

Abstract
Methamphetamine (meth) is a central nervous system (CNS) stimulant that results in psychological and physical dependency. The long-term effects of meth within the CNS include neuronal plasticity changes, blood–brain barrier compromise, inflammation, electrical dysfunction, neuronal/glial toxicity, and an increased risk to infectious diseases including HIV. Most of the reported meth effects in the CNS are related to dysregulation of chemical synapses by altering the release and uptake of neurotransmitters, especially dopamine, norepinephrine, and epinephrine. However, little is known about the effects of meth on connexin (Cx) containing channels, such as gap junctions (GJ) and hemichannels (HC). We examined the effects of meth on Cx expression, function, and its role in NeuroAIDS. We found that meth altered Cx expression and localization, decreased GJ communication between neurons and astrocytes, and induced the opening of Cx43/Cx36 HC. Furthermore, we found that these changes in GJ and HC induced by meth treatment were mediated by activation of dopamine receptors, suggesting that dysregulation of dopamine signaling induced by meth is essential for GJ and HC compromise. Meth-induced changes in GJ and HC contributed to amplified CNS toxicity by dysregulating glutamate metabolism and increasing the susceptibility of neurons and astrocytes to bystander apoptosis induced by HIV. Together, our results indicate that connexin containing channels, GJ and HC, are essential in the pathogenesis of meth and increase the sensitivity of the CNS to HIV CNS disease.
Keywords: apoptosis, cocaine, connexin, drugs, HIV
Methamphetamine (meth) is a highly addictive drug used worldwide. Its use is linked to risky behaviors and increased susceptibility to infectious diseases, including human immunodeficiency virus-1 (HIV), and its comorbidities. Epidemiological studies indicate that individuals with HIV who are substance abusers have a higher incidence of HIV-associated CNS disease (Shor-Posner 2000; Maragos et al. 2002; Bell et al. 2006; Burdo et al. 2006; Silverstein et al. 2011). In addition, several studies have found a strong correlation between substance abuse and non-adherence to antiretroviral treatment in HIV patients, thereby limiting the potential benefits of antiretroviral treatment. This results in lower rates of virological suppression and creates the potential for an enhanced disease progression (Tucker et al. 2003). Thus, there is a clear correlation between drugs of abuse, systemic inflammation/HIV replication, and CNS disease. However, the cellular and molecular mechanisms underlying this correlation are not known.
Meth has long lasting toxic effects on dopaminergic, noradrenergic, serotoninergic, and GABA-ergic terminals (Powrozek et al. 2004; Zhang et al. 2006; Ferrucci et al. 2013; Beardsley and Hauser 2014). The toxicity of meth in the CNS is associated with: (i) a reduction in vesicular monoamine transporter loading of synaptic vesicles (Volz 2006), resulting in the accumulation of monoamine neurotransmitters in the neuronal cytoplasm, and (ii) the prevention of the neurotransmitters’ reuptake leading to their accumulation in the synaptic cleft. This increased neurotransmitter concentration at the synaptic cleft in response to meth over-activates their receptors, resulting in profound behavioral and physiological effects (Zhang et al. 2001; Chiu and Schenk 2012). Most of the reports involving meth and synaptic signaling have been focused on chemical synapses. In contrast, the effects of meth on glial and neuronal connexin containing channels, gap junction (GJ), and hemichannels (HC), are still unclear.
GJ connect the cytoplasm of two cells, serving as a low-resistance bridge to coordinate electrical and metabolic function in tissues (Eugenin et al. 2012; Eugenin 2014). Each channel is formed by the docking of two unopposed connexin HC present on the surfaces of two contacting cells. In physiological conditions, GJ in glial cells help to clear high concentrations of neurotransmitters (i.e., glutamate) and ions (i.e., potassium) that accumulate in areas proximal to chemical synapses by regulating intracellular diffusion of these metabolites among connected cells (Pereda et al. 2003a,b; Cachope et al. 2007; Rash et al. 2013). In addition, it has recently been described that undocked HC on the surface of glial cells can open under stress and inflammatory conditions (Castellano and Eugenin 2014; Eugenin 2014; Velasquez and Eugenin 2014), resulting in the release of intracellular inflammatory factors, such as ATP, NADH, and PGE2 into the extracellular space, which leads to activation of ATP receptors and other inflammatory pathways (Pelegrin and Surprenant 2006, 2007; Locovei et al. 2007). In addition, in several disease models, reduction in GJ communication (GJC) and HC opening has been associated with severe problems in learning, memory, fear responses, and other behavioral issues (Frisch et al. 2005; Allen et al. 2011; Bissiere et al. 2011; Mercer 2012; Stehberg et al. 2012; Zlomuzica et al. 2012), all of which are observed in meth users (Logan 2002; Gettig et al. 2006; Darke et al. 2008). Furthermore, it has been shown that amphetamine and cocaine use compromises Cx36 expression and electrical synapse function (Onn and Grace 2000; McCracken et al. 2005a,b). However, despite the importance of both GJ and HC in normal CNS function and their roles in the development of drug-induced pathologies has not been investigated.
Our results show that meth compromises GJ communication and results in the opening of HC in neurons and astrocytes. These alterations in GJ and HC result in glutamate dysregulation but do not elicit apoptosis in the absence of HIV infection. However, when we treated HIV-infected cultures of neurons and astrocytes with meth, we observed a further increase in glutamate dysregulation as well as increased apoptosis of both cell types. Our results indicate that targeting GJ and HC may provide an alternative therapeutic route to reduce the physical and psychological damage caused by meth or associated diseases such as HIV and NeuroAIDS.
Materials and methods
Reagents
Dulbecco’s modified Eagle’s medium, fetal bovine serum, penicillin/streptomycin (P/S) and trypsin-EDTA were from Invitrogen (Grand Island, NY, USA). Monoclonal antibody to glial fribillary acid protein (GFAP), FITC, or Cy3-conjugated anti-rabbit IgG, and Cy3 or FITC coupled anti-mouse IgG antibodies were from Sigma (St Louis, MO, USA). Purified mouse IgG2B and IgG1 myeloma proteins were from Cappel Pharmaceuticals, Inc (Aurora, Ohio). Anti-Cx43, and secondary antibodies anti-mouse, anti-rabbit, and anti-goat immunoglobulins conjugated to Alexa-533, Alexa-633, and Alexa-588 were from Life Technologies (Eugene, OR, USA). Mimetic peptides targeting Cx43 (VCYDKSFPISHVR) or Cx36 (VCNTLQPGCNQAC) hemichannels were used to block dye uptake rate as previously characterized (Flores et al. 2012; Orellana et al. 2012a,b, 2013a,b; Abudara et al. 2014). Because of the limited characterization of Cx36 mimetic peptides, especially in human neurons, we characterized whether Cx36 mimetic peptides block Etd uptake induced by metabolic inhibitions as previously described (Contreras et al. 2002) using human neuronal cultures. Metabolic inhibition resulted in the opening of HC in human neurons. This HC opening in human neurons was only sensitive to human Cx36 mimetic peptides, and lanthanum, but not to scrambled peptides, or pannexin-1, Cx32, and Cx43 mimetic peptides (unpublished data).
Animal model of meth abuse
Meth users initially take small amounts of the drug intermittently before increasing the dose (Simon et al. 2002a,b; Melega et al. 2007). To simulate this pattern, increasing doses (2.5, 5, and 10 mg/kg of body weight/day at weeks 1, 2, and 3, respectively) of meth (Sigma) were intraperitoneally (i.p.) administered daily to female C57BL/6 mice (age, 6–8 weeks; Charles Rivers, Wilmington, MA, USA) over 21 days as previously described (Martinez et al. 2009; Patel et al. 2013). Phosphate-buffered saline (PBS)-treated animals were used as controls.
Ethics statement
All animal studies were conducted according to the experimental practices and standards approved by the Institutional Animal Care and Use Committee (IACUC) at NYIT College of Osteopathic Medicine (NYIT COM) (Protocol #: 2015-LM-01). Animal care for this study was approved by the Animal Welfare and Research Ethics Committee at Rutgers University and NYIT.
Hela Cx43-EGFP cells
Experiments were performed using HeLa cells (ATCC No. CCL-2, the parental cells) or HeLa cells transfected with cDNAs encoding rat Cx43 or rat Cx43 with EGFP attached to the C-terminus (Cx43-EGFP, a kind gift of Dr Michael V. Bennett, The Albert Einstein College of Medicine, Bronx, NY, USA) and were maintained in culture as described (Contreras et al. 2003).
Human neuronal and astrocyte cultures
Human fetal brain tissue was obtained as part of a research protocol approved by Rutgers University and all tissues are unidentified. The mixed cultures were prepared as previously described mainly from the frontal and occipital cortex and hippocampus (Eugenin and Berman 2003; Eugenin et al. 2007; King et al. 2010).
Immunofluorescence
Mixed cultures of human neurons and astrocytes were grown on coverslips, fixed, and permeabilized in 70% ethanol for 20 min at −20°C. Cells were incubated in blocking solution for 30 min at 22°C and then in diluted primary antibody (anti-Cx43, anti-Cx36, antimicrotubule-associated protein-2 (MAP-2) and anti-GFAP: 1 : 2000, 1 : 300; 1 : 2000 and 1 : 1000, respectively) overnight at 4°C. Cells were washed several times with PBS at 22°C and incubated with the appropriate secondary antibodies for at least 3 h at 22°C followed by another wash in PBS for 1 h. Cells were examined using an SP2 Leica or A1 Nikon confocal microscope (Leica Biosystems, Wetzlar, Germany or Nikon, Tokio, Japan). Antibody specificity was confirmed by replacing the primary antibody with a non-specific myeloma protein of the same isotype or non-immune serum as described (Rella et al. 2014; Subbian et al. 2014).
Western blot analysis
Relative levels of Cx43, Cx36, and tubulin were determined by immunoblot as described (Berthoud, et al. 1992; Eugenin et al. 2003). Brains obtained from untreated and treated animals with meth or mixed cultures of neurons and astrocytes were treated with the drug or vehicle, then, harvested in Tris buffer, 10 mM pH 7.4, containing protease and phosphatase inhibitors (20 mM; pyrophosphate, 20 mM; NaF, 100 mM; NaVO3, 200 μM; leupeptine, 500 μg/mL; aprotinine, 40 μg/mL; soybean trypsin inhibitor, 2 mg/mL; benzamidine, 1 mg/mL; ω-amino caproic acid, 1 mg/mL; phenylmethylsulfonyl fluoride, 3 mM and EDTA, 20 mM) (Eugenin, et al. 2001). Samples were lysed and the protein content of each cell lysate was determined using Bradford’s method (Bradford 1976) (Bio-Rad labs, Hercules, CA, USA). Samples containing 20 μg of protein were used to analyze Cx43, Cx36, and tubulin. Proteins were separated in 7.5% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and electrophoretically transferred to nitrocellulose, which were then incubated sequentially with blocking solution (5% non-fat milk in Tris-buffered saline), affinity purified rabbit polyclonal antibodies prepared against Cx43, Cx36, and tubulin (1 : 2000, 1 : 500, and 1 : 2000, respectively) and anti-rabbit IgG conjugated to horseradish peroxidase. Antigen-antibody complexes were detected by enhanced chemiluminescence (Perkin Elmer, Boston, MA, USA) and the resulting immunoblot signals were scanned and densitometric analysis was performed using NIH-image software. All results were normalized to the values obtained for control conditions.
