.
Despite these advances, there is a large unmet need for directed differentiation protocols for a broader set of cancer types, including breast, prostate and ovary. As directed differentiation techniques from PSCs continue to develop, the versatility of using PSCs in cancer modeling will also expand.
Organoid culture
Recent advances in three-dimensional (3D) culture techniques combined with existing differentiation protocols have enabled the generation of PSC-derived specific tissue or progenitor cells within a self-organized assembly known as an organoid [7, 8]. Compared to traditional two-dimensional (2D) culture systems, these 3D organoid cultures mimic better their in vivo PSC-derived counterparts, hence positioning the technology as a powerful tool for studying human development and modeling disease. Lancaster at al. [63] generated 3D cerebral organoids by differentiation of human PSCs. Matrigel droplets containing cerebral organoids were transferred into a spinning bioreactor, enabling a rapid, longer and more abundant formation of 3D brain tissue. These “mini brain” systems facilitate the study of human brain development and have been used to model microcephaly, among other neurodevelopmental disorders. With the introduction of appropriate mutations, these brain organoid systems have the potential to enhance our understanding of brain tumor biology.
3D gastrointestinal organoids were also derived from PSCs by Wells et al. [45] and closely mimic the in vivo intestinal epithelium, with both villus-like and crypt-like structures, consisting of differentiated enterocytes, goblet cells, as well as intestinal stem cells. Another protocol for generating PSC-derived gastric organoids [64] was later published by the same group. These 3D “mini-stomach” systems were used to model H. pylori infection, shedding light on the pathogenesis of this common disease. Organoids generated from human intestinal stem cells were also used to recapitulate the colorectal adenoma-to-carcinoma transition by Matano et al., highlighting the potential role of 3D organoid culture in studying gastrointestinal oncogenesis [65].
PSC-derived 3D culture and organoid systems have been used to model the liver bud [66, 67], lung [47], and pancreas [68] from endoderm; the kidney [69–72] from mesoderm; and the optic cup [73–75] from ectoderm. These methodologies hold substantial potential for investigating the distinct tumor types derived from these now-accessible cell lineages.
Concluding Remarks
PSC disease models have not only succeeded in replicating disease phenotypes but have also begun to find applications in the understanding of disease biology and in the development of novel therapies (see outstanding questions). Applications of PSCs to cancer will be advantageous to (1) model disease phenotypes; (2) elucidate pathological mechanisms; (3) predict patient survival; (4) identify potential biomarkers and therapeutic targets; (5) discover haploinsufficient genes by functional mapping of disease-associated chromosomal loss; and (6) apply drug screening to identify potential compounds to rescue particular disease phenotypes.
The high levels of genomic alterations already present in cancer cell lines and tumor-derived mouse models make to the elucidation of the initial steps of tumor development particularly challenging. Notably, iPSC-derived cells are free of cytogenetic rearrangements [4], allowing the study of early oncogenic mechanisms prior to the accumulation of secondary genomic alterations. A recent investigation comparing the mutational rate of somatic and pluripotent cell lines also demonstrated a 10-fold lower mutation rate in iPSCs with each generation compared to somatic cells [76], highlighting the advantages of using genetically stable iPSCs rather than somatic or even more error-prone cancer cells to expand a cell population with a given genetic trait. PSC disease models should be useful in identifying and characterizing the “second hit” during tumor development following a selective and stable introduction of the first one. Nonetheless, newly generated PSC lines should be fully characterized prior to experimental use to rule out chromosomal or genetic abnormalities.
Recent progress in genome editing tools, including optimizations of the TALEN and CRISPR/Cas9 systems, made the generation of tailored alterations in PSCs from isogenic backgrounds feasible. These powerful tools enable cancer researchers to easily build up a particular model system to investigate the role of specific gene alterations in tumorigenesis in various tissues and organs (Figure 1B). While a number of genetic cancer syndromes exhibit incomplete penetrance, the incidence of disease and the risk factor of developing disease frequently cannot be accurately estimated due to the relatively small number of affected patients. Genome-manipulated ESCs from a wider range of genetic backgrounds may help complement studies from patient-derived iPSCs to clarify the role of genetic backgrounds in cancer predisposition from familial cancer syndromes.
