

Abstract
Despite the inability of HIV-1 to infect neurons, over half of the HIV-1-infected population in the USA suffers from neurocognitive dysfunction. HIV-infected immune cells in the periphery enter the central nervous system by causing a breach in the blood–brain barrier. The damage to the neurons is mediated by viral and host toxic products released by activated and infected immune and glial cells. To evaluate the toxicity of any viral isolate, viral protein, or host inflammatory protein, we describe a protocol to assess the neuronal apoptosis and synaptic compromise in primary cultures of human neurons and astrocytes.
Keywords: HIV-1, Neuronal damage, Neuronal apoptosis, Neuro-inflammation, HIV-associated dementia, HAD, HIV-associated neurocognitive dysfunction, HAND, gp 120, Tat, Cytokines, Chemokines
1 Introduction
HIV-infected individuals on anti-retroviral therapy are living longer; but this can result in long-standing neurological damage [1] and HIV-associated neurocognitive disorders (HAND). HAND manifests in around 50 % of the HIV-infected population despite the effective use of ARV to control peripheral viremia and immunological decline [1]. Studies have demonstrated that HIV-1 enters the brain early in the infection [2]. The disease spectrum of HAND includes the milder forms such as asymptomatic neurocognitive impairment (ANI), mild neurocognitive disorder (MND), and the severe form, HIV-associated dementia (HAD). Even though the severity of damage caused by HIV to the brain in patients on anti-retroviral therapy has decreased as evidenced by the decreasing incidence of HAD, the overall prevalence of ANI and MND is increasing worldwide [3].
CNS homeostasis and immune privilege of the brain are primarily maintained by the impermeability of the blood–brain barrier (BBB) providing separation between the central nervous system (CNS) and the peripheral blood circulation. HIV-1 breaches the BBB early in the acute stages of infection [4]. HIV-1 infection of leukocytes increases expression of key proteins involved in transmigration and chemokine recognition, resulting in an enhanced invasion of the brain early during HIV infection by a mechanism named “Trojan horse” [5]. Once transmigrated leukocytes are inside the CNS, HIV-infected macrophages produce chemokines that recruit more immune cells from the circulation including T-cells and macrophages [6]. This influx of new transmigrated immune cells, over time, leads to an unusually high density of monocyte/macrophages in the brain including activated monocytes, macrophages, microglia resulting in enhanced inflammation, and brain tissue damage. Consequently, all these cells secrete neurotoxic viral proteins (Tat, gp 120) and inflammatory chemokines/cytokines (TNF-alpha, interferon alpha, CCL2, CXCL10) [7] that have the ability to interact with the cell surface receptors present on the neurons [7] triggering neuronal apoptosis. The recent increase in efforts to identify the genetic signatures of viral proteins that are responsible for neurovirulence necessitates appropriate methods to evaluate the relative neurotoxicity of different HIV-1 isolates to help identify the neurotoxic viral and host products responsible for neurotoxicity.
Most of the neuronal apoptosis assays documented in literature are currently performed using neuroblastoma cell lines (e.g., SH-SY5Y) and rodent primary neurons. However, these immortalized cell lines or primary neurons isolated from rodents fail to accurately represent the HIV neurotoxicity observed in vivo. Use of such cells also allows investigators to simply measure cell survival using MTT or WST-1 assays. In contrast, most human cultures of neurons tend to contain variable proportions of astrocytes and an even smaller proportion of glia, complicating the interpretation of the results in cell survival assays. Here we describe a neuronal apoptosis assay employing human primary neurons to quantify the number of neurons undergoing apoptosis to get a more accurate picture of the neuropathogenesis.
2 Materials
2.1 Culture of Primary Human Neurons
Primary human fetal brain tissue (obtained from fetal brain tissue repository).
Phosphate-buffered saline (PBS).
Trypsin-EDTA.
DNase.
Tweezers, razor blades.
250 and 150 μm Filters.
Minimal essential medium.
Fetal bovine serum.
Penicillin-streptomycin (10,000 U/ml).
Neurobasal media.
N2 Supplement—100×.
TMR in situ hybridization kit.
Anti-neurotubulin antibodies.
Alexa-conjugated secondary antibody—goat anti-rabbit IgG.