Dye coupling
To evaluate the function of GJs, intercellular transfer of Lucifer yellow (LY) (5% w/v in 150 mM LiCl) or neurobiotin (8%; Vector Laboratories, Burlingame, CA, US) was determined by microinjecting the dye into a single cell and measuring the diffusion of the dye into neighboring cells, as previously described (Meller 1992; Eugenin et al. 1998). Cells were scored as coupled if dye transfer occurred to one or more adjacent cells. Dye transfer was evaluated using confocal microscopy. Four independent experiments were performed, in which a minimum of 10 cells were microinjected per experiment. The incidence and index of dye coupling was scored as the percentage of injections that resulted in dye transfer and the numbers of cells coupled to a single microinjected cell, respectively.
Dye uptake and time-lapse microscopy
To characterize the functional state of Cx HCs, dye-uptake experiments using ethidium (EtBr) bromide were performed. Cells were washed twice in Hank’s balanced salt solution (HBSS) and then exposed to Locke’s solution (containing 154 mM NaCl, 5.4 mM KCl, 2.3 mM CaCl2, 5 mM HEPES, and pH 7.4) with 5 μM EtBr, followed by time-lapse microscopy. Phase-contrast and fluorescence microscopy with time-lapse imaging were used to record cell appearance and fluorescence-intensity changes in each condition. Fluorescence was recorded every 30 s. The NIH ImageJ software (Bethesta, MD, US) was used for off-line image analysis and fluorescence quantification. For data representation and calculation of EtBr uptake slopes, the average of two independent baseline fluorescent (FB) (expressed as arbitrary units, AU) was subtracted from mean fluorescent intensity (F1). Results of this calculation (F1–FB), for at least 20 cells, were averaged and plotted against time (expressed in minutes). Slopes were calculated using Microsoft Excel software and expressed, as AU/min. Microscope and camera settings remained the same in all experiments. Dead cells or cells with a damaged plasma membrane were clearly identified during the time-lapse microscopy as a result of their nonspecific Et uptake, determined by lack of time dependency and stability in dye uptake (not inhibited by HC blockers), and were not quantified.
Microinjection of astrocytes and neurons using brain tissue slices
Three hundred micrometers brain tissue slices were cut using a vibrotome (TPI series, model 1000) from mice injected with meth or saline. The sections were stained using HEPES solution containing sulforhodamine 101 to label astrocytes for 10 min, and subsequent washes in PBS. After staining, non-sulforhodamine cells with neuronal shape were microinjected with neurobiotin in the cortex. Positive cells for sulforhodamine 101, astrocytes, were injected with LY. After injection and staining (in case of neurobiotin using streptavidin-FITC; Sigma), cell to cell communication was analyzed and quantified in the presence and absence of meth. In all these studies we selected cortex and hippocampus since our human cultures are also from these areas in humans.
Image quantifications
Each marker or area examined was obtained from multiple optical sections or brain regions as well as dye coupling. Each area was analyzed after 3D reconstruction and deconvolution (Guan et al. 2006, 2008). To analyze expression of the proteins of interest, we use the 3D reconstructions described above followed by the generation of regions of interests to quantify the number of positive pixels as well as their intensity in MAP-2, GFAP, or sulforhodamine 101 positive cells. For each cell type analyzed a linear intensity histogram was generated to examine the expression of each cellular marker or Cx.
Measurement of extracellular glutamate
The glutamate concentrations in the mixed cultures of neurons and astrocyte medium were determined spectrophotometrically using a commercially available kit according to the manufacturer’s instructions (Sigma) with minimal modifications (Eugenin et al. 2003). The 20–50× concentrated medium obtained from mixed cultures of neurons and astrocytes have a level of glutamate of 2–10 nmol/sample.
Statistical analysis
Data were analyzed using Excel (Redmond, WA, US) and Origin 8.1 (Northamptom, MA, US). For single comparisons, Student’s t-test was performed. For multiple comparisons, mean differences were tested by non-parametric Kruskal–Wallis analysis and adjusted by use of the Bonferroni–Dunn correction. p values of < 0.05 were considered significant.
Results
Meth treatment alters expression and function of gap junction channels in the CNS in vivo
For all our studies, cortex and hippocampus tissues were analyzed because of their exquisite regulation of learning and memory by dopamine receptors and extensive reporting demonstrating the importance of Cx channels in these cells (Eugenin 2014; Hansen and Manahan-Vaughan 2014; Siekmeier and vanMaanen 2014; Werlen and Jones 2015). Brain tissue sections obtained from meth or PBS-treated animals were stained for Cx36 or Cx43, MAP-2 or GFAP, and DAPI and analyzed by confocal microscopy. First, we analyzed the expression and localization of neuronal Cx36, which is essential to maintain electrical connectivity between different populations of neurons (Deans et al. 2002; Pereda et al. 2003a; Zlomuzica et al. 2012). In control conditions (PBS injected mice), Cx36 was localized in MAP-2 neuronal cell bodies and processes (Fig. 1a, Control). In contrast, meth treatment resulted in decreased overall Cx36 staining in several brain regions, including cortex and hippocampus (Fig. 1a, a reduction in 68.26 ± 18.45% in positive pixels relative to control brains, p ≤ 0.05). Cx36 down-regulation induced by meth was confirmed by western blot analysis of total brain lysates (Fig. 1c). With respect to Cx43, analysis of brain tissue sections indicated that in PBS-treated mice, Cx43 was mainly localized between astrocytes (GFAP positive cells) as expected, whereas meth treatment induced relocalization of the protein into apparently intracellular vesicles (Fig. 1b). Imaging analysis did not show any difference in the total amount of Cx43 expression (data not shown) and western blot analysis corroborated this result (Fig. 1c). We also observed that the cell bodies of neurons from meth-treated animals were larger (by 14.5 ± 5.34%, n = 4 animals, p ≤ 0.05) and had shorter cellular processes (by 51.33 ± 15.97%, p ≤ 0.05) relative to PBS-treated animals. In addition, we observed increased GFAP expression in meth-treated conditions (29.34 ± 6.92% relative to control conditions, p ≤ 0.05), suggesting there is a presence of hypertrophic astrocytes (Fig. 1b). Thus, meth treatment altered the expression and localization of Cx36 and Cx43 containing GJ in vivo.
Fig. 1.
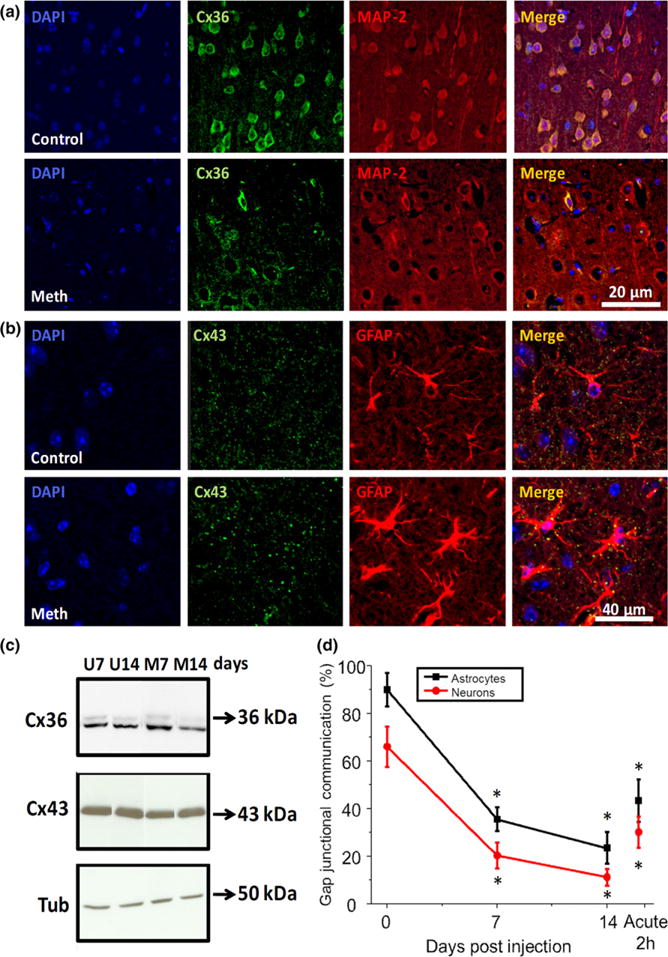
Meth injection into mice decreases gap junctional communication. (a) Murine brain sections were stained for Cx36 and microtubule-associated protein-2 (MAP-2), a neuronal marker, in control (upper panels, control) and meth-treated mice (lower panels, meth). In meth-treated conditions, Cx36 expression was reduced by 68.26 ± 18.45% in MAP-2+ cells. In addition, neuronal cell bodies of meth injected mice were larger by 14.5 ± 5.34% and the number of processes was reduced by 51.33 ± 15.97% compared to sham-injected mice. (b) Brain tissue sections were stained for Cx43 and glial fribillary acid protein (GFAP), a marker of astrocytes, in control (upper panels, control) and meth (lower panels, meth) treated conditions. Meth treatment increased the size of the punctate staining and these puncta were mainly localized in the intracellular areas of the astrocytes, suggesting internalization. No significant changes in total amount of Cx43 were detected in mean intensity measurements (data not represented). However, intensity of GFAP increased by 29.34 ± 6.92% in meth-injected mice, suggesting the drug induced a hypertrophic pathology. (c) Western blots for Cx36, Cx43, and tubulin (Tub) of untreated and meth-treated brain lysates obtained after 7 and 14 days post meth injection. Cx36 was decreased as denoted in Fig. 1a and Cx43 expression was not altered by meth treatment. (d) Dye coupling analysis using acute brain tissue slices stained with sulforhodamine to identify astrocytes. Microinjection of neurobiotin to examine neuron–neuron communication or Lucifer Yellow to examine astrocyte–astrocyte communication was performed. Meth-treated animals displayed a significant reduction in dye coupling between neurons (red line with circles) and astrocytes (black line with squares). Acute application of meth for 2 h into the fresh cut slices obtained from control animals (offset on right) displayed similar reduced dye transfer between neurons and astrocytes. (*p < 0.05, Student’s t-test for all comparisons, n = 4 mice).