Despite the versatility of PSC technology as a model for a number of cancer etiologies, the limited number of available protocols for tissue differentiation remains one of the major impediments precluding the wider application of this approach in cancer research. New developments in directed differentiation protocols and evolving 3D organoid culture techniques not only facilitate disease modeling in neuroscience, cardiology, and regenerative medicine – fields in which PSC models are currently frequently used – but open up opportunities in cancer research as more tissue types become experimentally available. We aim to see the development of new PSC-derived organoid methodologies to fill in the current gaps in our ability to produce 3D organoid models of prostate, breast, ovary, and many other currently inaccessible cancer cell types.
In conclusion, PSCs are destined for exciting future applications in the field of cancer biology. We look forward to the wide incorporation of PSC techniques into the toolkit of basic and clinical researchers seeking to efficiently recapitulate disease and/or dysregulated gene-associated cancer features and to have complementary cancer model systems.
Box 1. Tailoring the PSC Genome to Model Disease.
Advanced genome editing methodologies have now made it practical to tailor the human genome at the single-nucleotide level. ZFNs, TALENs and CRISPR/Cas9 have revolutionized the genome editing field by allowing site-directed mutagenesis. A combination of a genomic localization domain with a domain conferring nuclease activity permits the introduction of a directed double-stranded break (DSB) at any site of interest. Site-directed mutagenesis without DSBs has recently become feasible for certain (cytosine to thymidine) “base-editing” using a CRISPR/Cas9 fused to cytidine deaminase [92]. By applying any of these approaches, it is now feasible to generate PSCs harboring specific mutations from a PSC line of choice or to correct any genomic alteration in patient-derived iPSCs.
Correcting mutations in iPSCs
Genome editing has been effectively applied to correct the genetic alterations in patient-derived iPSCs for both research and clinical applications. Correction of a mutation in a patient-derived iPSC can be used to generate an isogenic control and demonstrate a mechanistic link between a mutation and its downstream disease state. Importantly, it permits study from a genetic background in which disease penetrance has been established. It also may serve as a promising solution for transplantation-based therapies. For instance, ZFN-piggyBac-mediated correction of SERPINA1 mutations in α1-antitrypsin deficiency patients-derived iPSCs was shown to lead to the rescue of SERPINA1 structure and biological function [23]. TALEN-mediated gene-corrected X-linked severe combined immunodeficiency (SCID-X1) iPSCs demonstrated a rescue of defective hematopoietic differentiation [93]. CRISPR/Cas9-piggyBac-mediated correction of human hemoglobin beta (HBB) mutation in β-thalassemia patient-derived iPSCs restored the expression of HBB expression [94]. Similarly, positive-negative drug selection strategies combined with gene targeting on chromosome 21 have been demonstrated to correct trisomy in Down syndrome iPSCs [81]. While not yet ready for clinical practice, transplantation of differentiated tissue from these engineered iPSC lines back into their patient sources may permit a cure of the underlying disease, particularly in single-gene disorders that can be rescued by only a small amount of functional enzyme.
Inducing mutations in wild-type PSCs
While iPSC models offer unique advantages for modeling a particular patient’s genetic background, the diversity of their genetic origins complicates the integration and comparison of findings across multiple iPSC lines. In contrast, genome-edited PSCs based on extensively characterized lines are likely to be less variable and provide a more useful resource to understand central disease pathogenesis at the genomic scale. For instance, generation of a polycystic kidney disease (PKD)-linked PKD1 or PKD2 knockout in human ESCs results in cyst formation in kidney tubules in an organoid model, recapitulating the human phenotype [70]. Introduction of a long-QT syndrome (LQTS)-associated KCNH2 mutation in human ESCs leads to reduced current conducted by the HERG channel and prolonged action potential duration, recapitulating the LQTS phenotype [95].
In summary, early studies applying genomic editing to PSCs highlight the potential of this system for investigating disease pathogenesis and developing clinical cell therapies.
Outstanding Questions.
Can the prognosis of and potential therapeutic strategies for cancers originating from differentiated lineages (e.g. osteoblasts, myelocytes, or astrocytes) be modeled reliably using patient iPSC-derived cells?
Can the generation and manipulation of patient-derived iPSCs become sufficiently standardized to allow for the use of iPSC disease models in the clinical management of familial cancer syndromes?
Do iPSCs derived from distinct patients with the same cancer syndrome have similar gene expression profiles? Does individual genetic background significantly affect the gene expression profiles of distinct iPSC-derived tumors?
Can PSCs be applied to identify and characterize the “second hit” during tumor development?
Are changes in patient-derived iPSCs equivalent to those in genome-manipulated ESCs in familial cancer syndromes?
Do in vitro reprogramming and differentiation processes trigger artificial tumor development in PSC disease models?