Prolong Gold anti-fade agent with DAPI.
2.2 Virus Production
293T cells.
Lipofectamine 2000 transfection reagent.
OptiMEM media.
DMEM tissue culture medium with L-glutamine, glucose, and sodium pyruvate.
Heat-inactivated pooled fetal bovine serum.
Penicillin-streptomycin (10,000 U/ml).
HIV-p24 ELISA kit.
2.3 Macrophage Differentiation and HIV Infection
Elutriated primary human monocytes.
Monocyte colony-stimulating factor (MCSF).
Pooled human serum.
24-Well polystyrene-treated tissue culture dishes.
DMEM tissue culture medium with glucose and sodium pyruvate.
L-Glutamine—200 mM.
Penicillin-streptomycin (10,000 U/ml).
3 Methods
The protocol described below includes culturing primary neurons, HIV infection of human macrophages, exposure of primary cultures of neurons to HIV-infected cell supernatants, staining of neuronal cultures by nuclear staining with DAPI, staining of apoptotic cells by TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling), and staining of neurons by neurotubulin staining. DAPI stain labels all nuclei, neurotubulin allows one to identify neurons, and TUNEL staining detects only the apoptotic nuclei; thus, one can quantitate the proportion of total cells constituted by the apoptotic neurons.
3.1 Culturing Fetal Primary Neurons
Human neuronal cultures are obtained from elective abortions. All the protocols must be approved for use by the Institutional Review Board offices. Most of the tissue employed corresponds to cortex and hippocampal tissue. In this section, we describe in detail the preparation and potential application of these cultures.
Place the tissue in a plate containing sterile PBS and remove the meninges using sterile forceps. Meninges contain blood and several cell types that can contaminate the purity of the CNS cell populations. Due to the fact that the tissue is of human origin, you need to use biosafety level 2+ (BL2+) handling conditions in case of contamination with human pathogens such as HIV, HCV, or CMV.
Mince the tissue with a razor to a fine grind, making it easy enough to pick it up with a pipet (see Note 1).
Tissue is cut into small sections and mixed with 1mL trypsin-EDTA and 100 μL DNase I.
Incubate for 45 min to an hour in a warm incubator in 50 ml Falcon tubes on a rotating shaker with trypsin-EDTA and DNase I (see Note 2).
After the incubation, pipet up and down to dissociate any remaining cell aggregates.
Then, pass the cell suspension solution through a 250 μm filter. The filtered cell suspension subsequently is filtered using a 150 μm filter to remove clumps of tissue and to obtain individual cells.
The cell suspension is centrifuged at 600 × g for 5 min at 18 °C (see Note 3). Wash two times and remove the supernatant with care.
Count the cells and plate in complete medium (minimal essential medium + 10 % FBS + PenStrep). Plate the 1.2 × 108 cells in a 150 ml tissue culture flask and place in the CO2 incubator. Do not disturb for 7–10 days.
After this period, observe the morphology to ensure that the cells display neuronal characteristics (see Note 4). Trypsinize with 8 ml trypsin for 1–2 min at 37 °C until you see the first evidence of adhesion loss to the plastic. Harvest the neurons and pellet the cells (quickly, 600 × g for 5 min) and count.
Plate 107 cells in 15 mL Neurobasal media, 1 % FBS, N2 supplement, and PenStrep, in a T-75 flask. Half media changes are done every alternate day. These cells can be used anytime from day 5 to day 12 post-splitting. Using this protocol, you can obtain enriched neuronal cultures containing 80–90 % of neurons and 10–20 % glia.
In these cultures, astrocytes grow in the bottom of the plate and neurons grow on the top of the astrocytes. For these cultures, approaches using confocal microscopy are extremely useful to evaluate cellular localization, neuronal processes, apoptosis, synaptic proteins, and glial-neuronal communication.
3.2 Virus Production
Infectious HIV-1 virus particles are produced by transient transfection of 293T cells using Lipofectamine reagent.
Seed 4 × 106 293T cells in 100 mm2 culture dishes 24 h prior to transfection.
Replace culture medium with ~8 ml fresh, pre-warmed 2.5 % FBS DMEM antibiotic-free medium 4 h prior to transfection to stimulate cell division and prevent toxicity.