To evaluate neuron–neuron and astrocyte–astrocyte communication, we used dye coupling in fresh brain tissue slices obtained from control- and meth-treated animals (Fig. 1d). Under control conditions, astrocytes were almost 100% coupled to other astrocytes when microinjected with LY (Fig. 1d) and 30–50% of neurons were coupled to other neurons when microinjected with neurobiotin (Fig. 1d). Meth treatment strongly reduced (by approximately 80% at 14 days after treatment) GJ communication among neurons and among astrocytes (Fig. 1d). In addition, acute treatment of control brains with meth (10 μM) for 120 min also resulted in reduction in dye transfer as compared to control conditions (Fig. 1d). Thus, our studies indicate that Cxs in neurons and astrocytes are compromised in meth-treated animals, supporting the hypothesis that meth alters electrical and metabolic communication.
Meth treatment of mixed cultures of human neurons and astrocytes reproduces the meth effects in Cxs observed in vivo
To examine the mechanisms by which meth reduces neuronal and glial GJ communication, we used primary human cultures of neurons and astrocytes treated with meth (1 or 10 μM – circulating concentrations observed in meth users) (Mendelson et al. 2006). In these cultures, neurons grow on top of an astrocyte monolayer, and by using confocal microscopy to visualize the different optical planes, astrocytes, and neurons can be distinguished from one another (Eugenin et al. 2007).
Confocal analysis of the top optical section of mixed cultures of neurons and astrocytes indicated that neuronal Cx36 was mainly localized in the cell body and neuronal processes (Fig. 2a, control). Treatment with meth for at least 6 h resulted in decreased Cx36 expression and collapse of neuronal processes (Fig. 2a, meth). Western blot analysis of these mixed cultures indicated that meth treatment decreased Cx36 expression at all time points analyzed (Fig. 2b, Cx36, at least −64.5 ± 11.54% for all time points relative to control conditions, p = 0.003, n = 4). Confocal analysis of astrocytes in the bottom optical section of the mixed cultures of neurons and astrocytes indicated that astrocytic Cx43 was mainly localized in cell to cell apposition membranes as well as in intracellular compartments (Fig. 2c, control). After meth treatment, the number and size of Cx43 containing plaques at the cell to cell apposition membranes was reduced (Fig. 2c, see arrows) and most of the Cx43 staining became intracellular, suggesting Cx43 internalization. Western blot analysis corroborated our confocal data – no changes in total Cx43 expression was detected after meth treatment (Fig. 2d). To further study the possibility that meth induced the internalization of Cx43 plaques, live cell imaging of HeLa cells transfected with Cx43-GFP was performed. Meth treatment (10 μM) induced a fast internalization (quantified as plaque instability) of Cx43 containing plaques present at the plasma membrane into intracellular compartments (Figure S1a and b). In summary, our data indicates that meth reduces Cx36 expression and induced the internalization of Cx43 channels.
Fig. 2.
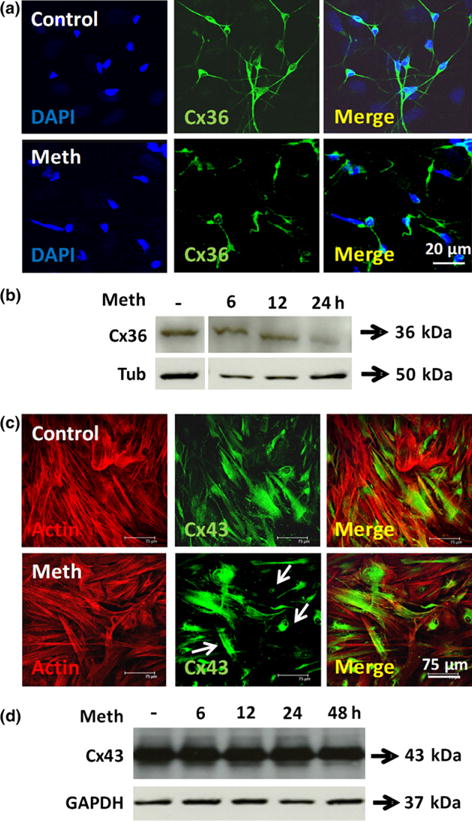
Gap junctions containing Cx36 and Cx43 in mixed cultures of human neurons and astrocytes are highly sensitive to meth treatment. (a) Human primary mixed cultures of neurons and astrocytes treated with meth (1 or 10 μM) and stained for Cx36 (green staining), and 4′,6-diamidino-2-phenylindole (DAPI) (blue staining) to identify nuclei. Neurons grow on top of the monolayer of astrocytes, thus, optical sectioning allows separation of both cell types. Meth treatment for 6, 12, 24 or 48 h decreased Cx36 expression at all times tested (figure correspond to 6 h post treatment). A 64.5 ± 11.45% in Cx36 expression and a reduction in the number of neuronal processes were observed in meth-treated conditions. (b) Western blot analysis indicates that Cx36 expression was decreased as soon as 6 h after meth treatment. Tubulin (tub) was used as a loading control. (c) Human primary mixed cultures of neurons and astrocytes treated with meth (1 or 10 μM) and stained for Cx43 (green staining), and actin (red staining) and DAPI (blue staining) to identify nuclei. Meth treatment for 6, 12, 24, or 48 h induced the internalization of Cx43 (figure corresponds to 6 h post treatment). But no significant differences in total green staining were detected between both conditions. (d) Western blot analysis of these cultures indicates that Cx43 expression was not altered by meth treatment. Tubulin (tub) was used as a loading control. n = 6.
Meth treatment alters GJ communication
To examine whether the changes in Cx36 and Cx43 localization result in alterations in GJ communication, dye coupling using neurobiotin (to evaluate neuronal GJ communication) and LY (to evaluate astrocyte GJ communication) was performed. Evaluation of neuronal GJ communication using confocal microscopy indicated that under normal conditions 60% of neurons were coupled to other neurons (Fig. 3a and b, 2.34 ± 0.47 neurons per microinjection). Meth treatment (1 μM or 10 μM) reduced coupling between neurons to 10% (Fig. 3b). Furthermore, meth treatment also resulted in near total shutdown of GJC between astrocytes (numbers after meth treatment) as compared to control conditions (11.76 ± 6.21 astrocytes per microinjection, Fig. 3c). Together, these results indicate that meth impairs GJ communication by modifying the expression and functional state of Cx containing channels in neurons and astrocytes.
Fig. 3.
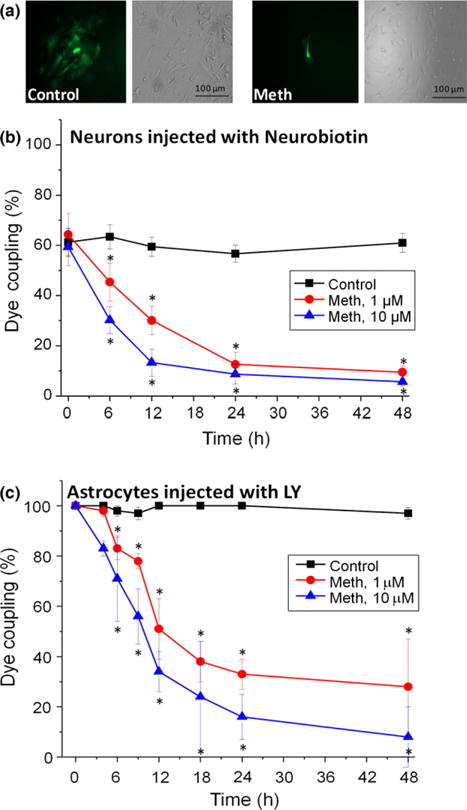
Meth treatment impairs gap junctional communication in human neurons and astrocytes. Mixed cultures of neurons and astrocytes were examined by dye coupling using neurobiotin for neurons and Lucifer yellow (LY) for astrocytes. (a) Corresponds to a representative example of dye transfer under control and meth (10 μM) treated astrocytes. (b) Corresponds to neuronal dye coupling in control and meth-treated conditions (1 and 10 μM). Dye coupling was examined after 6, 12, 24, and 48 h post treatment. (c) Corresponds to the results of astrocyte-astrocyte dye coupling under control and meth-treated conditions (1 and 10 μM). In both cases, neurons and astrocytes, meth treatment reduced gap junctional communication. (*p < 0.05, n = 5 different tissues).
Meth treatment induces the opening of hemichannels
While non-docking HC that do not participate in forming GJ are normally closed, they can become open under stress and inflammatory conditions (Kielian 2008; Eugenin et al. 2012; Eugenin 2014). To examine whether meth affects the opening of these channels, mixed cultures of neurons and astrocytes were subjected to Etd dye uptake to examine opening of HC as we described (Orellana et al. 2011, 2012b). In control conditions no HC opening was detected (Fig. 4a). In contrast, meth treatment induced a strong and fast HC opening (Fig. 4a, 5–15 min, * corresponds to p ≤ 0.002 relative to control, n = 4), as well as a lesser, but still significant (p = 0.007), HC opening after 18 to 48 h of treatment. To identify the type of HC opened in response to meth treatment, we used Cx HC blockers such as lanthanum (La+3, a connexin HC blocker) and Cx43 and Cx36 extracellular mimetic peptides (both Cx HC blockers). We found that all Cx HC blockers prevented Etd uptake induced by meth treatment (Fig. 4b, #p = 1.2 × 10−4, n = 4). These data suggest that Cx43 and Cx36 HC are opened in response to meth treatment.
Fig. 4.
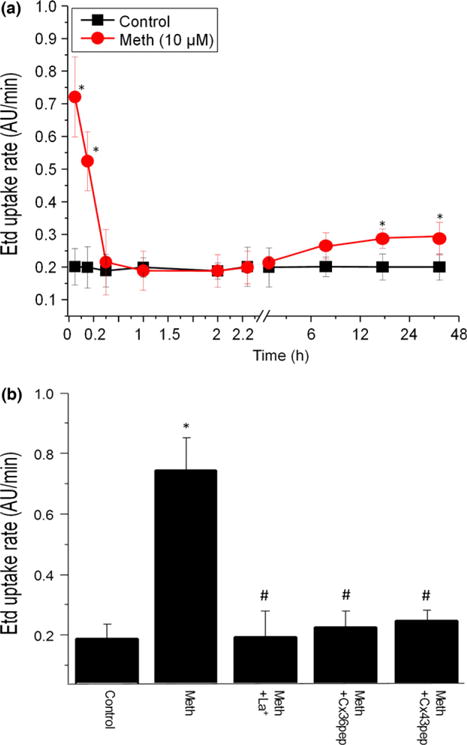
Meth induces the opening of hemichannels in mixed cultures of human neurons and astrocytes. (a) The rate of ethidium (Et) uptake by glial cells incubated in the absence or presence meth was measured up to 48 h. An increase in channel opening was evident in the first 5–15 min after meth (10 μM, lines with circles) treatment, followed by a smaller but significant opening between 6 and 48 h (lines with circles). No channel opening was observed in untreated cultures (control, lines with squares). (b) Connexin hemichannels blockers, but not pannexin-1 hemichannel blockers (data not shown), reduces the opening of the hemichannel induced by meth treatment. Lanthanum, 100 μM, La+3; Cx36 mimetic peptide, 500 μM; and Cx43 mimetic peptide, 500 μM) prevented Et uptake induced by meth. The time point analyzed correspond to 10 min after treatment. [*p < 0.05 as compare to control conditions (con, c), #p < 0.05 as compared to meth-treated conditions, n = 4].