Can patient iPSC-derived cells be used as part of cancer immunotherapy to train the immune system to recognize a cancer signature?
Trends Box.
Modeling cancer using PSCs overcomes several disadvantages of current model systems, including limited accessibility of patient samples, tumor heterogeneity and differences between species.
PSCs provide a powerful and unlimited source to generate differentiated cells that can be used to elucidate disease pathogenesis, drug discovery and development, and advance personalized healthcare.
Both patient-derived iPSCs and engineered ESCs completely phenocopy cancer features, suggesting that PSCs can serve as a useful in vitro human cancer model.
Currently evolving methodologies for gene expression manipulation, genome editing, and cell differentiation facilitate the application of PSCs to cancer research.
Acknowledgments
We sincerely apologize to authors whose work we could not include due to space limitations. I.R.L is supported by the NIH grant R01 HL119404. D.-F.L. is the CPRIT scholar in Cancer Research and supported by NIH Pathway to Independence Award R00 CA181496 and CPRIT Award RR160019.
Glossary Box
- Pluripotent stem cells (PSCs)
Cells with equivalent characteristics to the inner cell mass of the blastocyst-stage embryo. PSCs are capable of differentiating into any cell type and give rise to all adult tissues (pluripotency) and extensively replicate without differentiation and/or senescence (self-renewal)
- Embryonic stem cells (ESCs)
Pluripotent stem cells are derived from the inner cell mass of the blastocyst, an embryo at the pre-implantation stage. These cells are capable of proliferating and dividing without differentiating for a prolonged period in an in vitro tissue culture environment
- Induced pluripotent stem cells (iPSCs)
Pluripotent stem cells derived from differentiated somatic cells through somatic reprogramming by defined factors (e.g., OCT4, SOX2, KLF4 and c-MYC)
- Reprogram
The process of converting one specific cell type to another. It includes the conversion of somatic cells (e.g. dermal fibroblasts) to pluripotent stem cells and the conversion of one type of somatic cell to another
- Zinc finger nuclease (ZFN)
A genome editing methodology based on a fusion of the FokI restriction enzyme with a Cys2His2 zinc finger DNA-binding domain. ZFNs were among the first available tools to perform genome editing
- Transcription activator-like effector nucleases (TALENs)
A genome editing methodology based on fusions of the FokI DNA nuclease and a TAL effector DNA-binding domain derived from Xanthomonas bacteria. Engineered TAL effectors are able to bind to any specific DNA sequence, permitting FokI nuclease activity at a desired DNA location. TAL effectors contain highly conserved 33–34 amino acid repeat domains that recognize a specific base pair. TALENs induce target double-strand breaks to facilitate homologous recombination and enable customized genome alterations. TALENs are believed to have greater precision for genome editing than ZFN and CRISPR/Cas9
- Clustered, regularly interspaced, short palindromic repeat/Cas9 (CRISPR/Cas9)
A genome editing methodology based on the bacterial acquired immune system. Functioning in bacteria as a means of resistance to exogenous genetic elements similar to RNA interference in eukaryotic cells, it recognizes and cleaves DNAs based on a target RNA sequence. CRISPR systems depend on crRNA and tracrRNA for sequence-specific silencing. There are three types of CRISPR/Cas9 systems. The type II system used in Cas9 and its variants serves as an RNA-guided DNA nuclease or nickase that cleaves DNA upon crRNA-tracrRNA target recognition. The ease with which constructs targeting desired genomic loci can be generated has made this technology the tool of choice for many seeking to perform genome editing
- Organoid
A collection of multiple organ-specific cells cultured in a three-dimensional system that self-organize into organ-bud structures. 3D cultured organoids mimic the micro-anatomy of organs and are capable of recapitulating specific organ functions, enabling experimental study of otherwise inaccessible tissue
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thomson JA, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 4.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 5.Gonzalez F, et al. Methods for making induced pluripotent stem cells: reprogramming a la carte. Nat Rev Genet. 2011;12:231–242. doi: 10.1038/nrg2937. [DOI] [PubMed] [Google Scholar]
- 6.Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132:661–680. doi: 10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 7.Lancaster MA, Knoblich JA. Organogenesis in a dish: modeling development and disease using organoid technologies. Science. 2014;345:1247125. doi: 10.1126/science.1247125. [DOI] [PubMed] [Google Scholar]
- 8.Yin X, et al. Engineering Stem Cell Organoids. Cell Stem Cell. 2016;18:25–38. doi: 10.1016/j.stem.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grskovic M, et al. Induced pluripotent stem cells–opportunities for disease modelling and drug discovery. Nat Rev Drug Discov. 2011;10:915–929. doi: 10.1038/nrd3577. [DOI] [PubMed] [Google Scholar]