Prepare 24 μg DNA/Lipofectamine reagent mix in Opti-MEM media.
Add the DNA/Lipofectamine reagent mix in Opti-MEM to 100 mm culture dish with 293T cells by uniformly adding drops over the entire surface.
Incubate for 4 h at 37 °C with 5 % CO2.
Add 5 ml 20 % FBS DMEM medium to the 100 mm culture dish with 293T cells.
After 12 h, replace culture medium with 10 ml pre-warmed 10 % FBS DMEM.
Collect supernatants 48 h post-transfection. Centrifuge supernatants at 3000 × g for 5 min to clear the cell debris. Add 10 μl of 1 M HEPES buffer each to 1 ml aliquots of the supernatant and store at −80 °C for future use.
Quantitate the amount of virus obtained using HIV-p24 ELISA.
3.3 Macrophage Differentiation and Infection
Monocytes obtained by elutriating PBMCs are differentiated into macrophages using macrophage colony-stimulating factor (MCSF).
For differentiating monocytes into macrophages, primary human monocytes are incubated at 37 °C for 5 days in DMEM, 10 % human serum, PenStrep, L-glutamine, and MCSF (Sigma) at 6.6 ng/ml in 24-well plates with media changes every other day.
Following differentiation into macrophages, various viral isolates are used to infect macrophages in at least three different concentrations (see Note 5). (For full-length infectious molecular clones, virus obtained from the 293T cell transfection is used for infection and for clinical isolates, infectious PBMC supernatants are employed.)
Following infection, the macrophages are incubated for 5–10 days and half media changes are done every third day. Supernatants are collected every day to measuring HIV-p24 levels using ABL HIV-p24 ELISA kit. These supernatants are used to treat neuronal cultures once they exhibit equal infectivity (see Note 5).
3.4 Neurotoxicity Testing
If the sample to be tested in neurotoxicity is a protein/macromolecule such as viral proteins or chemokines/cytokines, it has to be directly added to the media, which is changed 2 days prior to treatment. If the sample to be tested for neurotoxicity is HIV-infected supernatant, then it has to be mixed with equal or greater volume of Neurobasal medium and added to the neurons. Some of these infected supernatants can be depleted of particular viral proteins, such as Tat and gp 120, using immune-depleted supernatants using specific antibodies [8, 9] and the toxicity of the medium with and without immune depletion can be compared.
Plate approximately 100,000 primary neurons on poly-lysine-coated MatTek plates in 2 mL Neurobasal media (without FBS) for a period of 6 days in the incubator and allow them to stabilize and differentiate with half media changes every 2 days.
Add either diluted HIV-infected macrophage supernatant or the protein/macromolecule of interest to the Neurobasal media and incubate with the neurons for a set amount of time. In Fig. 1, primary human neurons were incubated with the HIV-1ADA-infected macrophage supernatant for 24 h.
A dose–response curve as well as the kinetics of neuronal killing as a function of time should be performed the first time when neurotoxicity assay is performed (see Note 6).
Following the treatment, fix the neurons in 70 % ethanol for 15–20 min at −20 °C (if the apoptosis is massive or if the neuronal confluence is low, fix the neurons overnight).
Perform TUNEL assay using the TMR in situ hybridization kit (Roche; Cat No. 12156792910) (use 37 °C degree incubator with humidity control). Incubate for 1 h.
Following the TUNEL staining, wash the neurons two times in PBS and incubate overnight at 4 °C with anti-neurotubulin antibodies (Abeam cat. No. 21058) (primary antibody) (see Note 7).
Wash the neurons again and incubate with a suitable secondary antibody for 2–4 h at room temperature (low confluence/massive apoptosis again warrants overnight incubation at 4 °C).
Wash five times in 1× PBS with 5-min incubation between washes.
Add Prolong Gold anti-fade agent with DAPI (Invitrogen, cat number, P36931) to the neurons and place cover slips. Incubate overnight at 4 °C in a humid box.
Perform image capture and analysis using Zeiss Microscope and Zeiss Zen software. Image ten fields per treatment. For each field, you need to process all three fluorescence channels (red, green, and blue) and bright field. Each sample takes approximately an hour to process (ten fields total) (see Fig. 1).