Fig. 5.
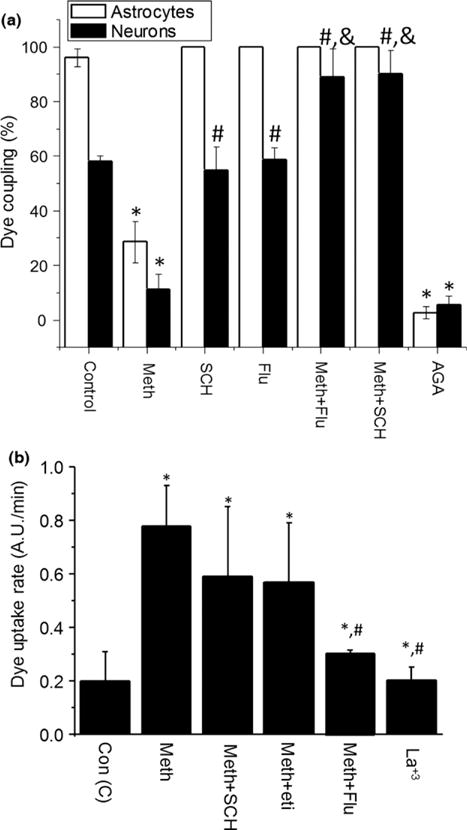
Activation of dopamine receptors in response to meth is involved in reduction in gap junctional communication and increased opening of connexin hemichannels. Mixed cultures of human neurons and astrocytes were analyzed for gap junctional communication by dye coupling and opening of hemichannels by dye uptake. (a) Dopamine receptor blockers SCH23390 (SCH, 10 μM; specific D1 antagonist) and flupenthixol (Flu, 1 μM; non-specific dopamine receptor blocker) prevented the decrease in gap junctional communication (GJC) induced by meth (meth+SCH or meth+Flu, respectively) in astrocytes (white bars) and neurons (bars with cross lines). Addition of SCH or Flu to control cells did not affect dye coupling. The gap junction/hemichannel blocker 18-alpha-glycyrrhetinic acid (AGA) was used as control (18-α-glycyrrhetinic acid, a GJ blocker). (b) D1 and D2 like dopamine receptors are involved in opening of connexin hemichannels induced for meth treatment. Opening of connexin hemichannels was not blocked by D1 antagonist (SCH23390, SCH) or D2 antagonist (Eticlopride, Eti). However, flupenthixol (Flu, 1 μM; nonspecific dopamine receptor blocker) blocked the opening of connexin hemichannels induced by meth. Lanthanum (La+3) was used as a positive control of blocking. (*p < 0.05 as compared to control conditions, #p < 0.05 as compared to meth-treated conditions, &p < 0.05 as compared to SCH or Flu–treated conditions. n = 4)
Meth-induced GJ communication compromise is mediated by activation of D1 like dopamine receptors
Several reports indicate that dopamine receptors can modulate GJ permeability (Baldridge et al. 1987, 1998; Hampson et al. 1992; Chiba and Saito 2000; Tibber et al. 2007; Kothmann et al. 2009; Bu et al. 2014), but it is unknown whether meth can alter GJC and HC opening indirectly by dysregulation of dopamine receptors. Thus, to examine whether dopamine and its receptors are involved in meth-mediated reduction in neuronal and glial GJ communication, we determined whether blocking D1 like or D2 like dopamine receptors affected dye transfer (GJ communication) or dye uptake (HC opening) in mixed neuron-astrocyte cultures. The pre-incubation of the mixed cultures with SCH23390 (1 and 10 μM, a selective D1 like dopamine receptor antagonist, SCH), eticlopride (10 and 50 μM, a selective D2 like receptor blocker, Eti) or Flupenthixol (1 μM, a non-selective dopamine receptor blocker, Flu) before meth treatment (10 μM) reduced the deleterious effects of meth on GJ communication (Fig. 5a). SCH and Flu alone did not affect GJ communication (Fig. 5a). In contrast to D1 like dopamine blockers, the D2 like receptor blocker, eticlopride, did not have any effect on astrocyte or neuronal GJ communication in the presence or absence of meth (data not shown). These results indicated that D1 like dopamine receptor opening was necessary for meth to compromise GJ communication.
Meth-induced HC opening was mediated by both D1 and D2 like dopamine receptors
To examine whether opening of HC in response to meth treatment was also dependent upon dopamine receptor activation, we tested the dopamine receptor blockers described above. Blocking D1 like dopamine receptors using SCH23390 (1 and 10 μM, n = 4) or D2 like dopamine receptors using eticlopride (10 or 50 μM, n = 3), did not alter HC opening (Fig. 5b). However, flupenthixol (1 μM, a non-selective dopamine blocker, Flu) prevented HC opening in response to meth treatment (Fig. 5b). These results suggest the involvement of an alternative dopamine receptor or a more complex receptor, such as that formed by hetero-oligomers of D1 and D2 receptors as previously described (So et al. 2009; Ferre et al. 2010). Alternatively, a different neurotransmitter may mediate meth-induced HC opening, as meth also alters the release of serotonin and epinephrine (Yu et al. 2015).
Meth treatment dysregulates glutamate release and increases bystander apoptosis in HIV-infected cultures
Meth accelerates the pathogenesis of several diseases including HIV-associated neurocognitive disorders (Loftis and Janowsky 2014; Borgmann and Ghorpade 2015; Passaro et al. 2015; Yu et al. 2015). Since GJ and HC are essential for clearing potentially toxic concentrations of glutamate from regions in the brain with high neuronal activity (Kielian 2008; Eugenin et al. 2012; Orellana et al. 2012b; Verkhratsky et al. 2012; Eugenin 2014), and meth use correlated with accelerated HIV-associated neurocognitive disorders pathogenesis, we asked if combined HIV infection and meth use could result in extracellular glutamate accumulation. Therefore, we assessed whether meth alone, or in combination with HIV, dysregulates the extracellular concentration of glutamate.
We found that HIV infection or meth treatment of astrocytes for 7, 14, or 21 days resulted in a significant increase in extracellular glutamate relative to control conditions (Fig. 6a). Meth treatment of HIV-infected astrocytes further increased the accumulation of extracellular levels of glutamate (Fig. 6a, HIV+meth). Moreover, blocking HC using La+3 or blocking GJ using 18-α-glycyrrhetinic acid or mimetic peptides totally abolished the glutamate dysregulation induced by meth, HIV infection, or the combination of both (Fig. 6a, HC/GJ blockers). Therefore, meth and HIV induce glutamate release by a mechanism involving HC and GJ channels.
Fig. 6.
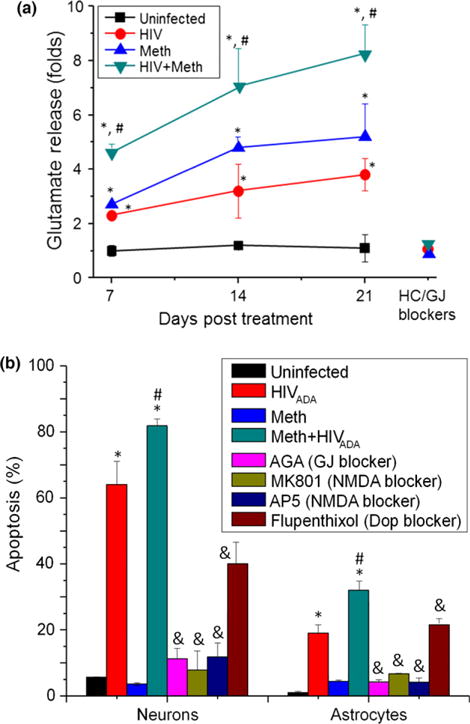
Meth and human immunodeficiency virus-1 (HIV) increase glutamate release and meth increases HIV-induced apoptosis. (a) Fold changes of extracellular glutamate were quantified in culture supernatant of astrocytes by ELISA. Control cultures have low amount of extracellular glutamate (lines with squares). HIV-infected or meth-treated cultures have increased extracellular accumulation of glutamate. The combination of HIV infection and meth treatment resulted in an additive effect and increased the release of glutamate 8 fold. In addition, of hemichannel blockers (mimetic peptides) and gap junction blockers (AGA) resulted in prevention of glutamate dysregulation. (*p < 0.05 as compared to control conditions, #p < 0.05 as compared to meth or HIV-treated conditions. n = 4). (b) Meth increased the bystander apoptosis induced by HIV infection of astrocytes. Apoptosis was evaluated by TUNEL staining. Basal apoptosis was minimal in neurons and astrocytes (black bars). HIV infection of astrocytes resulted in ~ 60% apoptosis in neurons and ~ 18% in uninfected astrocytes after 14 days post infection. HIV-infected astrocytes are protected from apoptosis (red bars) but most uninfected astrocytes undergo apoptosis. Meth treatment alone (1 and 10 μM) did not induce apoptosis in neurons and astrocytes (blue bars). The combination of HIV infection and meth treatment increased apoptosis of neurons to ~ 82% and astrocytes to ~ 33% (light blue bars). Addition of a gap junction (GJ) blocker, AGA, totally blocks spread of apoptosis from HIV-infected astrocytes into uninfected surrounding cells, neurons and astrocytes (pink bars). As we previously demonstrated, NMDAR activity in neurons is essential to trigger apoptosis in uninfected neurons and astrocytes induced by HIV infection of astrocytes. In agreement, blocking NMDAR activation using MK801 or AP-5, totally blocks apoptosis of neurons and astrocytes induced by HIV-infected astrocytes and meth (green and dark blue bars, respectively). Addition of flupenthixol, a pan dopamine receptor blocker, only partially reduced the apoptosis induced by HIV-infected astrocytes and meth treatment, suggesting a partial participation. Even when higher concentrations of this blocker were used (×100), no significant improvement in blocking apoptosis was observed (brown bars). Addition of blockers alone or in the presence of Meth did not altered apoptosis levels (data not shown). (*p < 0.05 as compared to control conditions, #p < 0.05 as compared to HIV infection alone, &p < 0.05 as compared to HIV-infected and meth-treated conditions. n = 4)
Meth increases the susceptibility of neurons and astrocytes to apoptosis
Our previous publications demonstrated a phenomenon termed, ‘bystander apoptosis’, where HIV-infected astrocytes spread apoptosis to uninfected astrocytes and neurons by a mechanism involving GJ and HC (Eugenin and Berman 2007, 2013; Eugenin et al. 2011). In addition, meth is known to increase CNS compromise (Tiwari et al. 2013; Bortell et al. 2015; Kesby et al. 2015). Thus, we analyzed whether meth treatment alters bystander apoptosis elicited by the few HIV-infected astrocytes into uninfected cells, as well as the role of GJ, N-methyl-D-aspartate receptor (NMDA), and dopamine receptors in this process. Consistent with our previous results (Eugenin and Berman 2007, 2013; Eugenin et al. 2007, 2011, 2012; King et al. 2010; Orellana et al. 2013a; Castellano and Eugenin 2014; Eugenin 2014), HIV infection of astrocytes resulted in bystander apoptosis of uninfected neurons and astrocytes in a GJ-dependent mechanism (Fig. 6b, HIVADA, *p ≤ 0.006, n = 4, relative to uninfected conditions). Pretreatment with meth (10 μM, 6 h prior to HIV infection) further enhanced neuronal and astrocyte apoptosis by 15 to 20% (Fig. 6b, meth+HIVADA, #, p ≤ 0.003, n = 4, as compared to HIVADA condition). Meth treatment in the absence of HIV infection did not induce apoptosis (Fig. 6b, meth). Furthermore, blocking GJ channels using 18-alpha-glycyrrhetinic acid (32 μM), blocking NMDA receptor activation using MK801 or (2R)-amino-5-phosphonovaleric acid; (2R)-amino-5-phosphonopentanoate (5 μM), or blocking dopamine receptor activation using flupenthixol (1 μM) all reduced bystander apoptosis (&, p ≤ 0.007, n = 4 as compared to meth+HIVADA). Together, these results indicate that meth enhances bystander apoptosis elicited by HIV-infected astrocytes in a manner dependent on GJ, NMDA receptor, and dopamine receptor signaling.