- 10.Dimos JT, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321:1218–1221. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- 11.Park IH, et al. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yagi T, et al. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet. 2011;20:4530–4539. doi: 10.1093/hmg/ddr394. [DOI] [PubMed] [Google Scholar]
- 13.Brennand KJ, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473:221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carvajal-Vergara X, et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature. 2010;465:808–812. doi: 10.1038/nature09005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pasca SP, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. 2011;17:1657–1662. doi: 10.1038/nm.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Itzhaki I, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–229. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- 17.Moretti A, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363:1397–1409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- 18.Rashid ST, et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J Clin Invest. 2010;120:3127–3136. doi: 10.1172/JCI43122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu GH, et al. Targeted gene correction of laminopathy-associated LMNA mutations in patient-specific iPSCs. Cell Stem Cell. 2011;8:688–694. doi: 10.1016/j.stem.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith-Arica JR, et al. Infection efficiency of human and mouse embryonic stem cells using adenoviral and adeno-associated viral vectors. Cloning Stem Cells. 2003;5:51–62. doi: 10.1089/153623003321512166. [DOI] [PubMed] [Google Scholar]
- 21.Ivanova N, et al. Dissecting self-renewal in stem cells with RNA interference. Nature. 2006;442:533–538. doi: 10.1038/nature04915. [DOI] [PubMed] [Google Scholar]
- 22.Lee DF, et al. Regulation of embryonic and induced pluripotency by aurora kinase-p53 signaling. Cell Stem Cell. 2012;11:179–194. doi: 10.1016/j.stem.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yusa K, et al. Targeted gene correction of alpha1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478:391–394. doi: 10.1038/nature10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zou J, et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell. 2009;5:97–110. doi: 10.1016/j.stem.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hockemeyer D, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo Y, et al. Generation of GFP Reporter Human Induced Pluripotent Stem Cells Using AAVS1 Safe Harbor Transcription Activator-Like Effector Nuclease. Curr Protoc Stem Cell Biol. 2014;29:5A 7 1–18. doi: 10.1002/9780470151808.sc05a07s29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding Q, et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell. 2013;12:238–251. doi: 10.1016/j.stem.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee DF, et al. Modeling familial cancer with induced pluripotent stem cells. Cell. 2015;161:240–254. doi: 10.1016/j.cell.2015.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mulero-Navarro S, et al. Myeloid Dysregulation in a Human Induced Pluripotent Stem Cell Model of PTPN11-Associated Juvenile Myelomonocytic Leukemia. Cell Rep. 2015;13:504–515. doi: 10.1016/j.celrep.2015.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kotini AG, et al. Functional analysis of a chromosomal deletion associated with myelodysplastic syndromes using isogenic human induced pluripotent stem cells. Nat Biotechnol. 2015;33:646–655. doi: 10.1038/nbt.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Funato K, et al. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science. 2014;346:1529–1533. doi: 10.1126/science.1253799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duan S, et al. PTEN deficiency reprogrammes human neural stem cells towards a glioblastoma stem cell-like phenotype. Nat Commun. 2015;6:10068. doi: 10.1038/ncomms10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li FP, et al. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988;48:5358–5362. [PubMed] [Google Scholar]
- 36.Dugimont T, et al. The H19 TATA-less promoter is efficiently repressed by wild-type tumor suppressor gene product p53. Oncogene. 1998;16:2395–2401. doi: 10.1038/sj.onc.1201742. [DOI] [PubMed] [Google Scholar]