Transfer images to Nikon NIS software for further processing. Determine the percent apoptosis/survival based on the proportion of cells with DAPI-neurotubulin stain that are TUNEL positive (Fig. 2).
Measure the dendrite width and length using Nikon (NIS elements) advanced research software and calculate the median dendrite width using 20–30 measurements/cell for 50–100 different cells in the field.
Fig. 1.
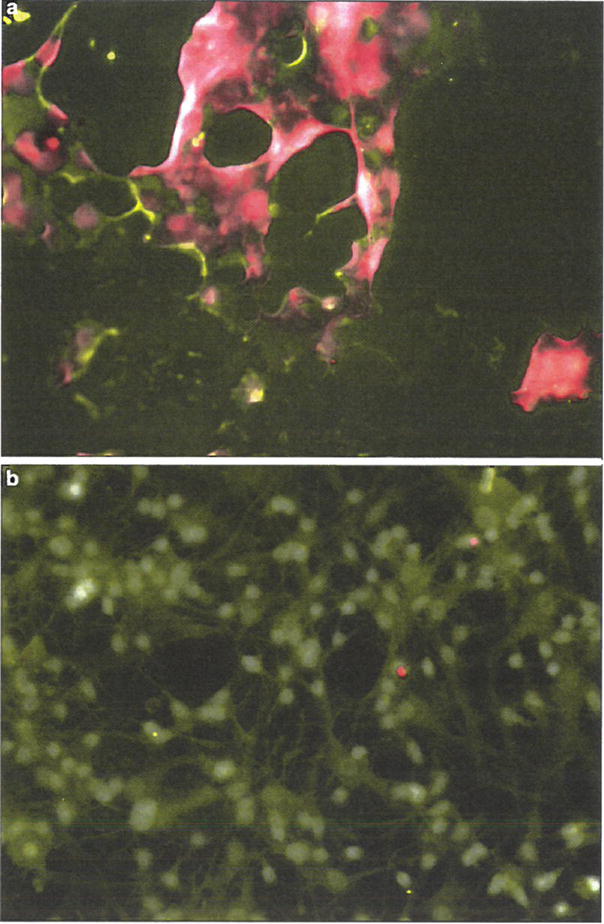
Neurotubulin/TUNEL staining of primary human neurons treated with either HIV-1-infected or uninfected MDM supernatants. TUNEL-stained primary human neurons that were treated with HIV-1ADA-infected or uninfected macrophage supernatant, then co-stained with anti-neurotubulin antibodies and DAPI (Prolong Gold anti-fade with DAPI, Invitrogen). Neurons treated with HIV-1-infected supernatant exhibit massive apoptosis as demonstrated by TUNEL staining.
Fig. 2.
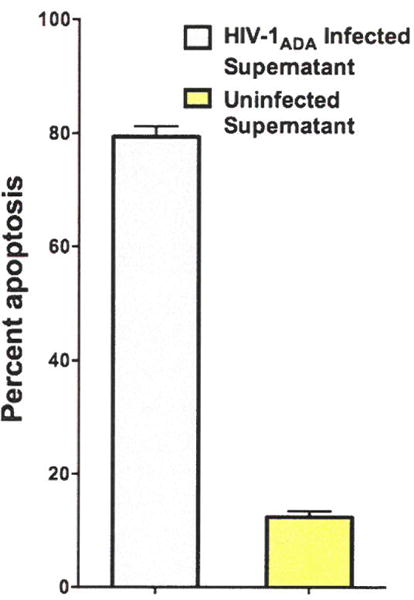
HIV-1ADA-infected supernatant causes neuronal apoptosis in primary human neurons. HIV-1ADA-infected macrophage supernatant and uninfected macrophage supernatant were diluted with 100 μl of Neurobasal medium and incubated with primary human neurons in MatTek plates for 18 h. Percent apoptosis/survival was determined based on the proportion of cells with DAPI-neurotubulin stain that are TUNEL positive. HIV-1ADA supernatant leads to a greater loss of neuronal viability when compared to uninfected supernatant.
Acknowledgments
This work was supported by NIH R01 MH083579 and R21 MH101003 (to V.R.P.). The authors would like to thank Arthur Ruiz for critically reading the manuscript.