Discussion
In this study, we demonstrate that meth has devastating effects on neurons and astrocytes by dysregulating connexin containing channels by dopamine receptor mediated mechanism. In addition, we showed that meth increased glutamate deregulation release and increased susceptibility of uninfected neurons and astrocytes to HIV-mediated bystander apoptosis. Together, these results indicate that meth has profound negative effects in GJ communication and opening of HC. These changes in connexin containing channels, GJ and HC, alter glutamate metabolism and increase the sensitivity of neurons and astrocytes to HIV-induced apoptosis. Thus, meth not only has devastating effects in uninfected individuals but also in HIV-infected individuals where accelerated CNS disease is observed.
Most drugs of abuse generate a lack of electrical coordination between different areas of the brain, but the mechanism of this dysregulation is unknown. On the basis of our data, we propose that lack of electrical coordination in meth users is due to reduced GJC. Currently, several treatments to re-coordinate this deficiency in drug users have been attempted, such as deep brain stimulation in the affected areas (see reviews Feil and Zangen 2010; Luigjes et al. 2012). Interestingly, most of these changes in humans can be recapitulated in animals lacking Cxs. For example, deletion of Cx36 impairs learning and memory by altering electrotonic coupling without significant changes in monoamine expression (Frisch et al. 2005). Deletion of Cx43 results in accelerated spreading depression, accelerated locomotor activity (Theis et al. 2003), and increased exploratory behavior (Frisch et al. 2003), whereas deletion of Cx30 results in increased emotionality (Dere et al. 2003). Furthermore, down-regulation of GJ communication causes gamma frequency oscillations in the hippocampus (Hormuzdi et al. 2001), impairs cerebellar motor learning (van de Giessen et al. 2009) and high gain rod photoreceptors (Deans et al. 2002; Volgyi et al. 2004) in association with dephosphorylation of Cx36, suggesting that Cxs containing channels are essential for all these functions. All these changes are key behavioral features also observed in meth users and not associated directly with large changes in monoamines. Thus, electrical deficiency or coordination is a key feature often observed in drug users and may be related to alterations in Cxs containing channels.
In physiological conditions, it is known that dopamine down-regulates GJ communication in the retina by activation of D1 like receptors (McMahon et al. 1989; Hampson et al. 1992; Velazquez et al. 1997; Weiler et al. 2000; Nolan et al. 2007; Li et al. 2013). Recently, Pereda and collaborators, demonstrated that opiods potentiate electrical transmission in mixed synapses (chemical and electrical) on the Mauthner cells (Cachope et al. 2007; Cachope and Pereda 2015), suggesting that other drugs act in a different manner within the CNS. Our data indicate that meth induces down-regulation of GJ communication and opening of HC, which is mediated by activation of D1 like dopamine receptors. We propose that problems in electrical coordination in cases of drug abuse involves the dysregulation of GJC and HC opening because of activation of specific dopamine receptors. Therefore, several problems of electrical coordination in response to drug abuse may be prevented by drugs that target dopamine receptors. This also indicates that individuals taking medications that target the dopamine receptors may be at risk of having unwanted effects in GJC and HC opening, which contributes to additional CNS problems related to cognition and increased susceptibility to infectious diseases in the CNS.
In the last decade, the role of HC has been described in several pathological conditions. The opening of Cx HC results in the release of ATP, glutamate, and other intracellular molecules leading to activation of ATP receptors and inflammatory pathways (Pelegrin and Surprenant 2006, 2007; Locovei et al. 2007). Normally, HC are closed because of their high permeability to prevent release of pro-inflammatory molecules, cellular activation, and apoptosis. However, we show here that meth treatment induces HC opening, which is likely to result in synaptic compromise, inflammation, neurotransmitter recycling dysregulation, and secretion of immune/CNS factors such as glutamate and ATP. Indeed, HC opening and reduction in GJC has been associated with severe problems in learning and memory as well as fear and other behavioral issues (Frisch et al. 2005; Allen et al. 2011; Bissiere et al. 2011; Mercer 2012; Stehberg et al. 2012; Zlomuzica et al. 2012). Our data also indicate that opening of HC contributes to glutamate dysregulation and to the amplification of apoptosis in response to HIV. These results provide mechanistic support for several reports describing the potentiation of drug abuse and cognitive deficits in the HIV-infected population (Scott et al. 2007; Watkins and Treisman 2012; Alfahad and Nath 2013; Purohit et al. 2013; Anderson et al. 2015).
Interestingly, we found that in the context of HIV, meth increases the percentage of HIV-infected astrocytes (data not shown, Eugenin E.A. and Martinez L.R. unpublished data), suggesting a regulation of HIV entry and/or replication. Currently, whether meth increases HIV infection/replication is controversial, but several reports have indicated that meth increases the infectivity of CD4+ T cells and monocyte-derived macrophages (Liang et al. 2008; Nair et al. 2009). Others have reported that meth does not affect viral replication in CD4+ T cells (Mantri et al. 2014). Experiments using meth-treated rats injected with HIV-tat show synergistic reduction in levels of dopamine and its metabolites, probably because of destruction of dopaminergic terminals (Flora et al. 2003). Similar results of dopaminergic compromise and monocyte recruitment into the brain were found in monkeys (Theodore et al. 2006a,b). In agreement, studies in patients indicate that meth correlates with higher plasma HIV replication (Ellis et al. 2003; Carrico et al. 2007). Furthermore, recent studies indicate that dopamine increases HIV entry into monocyte-derived macrophages, suggesting a regulation of HIV receptors on the surface of these cells (Gaskill et al. 2009). However, there are no studies of meth-induced HIV potentiation in the CNS. Future studies will identify the mechanism(s) by which meth increases HIV infection of astrocytes.
Our previous publications demonstrated that HIV-infected astrocytes induced bystander apoptosis of uninfected cells by several mechanisms, including: maintenance of Cx43 between HIV-infected cells and uninfected cells; transfer of toxic signals from HIV-infected astrocytes into uninfected cells (mostly IP3 and calcium) by GJ; extended survival of HIV-infected cells; and activation of NMDA receptor in neurons (Eugenin and Berman 2007; Eugenin et al. 2011). In this study, we demonstrated that meth treatment of mixed cultures of human neurons and astrocytes enhanced HIV-mediated bystander apoptosis by a NMDA receptor-mediated mechanism (Fig. 6b). This enhancement of apoptosis may be because of the extracellular accumulation of glutamate; however, we also demonstrated that enhanced apoptosis induced by meth was GJ/HC dependent and mediated by dopamine receptors. In agreement, it was shown that cocaine-enhanced T-cell apoptosis in response to HIV infection (Pandhare et al. 2014), suggesting that drugs that alter the dopaminergic system contribute to the pathogenesis of HIV and NeuroAIDS by increasing apoptosis. Interestingly, while our previous studies show that bystander apoptosis from HIV infection is GJ-dependent, in this study we show meth reduces GJ communication. Therefore, it is possible that HIV-infected astrocytes maintain GJ communication despite meth treatment, which would allow for the amplification of apoptosis from bystander killing and HC opening.
On the basis of our published and new data, we propose the following sequence of events: (i) meth results in an increase in the release of dopamine in dopaminergic terminals; (ii) increased extracellular levels of dopamine overactivates D1 and D2 like dopamine receptors; (iii) resulting in the reduction in neuronal and glial GJ communication and HC opening; (iv) dysregulation of Cx containing channels results in glutamate dysregulation; (v) which in turn results in activation of glutamate receptors, including NMDA receptors; (vi) these changes result in increased sensitivity of neurons and astrocytes to apoptosis, which is further increased in the presence of HIV infection. We also propose that HIV-infected glial cells maintain GJ communication necessary for bystander apoptosis, and that these cells may be protected from glutamate toxicity by an unknown mechanism.
In conclusion, our study provides insight into the understanding of the pathological effects of meth and HIV on the CNS. These data represent a novel mechanism in drug abuse in NeuroAIDS pathology and indicate potential new targets for therapeutic interventions to reduce the CNS effects of meth use and HIV.
Supplementary Material
Figure S1. Meth induces internalization of Cx43 plaques in HeLa cells. HeLa cells stably transfected with Cx43-GFP were grown to confluence, treated with meth, and subjected to time-lapse imaging. Meth reduced plaque stability by inducing internalization of Cx43. (a) representative example of time-lapse image sequence of Cx43 plaque internalization. (b) Quantification of plaque stability in control and meth-treated cultures over 12 h. (*p < 0.05 as compared to control conditions).
Acknowledgments
We thank the Fetal Tissue Repository at the Albert Einstein College of Medicine, Bronx, NY and the Alfred P. Sloan Foundation Minority fellowship (to P.C.). This work was supported by the National Institutes of Mental Health grant, MH096625, and PHRI funding (to E. A. E.). L.R.M. was supported by the NYIT COM start-up funds.
All experiments were conducted in compliance with the ARRIVE guidelines.
Abbreviations
- 5HT
serotonin
- AGA
18-alpha-glycyrrhetinic acid
- AP-5
(2R)-amino-5-phosphono valeric acid; (2R)-amino-5-phosphonopentanoate
- ART
antiretroviral treatment
- ATP
adenosine triphosphate
- CNS
central nervous system
- Cx
connexin
- DAPI
4′,6-diamidino-2-phenylindole
- Dop
dopamine
- ECT
ecticlopride
- Flu
flupenthixol
- GFAP
glial fribillary acid protein
- GJC
gap junctional communication
- GJ
gap junction
- HC
hemichannels
- HIV
human immunodeficiency virus-1
- LY
Lucifer yellow
- MAP-2
microtubule-associated protein-2
- meth
methamphetamine
- NADH
nicotinamide adenine dinucleotide
- NE
norepinephrine
- NMDA
N-methyl-D-aspartate receptor
- PGE2
prostaglandin E2
- SCH
SCH23390
Footnotes
Conflict of interest disclosure
The authors declare no conflict of interest.