- 37.Roberts AE, et al. Noonan syndrome. Lancet. 2013;381:333–342. doi: 10.1016/S0140-6736(12)61023-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindsley RC, Ebert BL. Molecular pathophysiology of myelodysplastic syndromes. Annu Rev Pathol. 2013;8:21–47. doi: 10.1146/annurev-pathol-011811-132436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu G, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwartzentruber J, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 41.Furnari FB, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 42.Si-Tayeb K, et al. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology. 2010;51:297–305. doi: 10.1002/hep.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goldman O, et al. KDR identifies a conserved human and murine hepatic progenitor and instructs early liver development. Cell Stem Cell. 2013;12:748–760. doi: 10.1016/j.stem.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hannan NR, et al. Production of hepatocyte-like cells from human pluripotent stem cells. Nat Protoc. 2013;8:430–437. doi: 10.1038/nprot.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spence JR, et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature. 2011;470:105–109. doi: 10.1038/nature09691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Antonica F, et al. Generation of functional thyroid from embryonic stem cells. Nature. 2012;491:66–71. doi: 10.1038/nature11525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dye BR, et al. In vitro generation of human pluripotent stem cell derived lung organoids. Elife. 2015;4 doi: 10.7554/eLife.05098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang SX, et al. Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat Biotechnol. 2014;32:84–91. doi: 10.1038/nbt.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang SX, et al. The in vitro generation of lung and airway progenitor cells from human pluripotent stem cells. Nat Protoc. 2015;10:413–425. doi: 10.1038/nprot.2015.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xia Y, et al. Directed differentiation of human pluripotent cells to ureteric bud kidney progenitor-like cells. Nat Cell Biol. 2013;15:1507–1515. doi: 10.1038/ncb2872. [DOI] [PubMed] [Google Scholar]
- 51.Takasato M, et al. Directing human embryonic stem cell differentiation towards a renal lineage generates a self-organizing kidney. Nat Cell Biol. 2014;16:118–126. doi: 10.1038/ncb2894. [DOI] [PubMed] [Google Scholar]
- 52.Osakada F, et al. Stepwise differentiation of pluripotent stem cells into retinal cells. Nat Protoc. 2009;4:811–824. doi: 10.1038/nprot.2009.51. [DOI] [PubMed] [Google Scholar]
- 53.Nakano T, et al. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell. 2012;10:771–785. doi: 10.1016/j.stem.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 54.Mellough CB, et al. Efficient stage-specific differentiation of human pluripotent stem cells toward retinal photoreceptor cells. Stem Cells. 2012;30:673–686. doi: 10.1002/stem.1037. [DOI] [PubMed] [Google Scholar]
- 55.Lamba DA, et al. Efficient generation of retinal progenitor cells from human embryonic stem cells. Proc Natl Acad Sci USA. 2006;103:12769–12774. doi: 10.1073/pnas.0601990103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang R, et al. Generation of folliculogenic human epithelial stem cells from induced pluripotent stem cells. Nat Commun. 2014;5:3071. doi: 10.1038/ncomms4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kidwai FK, et al. Differentiation of epidermal keratinocytes from human embryonic stem cells. Methods Mol Biol. 2014;1195:13–22. doi: 10.1007/7651_2013_46. [DOI] [PubMed] [Google Scholar]
- 58.Grigoriadis AE, et al. Directed differentiation of hematopoietic precursors and functional osteoclasts from human ES and iPS cells. Blood. 2010;115:2769–2776. doi: 10.1182/blood-2009-07-234690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kennedy M, et al. Development of the hemangioblast defines the onset of hematopoiesis in human ES cell differentiation cultures. Blood. 2007;109:2679–2687. doi: 10.1182/blood-2006-09-047704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chambers SM, et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lian Q, et al. Derivation of clinically compliant MSCs from CD105+, CD24-differentiated human ESCs. Stem Cells. 2007;25:425–436. doi: 10.1634/stemcells.2006-0420. [DOI] [PubMed] [Google Scholar]
- 62.Fang D, et al. Defining the conditions for the generation of melanocytes from human embryonic stem cells. Stem Cells. 2006;24:1668–1677. doi: 10.1634/stemcells.2005-0414. [DOI] [PubMed] [Google Scholar]
- 63.Lancaster MA, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McCracken KW, et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature. 2014;516:400–404. doi: 10.1038/nature13863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Matano M, et al. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat Med. 2015;21:256–262. doi: 10.1038/nm.3802. [DOI] [PubMed] [Google Scholar]
- 66.Takebe T, et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature. 2013;499:481–484. doi: 10.1038/nature12271. [DOI] [PubMed] [Google Scholar]
- 67.Takebe T, et al. Generation of a vascularized and functional human liver from an iPSC-derived organ bud transplant. Nat Protoc. 2014;9:396–409. doi: 10.1038/nprot.2014.020. [DOI] [PubMed] [Google Scholar]
- 68.Boj SF, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324–338. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xia Y, et al. The generation of kidney organoids by differentiation of human pluripotent cells to ureteric bud progenitor-like cells. Nat Protoc. 2014;9:2693–2704. doi: 10.1038/nprot.2014.182. [DOI] [PubMed] [Google Scholar]