Footnotes
If the tissue is bloody, rinse it well with PBS prior to further processing. Use tweezers to separate the thick white tissue from the cortical tissue.
Incubations greater than an hour will lead to loss of tissue. Limit the incubations to less than 1 h.
The pellet at this juncture is held together loosely; therefore it should be handled with care.
Two distinct cell layers can be observed—a bottom layer of glial cells and a top layer of neurons. Constantly monitor trypsinized neurons for first signs of loss of adhesion.
Different virus isolates have varied replication rates; therefore it is critical to titer the doses of initial infection to generate infected cell cultures whose supernatants contain similar amounts of p24. Typically 10ng, 100ng and 500ng virus supernatants are used to titer the infectivity. Macrophages are plated in either 12 or 24 well plates and allowed to differentiate for a period of 72 hours in media containing MCSF (Macrophage colony stimulating factor). Once the macrophages are completely adherent and differentiated, viral isolates are tittered in various concentrations to obtain similar infectivity.
It is recommended to use a positive control such as LPS-treated macrophage supernatant where robust neuronal cell death can be observed.
Primary antibody incubation needs to be at 4 °C to preserve the TUNEL staining. Incubation at 37 °C will lead to false-positive TUNEL results.
References
- 1.Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, Simpson DM, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I, Group C, Group H HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol. 2011;17:3–16. doi: 10.1007/s13365-010-0006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Resnick L, Berger JR, Shapshak P, Tourtellotte WW. Early penetration of the blood-brain-barrier by HIV. Neurology. 1988;38:9–14. doi: 10.1212/wnl.38.1.9. [DOI] [PubMed] [Google Scholar]
- 3.Airoldi M, Bandera A, Trabattoni D, Tagliabue B, Arosio B, Soria A, Rainone V, Lapadula G, Annoni G, Clerici M, Gori A. Neurocognitive impairment in HIV-infected naïve patients with advanced disease: the role of virus and intrathecal immune activation. Clin Dev Immunol. 2012;2012:467154. doi: 10.1155/2012/467154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.An SF, Groves M, Gray F, Scaravilli F. Early entry and widespread cellular involvement of HIV-1 DNA in brains of HIV-1 positive asymptomatic individuals. J Neuropathol Exp Neurol. 1999;58:1156–1162. doi: 10.1097/00005072-199911000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Meltzer MS, Skillman DR, Gomatos PJ, Kalter DC, Gendelman HE. Role of mononuclear phagocytes in the pathogenesis of human immunodeficiency virus infection. Annu Rev Immunol. 1990;8:169–194. doi: 10.1146/annurev.iv.08.040190.001125. [DOI] [PubMed] [Google Scholar]
- 6.Toborek M, Lee YW, Flora G, Pu H, Andras IE, Wylegala E, Hennig B, Nath A. Mechanisms of the blood-brain barrier disruption in HIV-1 infection. Cell Mol Neurobiol. 2005;25:181–199. doi: 10.1007/s10571-004-1383-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rao VR, Ruiz AP, Prasad VR. Viral and cellular factors underlying neuropathogenesis in HIV associated neurocognitive disorders (HAND) AIDS Res Ther. 2014;11:13. doi: 10.1186/1742-6405-11-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rao VR, Neogi U, Eugenin E, Prasad VR. The gp120 protein is a second determinant of decreased neurovirulence of Indian HIV-1C isolates compared to Southern African HIV-1C isolates. PLoS One. 2014;9:e107074. doi: 10.1371/journal.pone.0107074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rao VR, Neogi U, Talboom JS, Padilla L, Rahman M, Fritz-French C, Gonzalez-Ramirez S, Verma A, Wood C, Ruprecht RM, Ranga U, Azim T, Joska J, Eugenin E, Shet A, Bimonte-Nelson H, Tyor WR, Prasad VR. Clade C HIV-1 isolates circulating in Southern Africa exhibit a greater frequency of dicysteine motif-containing Tat variants than those in Southeast Asia and cause increased neurovirulence. Retrovirology. 2013;10:61. doi: 10.1186/1742-4690-10-61. [DOI] [PMC free article] [PubMed] [Google Scholar]