Author contributions
All the authors were involved in the preparation of the manuscript, design of the experiments, analysis of the data, and intellectual content.
Supporting information
Additional Supporting Information may be found online in the supporting information tab for this article.
References
- Abudara V, Bechberger J, Freitas-Andrade M, DeBock M, Wang N, Bultynck G, Naus CC, Leybaert L, Giaume C. The connexin43 mimetic peptide Gap19 inhibits hemichannels without altering gap junctional communication in astrocytes. Front Cell Neurosci. 2014;8:306. doi: 10.3389/fncel.2014.00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfahad TB, Nath A. Update on HIV-associated neurocognitive disorders. Curr Neurol Neurosci Rep. 2013;13:387. doi: 10.1007/s11910-013-0387-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen K, Fuchs EC, Jaschonek H, Bannerman DM, Monyer H. Gap junctions between interneurons are required for normal spatial coding in the hippocampus and short-term spatial memory. J Neurosci. 2011;31:6542–6552. doi: 10.1523/JNEUROSCI.6512-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson AM, Higgins MK, Ownby RL, Waldrop-Valverde D. Changes in neurocognition and adherence over six months in HIV-infected individuals with cocaine or heroin dependence. AIDS Care. 2015;27:333–337. doi: 10.1080/09540121.2014.985183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge WH, Ball AK, Miller RG. Dopaminergic regulation of horizontal cell gap junction particle density in goldfish retina. J Comp Neurol. 1987;265:428–436. doi: 10.1002/cne.902650310. [DOI] [PubMed] [Google Scholar]
- Baldridge WH, Vaney DI, Weiler R. The modulation of intercellular coupling in the retina. Semin Cell Dev Biol. 1998;9:311–318. doi: 10.1006/scdb.1998.0235. [DOI] [PubMed] [Google Scholar]
- Beardsley PM, Hauser KF. Glial modulators as potential treatments of psychostimulant abuse. Adv Pharmacol. 2014;69:1–69. doi: 10.1016/B978-0-12-420118-7.00001-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JE, Arango JC, Anthony IC. Neurobiology of multiple insults: HIV-1-associated brain disorders in those who use illicit drugs. J Neuroimmune Pharmacol. 2006;1:182–191. doi: 10.1007/s11481-006-9018-2. [DOI] [PubMed] [Google Scholar]
- Berthoud VM, Ledbetter ML, Hertzberg EL, Sáez JC. Connexin43 in MDCK cells: regulation by a tumor-promoting phorbol ester and Ca2+ Eur J Cell Biol. 1992;57:40–50. [PubMed] [Google Scholar]
- Bissiere S, Zelikowsky M, Ponnusamy R, Jacobs NS, Blair HT, Fanselow MS. Electrical synapses control hippocampal contributions to fear learning and memory. Science. 2011;331:87–91. doi: 10.1126/science.1193785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgmann K, Ghorpade A. HIV-1, methamphetamine and astrocytes at neuroinflammatory Crossroads. Front Microbiol. 2015;6:1143. doi: 10.3389/fmicb.2015.01143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortell N, Morsey B, Basova L, Fox HS, Marcondes MC. Phenotypic changes in the brain of SIV-infected macaques exposed to methamphetamine parallel macrophage activation patterns induced by the common gamma-chain cytokine system. Front Microbiol. 2015;6:900. doi: 10.3389/fmicb.2015.00900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Bu JY, Li H, Gong HQ, Liang PJ, Zhang PM. Gap junction permeability modulated by dopamine exerts effects on spatial and temporal correlation of retinal ganglion cells’ firing activities. J Comput Neurosci. 2014;36:67–79. doi: 10.1007/s10827-013-0469-1. [DOI] [PubMed] [Google Scholar]
- Burdo TH, Katner SN, Taffe MA, Fox HS. Neuroimmunity, drugs of abuse, and neuroAIDS. J Neuroimmune Pharmacol. 2006;1:41–49. doi: 10.1007/s11481-005-9001-3. [DOI] [PubMed] [Google Scholar]
- Cachope R, Pereda AE. Opioids potentiate electrical transmission at mixed synapses on the Mauthner cell. J Neurophysiol. 2015;114:689–697. doi: 10.1152/jn.00165.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachope R, Mackie K, Triller A, O’Brien J, Pereda AE. Potentiation of electrical and chemical synaptic transmission mediated by endocannabinoids. Neuron. 2007;56:1034–1047. doi: 10.1016/j.neuron.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrico AW, Johnson MO, Moskowitz JT, et al. Affect regulation, stimulant use, and viral load among HIV-positive persons on anti-retroviral therapy. Psychosom Med. 2007;69:785–792. doi: 10.1097/PSY.0b013e318157b142. [DOI] [PubMed] [Google Scholar]
- Castellano P, Eugenin EA. Regulation of gap junction channels by infectious agents and inflammation in the CNS. Front Cell Neurosci. 2014;8:122. doi: 10.3389/fncel.2014.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba C, Saito T. Gap junctional coupling between progenitor cells of regenerating retina in the adult newt. J Neurobiol. 2000;42:258–269. doi: 10.1002/(sici)1097-4695(20000205)42:2<258::aid-neu9>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Chiu VM, Schenk JO. Mechanism of action of methamphetamine within the catecholamine and serotonin areas of the central nervous system. Curr Drug Abuse Rev. 2012;5:227–242. doi: 10.2174/1874473711205030227. [DOI] [PubMed] [Google Scholar]
- Contreras JE, Sanchez HA, Eugenin EA, Speidel D, Theis M, Willecke K, Bukauskas FF, Bennett MV, Saez JC. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc Natl Acad Sci USA. 2002;99:495–500. doi: 10.1073/pnas.012589799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras JE, Saez JC, Bukauskas FF, Bennett MV. Gating and regulation of connexin 43 (Cx43) hemichannels. Proc Natl Acad Sci USA. 2003;100:11388–11393. doi: 10.1073/pnas.1434298100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darke S, Kaye S, McKetin R, Duflou J. Major physical and psychological harms of methamphetamine use. Drug Alcohol Rev. 2008;27:253–262. doi: 10.1080/09595230801923702. [DOI] [PubMed] [Google Scholar]
- Deans MR, Volgyi B, Goodenough DA, Bloomfield SA, Paul DL. Connexin36 is essential for transmission of rod-mediated visual signals in the mammalian retina. Neuron. 2002;36:703–712. doi: 10.1016/s0896-6273(02)01046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dere E, De Souza-Silva MA, Frisch C, Teubner B, Sohl G, Willecke K, Huston JP. Connexin30-deficient mice show increased emotionality and decreased rearing activity in the open-field along with neurochemical changes. Eur J Neurosci. 2003;18:629–638. doi: 10.1046/j.1460-9568.2003.02784.x. [DOI] [PubMed] [Google Scholar]
- Ellis RJ, Childers ME, Cherner M, Lazzaretto D, Letendre S, Grant I. Increased human immunodeficiency virus loads in active methamphetamine users are explained by reduced effectiveness of antiretroviral therapy. J Infect Dis. 2003;188:1820–1826. doi: 10.1086/379894. [DOI] [PubMed] [Google Scholar]
- Eugenin EA. Role of connexin/pannexin containing channels in infectious diseases. FEBS Lett. 2014;588:1389–1395. doi: 10.1016/j.febslet.2014.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, Berman JW. Chemokine-dependent mechanisms of leukocyte trafficking across a model of the blood-brain barrier. Methods. 2003;29:351–361. doi: 10.1016/s1046-2023(02)00359-6. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, Berman JW. Gap junctions mediate human immunodeficiency virus-bystander killing in astrocytes. J Neurosci. 2007;27:12844–12850. doi: 10.1523/JNEUROSCI.4154-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, Berman JW. Cytochrome C dysregulation induced by HIV infection of astrocytes results in bystander apoptosis of uninfected astrocytes by an IP and Calcium dependent mechanism. J Neurochem. 2013;127:644–651. doi: 10.1111/jnc.12443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, Gonzalez H, Saez CG, Saez JC. Gap junctional communication coordinates vasopressin-induced glycogenolysis in rat hepatocytes. Am J Physiol. 1998;274:G1109–G1116. doi: 10.1152/ajpgi.1998.274.6.G1109. [DOI] [PubMed] [Google Scholar]
- Eugenín EA, Eckardt D, Theis M, Willecke K, Bennett MV, Saez JC. Microglia at brain stab wounds express connexin43 and in vitro form functional gap junctions after treatment with interferon-gamma and tumor necrosis factor-alpha. Proc Natl Acad Sci USA. 2001;98:4190–4195. doi: 10.1073/pnas.051634298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, D’Aversa TG, Lopez L, Calderon TM, Berman JW. MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J Neurochem. 2003;85:1299–1311. doi: 10.1046/j.1471-4159.2003.01775.x. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, King JE, Nath A, Calderon TM, Zukin RS, Bennett MV, Berman JW. HIV-tat induces formation of an LRP-PSD-95- NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc Natl Acad Sci USA. 2007;104:3438–3443. doi: 10.1073/pnas.0611699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, Clements JE, Zink MC, Berman JW. Human immunodeficiency virus infection of human astrocytes disrupts blood-brain barrier integrity by a gap junction-dependent mechanism. J Neurosci. 2011;31:9456–9465. doi: 10.1523/JNEUROSCI.1460-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, Basilio D, Saez JC, Orellana JA, Raine CS, Bukauskas F, Bennett MV, Berman JW. The role of gap junction channels during physiologic and pathologic conditions of the human central nervous system. J Neuroimmune Pharmacol. 2012;7:499–518. doi: 10.1007/s11481-012-9352-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil J, Zangen A. Brain stimulation in the study and treatment of addiction. Neurosci Biobehav Rev. 2010;34:559–574. doi: 10.1016/j.neubiorev.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Ferre S, Lluis C, Lanciego JL, Franco R. Prime time for G-protein-coupled receptor heteromers as therapeutic targets for CNS disorders: the dopamine D(1)-D(3) receptor heteromer. CNS Neurol Disord Drug Targets. 2010;9:596–600. doi: 10.2174/187152710793361603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrucci M, Giorgi FS, Bartalucci A, Busceti CL, Fornai F. The effects of locus coeruleus and norepinephrine in methamphetamine toxicity. Curr Neuropharmacol. 2013;11:80–94. doi: 10.2174/157015913804999522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flora G, Lee YW, Nath A, Hennig B, Maragos W, Toborek M. Methamphetamine potentiates HIV-1 Tat protein-mediated activation of redox-sensitive pathways in discrete regions of the brain. Exp Neurol. 2003;179:60–70. doi: 10.1006/exnr.2002.8048. [DOI] [PubMed] [Google Scholar]