- 70.Freedman BS, et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat Commun. 2015;6:8715. doi: 10.1038/ncomms9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morizane R, et al. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat Biotechnol. 2015;33:1193–1200. doi: 10.1038/nbt.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Takasato M, et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature. 2015;526:564–568. doi: 10.1038/nature15695. [DOI] [PubMed] [Google Scholar]
- 73.Eiraku M, et al. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature. 2011;472:51–56. doi: 10.1038/nature09941. [DOI] [PubMed] [Google Scholar]
- 74.Volkner M, et al. Retinal Organoids from Pluripotent Stem Cells Efficiently Recapitulate Retinogenesis. Stem Cell Reports. 2016;6:525–538. doi: 10.1016/j.stemcr.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kuwahara A, et al. Generation of a ciliary margin-like stem cell niche from self-organizing human retinal tissue. Nat Commun. 2015;6:6286. doi: 10.1038/ncomms7286. [DOI] [PubMed] [Google Scholar]
- 76.Rouhani FJ, et al. Mutational History of a Human Cell Lineage from Somatic to Induced Pluripotent Stem Cells. PLoS Genet. 2016;12:e1005932. doi: 10.1371/journal.pgen.1005932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Somers A, et al. Generation of transgene-free lung disease-specific human induced pluripotent stem cells using a single excisable lentiviral stem cell cassette. Stem Cells. 2010;28:1728–1740. doi: 10.1002/stem.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fukawatase Y, et al. Ataxia telangiectasia derived iPS cells show preserved x-ray sensitivity and decreased chromosomal instability. Sci Rep. 2014;4:5421. doi: 10.1038/srep05421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Garcon L, et al. Ribosomal and hematopoietic defects in induced pluripotent stem cells derived from Diamond Blackfan anemia patients. Blood. 2013;122:912–921. doi: 10.1182/blood-2013-01-478321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ge J, et al. Dysregulation of the Transforming Growth Factor beta Pathway in Induced Pluripotent Stem Cells Generated from Patients with Diamond Blackfan Anemia. PLoS One. 2015;10:e0134878. doi: 10.1371/journal.pone.0134878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li LB, et al. Trisomy correction in Down syndrome induced pluripotent stem cells. Cell Stem Cell. 2012;11:615–619. doi: 10.1016/j.stem.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Agarwal S, et al. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature. 2010;464:292–296. doi: 10.1038/nature08792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Batista LF, et al. Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nature. 2011;474:399–402. doi: 10.1038/nature10084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Itoh M, et al. Generation of keratinocytes from normal and recessive dystrophic epidermolysis bullosa-induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2011;108:8797–8802. doi: 10.1073/pnas.1100332108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tolar J, et al. Induced pluripotent stem cells from individuals with recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2011;131:848–856. doi: 10.1038/jid.2010.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Raya A, et al. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature. 2009;460:53–59. doi: 10.1038/nature08129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ye Z, et al. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood. 2009;114:5473–5480. doi: 10.1182/blood-2009-04-217406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tulpule A, et al. Pluripotent stem cell models of Shwachman-Diamond syndrome reveal a common mechanism for pancreatic and hematopoietic dysfunction. Cell Stem Cell. 2013;12:727–736. doi: 10.1016/j.stem.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cheung HH, et al. Telomerase protects werner syndrome lineage-specific stem cells from premature aging. Stem Cell Reports. 2014;2:534–546. doi: 10.1016/j.stemcr.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shimamoto A, et al. Reprogramming suppresses premature senescence phenotypes of Werner syndrome cells and maintains chromosomal stability over long-term culture. PLoS One. 2014;9:e112900. doi: 10.1371/journal.pone.0112900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thatava T, et al. Successful disease-specific induced pluripotent stem cell generation from patients with kidney transplantation. Stem Cell Res Ther. 2011;2:48. doi: 10.1186/scrt89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Komor AC, et al. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Menon T, et al. Lymphoid regeneration from gene-corrected SCID-X1 subject-derived iPSCs. Cell Stem Cell. 2015;16:367–372. doi: 10.1016/j.stem.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xie F, et al. Seamless gene correction of beta-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. 2014;24:1526–1533. doi: 10.1101/gr.173427.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bellin M, et al. Isogenic human pluripotent stem cell pairs reveal the role of a KCNH2 mutation in long-QT syndrome. EMBO J. 2013;32:3161–3175. doi: 10.1038/emboj.2013.240. [DOI] [PMC free article] [PubMed] [Google Scholar]