- Flores CE, Nannapaneni S, Davidson KG, Yasumura T, Bennett MV, Rash JE, Pereda AE. Trafficking of gap junction channels at a vertebrate electrical synapse in vivo. Proc Natl Acad Sci USA. 2012;109:E573–E582. doi: 10.1073/pnas.1121557109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch C, Theis M, De Souza Silva MA, Dere E, Sohl G, Teubner B, Namestkova K, Willecke K, Huston JP. Mice with astrocyte-directed inactivation of connexin43 exhibit increased exploratory behaviour, impaired motor capacities, and changes in brain acetylcholine levels. Eur J Neurosci. 2003;18:2313–2318. doi: 10.1046/j.1460-9568.2003.02971.x. [DOI] [PubMed] [Google Scholar]
- Frisch C, De Souza-Silva MA, Sohl G, Guldenagel M, Willecke K, Huston JP, Dere E. Stimulus complexity dependent memory impairment and changes in motor performance after deletion of the neuronal gap junction protein connexin36 in mice. Behav Brain Res. 2005;157:177–185. doi: 10.1016/j.bbr.2004.06.023. [DOI] [PubMed] [Google Scholar]
- Gaskill PJ, Calderon TM, Luers AJ, Eugenin EA, Javitch JA, Berman JW. Human immunodeficiency virus (HIV) infection of human macrophages is increased by dopamine: a bridge between HIV-associated neurologic disorders and drug abuse. Am J Pathol. 2009;175:1148–1159. doi: 10.2353/ajpath.2009.081067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gettig JP, Grady SE, Nowosadzka I. Methamphetamine: putting the brakes on speed. J Sch Nurs. 2006;22:66–73. [PubMed] [Google Scholar]
- van de Giessen E, de Win MM, Tanck MW, van den Brink W, Baas F, Booij J. Striatal dopamine transporter availability associated with polymorphisms in the dopamine transporter gene SLC6A3. J Nucl Med. 2009;50:45–52. doi: 10.2967/jnumed.108.053652. [DOI] [PubMed] [Google Scholar]
- Guan YQ, Cai YY, Lee YT, Opas M. An automatic method for identifying appropriate gradient magnitude for 3D boundary detection of confocal image stacks. J Microsc. 2006;223:66–72. doi: 10.1111/j.1365-2818.2006.01600.x. [DOI] [PubMed] [Google Scholar]
- Guan YQ, Cai YY, Zhang X, Lee YT, Opas M. Adaptive correction technique for 3D reconstruction of fluorescence microscopy images. Microsc Res Tech. 2008;71:146–157. doi: 10.1002/jemt.20536. [DOI] [PubMed] [Google Scholar]
- Hampson EC, Vaney DI, Weiler R. Dopaminergic modulation of gap junction permeability between amacrine cells in mammalian retina. J Neurosci. 1992;12:4911–4922. doi: 10.1523/JNEUROSCI.12-12-04911.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen N, Manahan-Vaughan D. Dopamine D1/D5 receptors mediate informational saliency that promotes persistent hippocampal long-term plasticity. Cereb Cortex. 2014;24:845–858. doi: 10.1093/cercor/bhs362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormuzdi SG, Pais I, LeBeau FE, Towers SK, Rozov A, Buhl EH, Whittington MA, Monyer H. Impaired electrical signaling disrupts gamma frequency oscillations in connexin 36-deficient mice. Neuron. 2001;31:487–495. doi: 10.1016/s0896-6273(01)00387-7. [DOI] [PubMed] [Google Scholar]
- Kesby JP, Heaton RK, Young JW, Umlauf A, Woods SP, Letendre SL, Markou A, Grant I, Semenova S. Methamphetamine exposure combined with HIV-1 disease or gp120 expression: comparison of learning and executive functions in humans and mice. Neuropsychopharmacology. 2015;40:1899–1909. doi: 10.1038/npp.2015.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T. Glial connexins and gap junctions in CNS inflammation and disease. J Neurochem. 2008;106:1000–1016. doi: 10.1111/j.1471-4159.2008.05405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King JE, Eugenin EA, Hazleton JE, Morgello S, Berman JW. Mechanisms of HIV-tat-induced phosphorylation of N-methyl-D-aspartate receptor subunit 2A in human primary neurons: implications for neuroAIDS pathogenesis. Am J Pathol. 2010;176:2819–2830. doi: 10.2353/ajpath.2010.090642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothmann WW, Massey SC, O’Brien J. Dopamine-stimulated dephosphorylation of connexin 36 mediates AII amacrine cell uncoupling. J Neurosci. 2009;29:14903–14911. doi: 10.1523/JNEUROSCI.3436-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Zhang Z, Blackburn MR, Wang SW, Ribelayga CP, O’Brien J. Adenosine and dopamine receptors coregulate photoreceptor coupling via gap junction phosphorylation in mouse retina. J Neurosci. 2013;33:3135–3150. doi: 10.1523/JNEUROSCI.2807-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Wang X, Chen H, Song L, Ye L, Wang SH, Wang YJ, Zhou L, Ho WZ. Methamphetamine enhances HIV infection of macrophages. Am J Pathol. 2008;172:1617–1624. doi: 10.2353/ajpath.2008.070971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locovei S, Scemes E, Qiu F, Spray DC, Dahl G. Pannexin1 is part of the pore forming unit of the P2X(7) receptor death complex. FEBS Lett. 2007;581:483–488. doi: 10.1016/j.febslet.2006.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftis JM, Janowsky A. Neuroimmune basis of methamphetamine toxicity. Int Rev Neurobiol. 2014;118:165–197. doi: 10.1016/B978-0-12-801284-0.00007-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan BK. Methamphetamine - effects on human performance and behavior. Forensic Sci Rev. 2002;14:133–151. [PubMed] [Google Scholar]
- Luigjes J, van den Brink W, Feenstra M, van den Munckhof P, Schuurman PR, Schippers R, Mazaheri A, De Vries TJ, Denys D. Deep brain stimulation in addiction: a review of potential brain targets. Mol Psychiatry. 2012;17:572–583. doi: 10.1038/mp.2011.114. [DOI] [PubMed] [Google Scholar]
- Mantri CK, Mantri JV, Pandhare J, Dash C. Methamphetamine inhibits HIV-1 replication in CD4+ T cells by modulating anti-HIV-1 miRNA expression. Am J Pathol. 2014;184:92–100. doi: 10.1016/j.ajpath.2013.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maragos WF, Young KL, Turchan JT, Guseva M, Pauly JR, Nath A, Cass WA. Human immunodeficiency virus-1 Tat protein and methamphetamine interact synergistically to impair striatal dopaminergic function. J Neurochem. 2002;83:955–963. doi: 10.1046/j.1471-4159.2002.01212.x. [DOI] [PubMed] [Google Scholar]
- Martinez LR, Mihu MR, Gacser A, Santambrogio L, Nosanchuk JD. Methamphetamine enhances histoplasmosis by immunosuppression of the host. J Infect Dis. 2009;200:131–141. doi: 10.1086/599328. [DOI] [PubMed] [Google Scholar]
- McCracken CB, Hamby SM, Patel KM, Morgan D, Vrana KE, Roberts DC. Extended cocaine self-administration and deprivation produces region-specific and time-dependent changes in connexin36 expression in rat brain. Synapse. 2005a;58:141–150. doi: 10.1002/syn.20194. [DOI] [PubMed] [Google Scholar]
- McCracken CB, Patel KM, Vrana KE, Paul DL, Roberts DC. Amphetamine withdrawal produces region-specific and time-dependent changes in connexin36 expression in rat brain. Synapse. 2005b;56:39–44. doi: 10.1002/syn.20127. [DOI] [PubMed] [Google Scholar]
- McMahon DG, Knapp AG, Dowling JE. Horizontal cell gap junctions: single-channel conductance and modulation by dopamine. Proc Natl Acad Sci USA. 1989;86:7639–7643. doi: 10.1073/pnas.86.19.7639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melega WP, Cho AK, Harvey D, Lacan G. Methamphetamine blood concentrations in human abusers: application to pharmacokinetic modeling. Synapse. 2007;61:216–220. doi: 10.1002/syn.20365. [DOI] [PubMed] [Google Scholar]
- Meller K. Axoplasmic transport of horseradish peroxidase in single neurons of the dorsal root ganglion studied in vitro by microinjection. Cell Tissue Res. 1992;270:139–148. doi: 10.1007/BF00381888. [DOI] [PubMed] [Google Scholar]
- Mendelson J, Uemura N, Harris D, Nath RP, Fernandez E, Jacob P, 3rd, Everhart ET, Jones RT. Human pharmacology of the methamphetamine stereoisomers. Clin Pharmacol Ther. 2006;80:403–420. doi: 10.1016/j.clpt.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Mercer A. Electrically coupled excitatory neurones in cortical regions. Brain Res. 2012;1487:192–197. doi: 10.1016/j.brainres.2012.03.069. [DOI] [PubMed] [Google Scholar]
- Nair MP, Saiyed ZM, Nair N, Gandhi NH, Rodriguez JW, Boukli N, Provencio-Vasquez E, Malow RM, Miguez-Burbano MJ. Methamphetamine enhances HIV-1 infectivity in monocyte derived dendritic cells. J Neuroimmune Pharmacol. 2009;4:129–139. doi: 10.1007/s11481-008-9128-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan EB, Harrison LM, Lahoste GJ, Ruskin DN. Behavioral synergism between D(1) and D(2) dopamine receptors in mice does not depend on gap junctions. Synapse. 2007;61:279–287. doi: 10.1002/syn.20371. [DOI] [PubMed] [Google Scholar]
- Onn SP, Grace AA. Amphetamine withdrawal alters bistable states and cellular coupling in rat prefrontal cortex and nucleus accumbens neurons recorded in vivo. J Neurosci. 2000;20:2332–2345. doi: 10.1523/JNEUROSCI.20-06-02332.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orellana JA, Froger N, Ezan P, Jiang JX, Bennett MV, Naus CC, Giaume C, Saez JC. ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. J Neurochem. 2011;118:826–840. doi: 10.1111/j.1471-4159.2011.07210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orellana JA, Saez PJ, Cortes-Campos C, et al. Glucose increases intracellular free Ca(2 +) in tanycytes via ATP released through connexin 43 hemichannels. Glia. 2012a;60:53–68. doi: 10.1002/glia.21246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orellana JA, von Bernhardi R, Giaume C, Saez JC. Glial hemichannels and their involvement in aging and neurodegenerative diseases. Rev Neurosci. 2012b;23:163–177. doi: 10.1515/revneuro-2011-0065. [DOI] [PubMed] [Google Scholar]
- Orellana JA, Saez JC, Bennett MV, Berman JW, Morgello S, Eugenin EA. HIV increases the release of dickkopf-1 protein from human astrocytes by a Cx43 hemichannel-dependent mechanism. J Neurochem. 2013a;128:752–763. doi: 10.1111/jnc.12492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orellana JA, Velasquez S, Williams DW, Saez JC, Berman JW, Eugenin EA. Pannexin1 hemichannels are critical for HIV infection of human primary CD4+ T lymphocytes. J Leukoc Biol. 2013b;94:399–407. doi: 10.1189/jlb.0512249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandhare J, Addai AB, Mantri CK, Hager C, Smith RM, Barnett L, Villalta F, Kalams SA, Dash C. Cocaine enhances HIV-1-induced CD4(+) T-cell apoptosis: implications in disease progression in cocaine-abusing HIV-1 patients. Am J Pathol. 2014;184:927–936. doi: 10.1016/j.ajpath.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passaro RC, Pandhare J, Qian HZ, Dash C. The complex interaction between methamphetamine abuse and HIV-1 pathogenesis. J Neuroimmune Pharmacol. 2015;10:477–486. doi: 10.1007/s11481-015-9604-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D, Desai GM, Frases S, Cordero RJ, DeLeon-Rodriguez CM, Eugenin EA, Nosanchuk JD, Martinez LR. Methamphetamine enhances Cryptococcus neoformans pulmonary infection and dissemination to the brain. MBio. 2013;4:1–10. doi: 10.1128/mBio.00400-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006;25:5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelegrin P, Surprenant A. Pannexin-1 couples to maitotoxin- and nigericin-induced interleukin-1beta release through a dye uptake-independent pathway. J Biol Chem. 2007;282:2386–2394. doi: 10.1074/jbc.M610351200. [DOI] [PubMed] [Google Scholar]
- Pereda A, O’Brien J, Nagy JI, Bukauskas F, Davidson KG, Kamasawa N, Yasumura T, Rash JE. Connexin35 mediates electrical transmission at mixed synapses on Mauthner cells. J Neurosci. 2003a;23:7489–7503. doi: 10.1523/JNEUROSCI.23-20-07489.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereda A, O’Brien J, Nagy JI, Smith M, Bukauskas F, Davidson KG, Kamasawa N, Yasumura T, Rash JE. Short-range functional interaction between connexin35 and neighboring chemical synapses. Cell Commun Adhes. 2003b;10:419–423. doi: 10.1080/15419060390263254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powrozek TA, Sari Y, Singh RP, Zhou FC. Neurotransmitters and substances of abuse: effects on adult neurogenesis. Curr Neurovasc Res. 2004;1:251–260. doi: 10.2174/1567202043362225. [DOI] [PubMed] [Google Scholar]
- Purohit V, Rapaka R, Frankenheim J, Avila A, Sorensen R, Rutter J. National Institute on Drug Abuse symposium report: drugs of abuse, dopamine, and HIV-associated neurocognitive disorders/HIV-associated dementia. J Neurovirol. 2013;19:119–122. doi: 10.1007/s13365-013-0153-2. [DOI] [PubMed] [Google Scholar]
- Rash JE, Curti S, Vanderpool KG, et al. Molecular and functional asymmetry at a vertebrate electrical synapse. Neuron. 2013;79:957–969. doi: 10.1016/j.neuron.2013.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rella CE, Ruel N, Eugenin EA. Development of imaging techniques to study the pathogenesis of biosafety level 2/3 infectious agents. Pathog Dis. 2014;72:167–173. doi: 10.1111/2049-632X.12199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JC, Woods SP, Matt GE, Meyer RA, Heaton RK, Atkinson JH, Grant I. Neurocognitive effects of methamphetamine: a critical review and meta-analysis. Neuropsychol Rev. 2007;17:275–297. doi: 10.1007/s11065-007-9031-0. [DOI] [PubMed] [Google Scholar]
- Shor-Posner G. Cognitive function in HIV-1-infected drug users. J Acquir Immune Defic Syndr. 2000;25(Suppl 1):S70–S73. doi: 10.1097/00042560-200010001-00011. [DOI] [PubMed] [Google Scholar]
- Siekmeier PJ, vanMaanen DP. Dopaminergic contributions to hippocampal pathophysiology in schizophrenia: a computational study. Neuropsychopharmacology. 2014;39:1713–1721. doi: 10.1038/npp.2014.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstein PS, Shah A, Gupte R, Liu X, Piepho RW, Kumar S, Kumar A. Methamphetamine toxicity and its implications during HIV-1 infection. J Neurovirol. 2011;17:401–415. doi: 10.1007/s13365-011-0043-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon SL, Domier CP, Sim T, Richardson K, Rawson RA, Ling W. Cognitive performance of current methamphetamine and cocaine abusers. J Addict Dis. 2002a;21:61–74. doi: 10.1300/j069v21n01_06. [DOI] [PubMed] [Google Scholar]
- Simon SL, Richardson K, Dacey J, Glynn S, Domier CP, Rawson RA, Ling W. A comparison of patterns of methamphetamine and cocaine use. J Addict Dis. 2002b;21:35–44. doi: 10.1300/j069v21n01_04. [DOI] [PubMed] [Google Scholar]
- So CH, Verma V, Alijaniaram M, Cheng R, Rashid AJ, O’Dowd BF, George SR. Calcium signaling by dopamine D5 receptor and D5-D2 receptor hetero-oligomers occurs by a mechanism distinct from that for dopamine D1-D2 receptor hetero-oligomers. Mol Pharmacol. 2009;75:843–854. doi: 10.1124/mol.108.051805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehberg J, Moraga-Amaro R, Salazar C, et al. Release of gliotransmitters through astroglial connexin 43 hemichannels is necessary for fear memory consolidation in the basolateral amygdala. FASEB J. 2012;26:3649–3657. doi: 10.1096/fj.11-198416. [DOI] [PubMed] [Google Scholar]
- Subbian S, Eugenin E, Kaplan G. Detection of Mycobacterium tuberculosis in latently infected lungs by immunohistochemistry and confocal microscopy. J Med Microbiol. 2014;63:1432–1435. doi: 10.1099/jmm.0.081091-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis M, Jauch R, Zhuo L, et al. Accelerated hippocampal spreading depression and enhanced locomotory activity in mice with astrocyte-directed inactivation of connexin43. J Neurosci. 2003;23:766–776. doi: 10.1523/JNEUROSCI.23-03-00766.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodore S, Cass WA, Maragos WF. Methamphetamine and human immunodeficiency virus protein Tat synergize to destroy dopaminergic terminals in the rat striatum. Neuroscience. 2006a;137:925–935. doi: 10.1016/j.neuroscience.2005.10.056. [DOI] [PubMed] [Google Scholar]
- Theodore S, Stolberg S, Cass WA, Maragos WF. Human immunodeficiency virus-1 protein tat and methamphetamine interactions. Ann N Y Acad Sci. 2006b;1074:178–190. doi: 10.1196/annals.1369.018. [DOI] [PubMed] [Google Scholar]
- Tibber MS, Becker D, Jeffery G. Levels of transient gap junctions between the retinal pigment epithelium and the neuroblastic retina are influenced by catecholamines and correlate with patterns of cell production. J Comp Neurol. 2007;503:128–134. doi: 10.1002/cne.21388. [DOI] [PubMed] [Google Scholar]
- Tiwari S, Nair MP, Saxena SK. Latest trends in drugs of abuse - HIV infection and neuroAIDS. Future Virol. 2013;8:121–127. doi: 10.2217/fvl.12.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker JS, Burnam MA, Sherbourne CD, Kung FY, Gifford AL. Substance use and mental health correlates of nonadherence to antiretroviral medications in a sample of patients with human immunodeficiency virus infection. Am J Med. 2003;114:573–580. doi: 10.1016/s0002-9343(03)00093-7. [DOI] [PubMed] [Google Scholar]
- Velasquez S, Eugenin EA. Role of pannexin-1 hemichannels and purinergic receptors in the pathogenesis of human diseases. Front Physiol. 2014;5:96. doi: 10.3389/fphys.2014.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez JL, Han D, Carlen PL. Neurotransmitter modulation of gap junctional communication in the rat hippocampus. Eur J Neurosci. 1997;9:2522–2531. doi: 10.1111/j.1460-9568.1997.tb01681.x. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Rodriguez JJ, Parpura V. Calcium signalling in astroglia. Mol Cell Endocrinol. 2012;353:45–56. doi: 10.1016/j.mce.2011.08.039. [DOI] [PubMed] [Google Scholar]
- Volgyi B, Deans MR, Paul DL, Bloomfield SA. Convergence and segregation of the multiple rod pathways in mammalian retina. J Neurosci. 2004;24:11182–11192. doi: 10.1523/JNEUROSCI.3096-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volz TJ, Hanson GR, Fleckenstein AE. Kinetic analysis of developmental changes in vesicular monoamine transporter-2 function. Synapse. 2006;60:474–477. doi: 10.1002/syn.20321. [DOI] [PubMed] [Google Scholar]
- Watkins CC, Treisman GJ. Neuropsychiatric complications of aging with HIV. J Neurovirol. 2012;18:277–290. doi: 10.1007/s13365-012-0108-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler R, Pottek M, He S, Vaney DI. Modulation of coupling between retinal horizontal cells by retinoic acid and endogenous dopamine. Brain Res Brain Res Rev. 2000;32:121–129. doi: 10.1016/s0165-0173(99)00071-5. [DOI] [PubMed] [Google Scholar]
- Werlen E, Jones MW. Modulating the map: dopaminergic tuning of hippocampal spatial coding and interactions. Prog Brain Res. 2015;219:187–216. doi: 10.1016/bs.pbr.2015.03.002. [DOI] [PubMed] [Google Scholar]
- Yu S, Zhu L, Shen Q, Bai X, Di X. Recent advances in methamphetamine neurotoxicity mechanisms and its molecular pathophysiology. Behav Neurol. 2015;2015:103969. doi: 10.1155/2015/103969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Loonam TM, Noailles PA, Angulo JA. Comparison of cocaine- and methamphetamine-evoked dopamine and glutamate overflow in somatodendritic and terminal field regions of the rat brain during acute, chronic, and early withdrawal conditions. Ann N Y Acad Sci. 2001;937:93–120. doi: 10.1111/j.1749-6632.2001.tb03560.x. [DOI] [PubMed] [Google Scholar]
- Zhang X, Lee TH, Xiong X, Chen Q, Davidson C, Wetsel WC, Ellinwood EH. Methamphetamine induces long-term changes in GABAA receptor alpha2 subunit and GAD67 expression. Biochem Biophys Res Commun. 2006;351:300–305. doi: 10.1016/j.bbrc.2006.10.046. [DOI] [PubMed] [Google Scholar]
- Zlomuzica A, Viggiano D, Degen J, Binder S, Ruocco LA, Sadile AG, Willecke K, Huston JP, Dere E. Behavioral alterations and changes in Ca/calmodulin kinase II levels in the striatum of connexin36 deficient mice. Behav Brain Res. 2012;226:293–300. doi: 10.1016/j.bbr.2011.08.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Meth induces internalization of Cx43 plaques in HeLa cells. HeLa cells stably transfected with Cx43-GFP were grown to confluence, treated with meth, and subjected to time-lapse imaging. Meth reduced plaque stability by inducing internalization of Cx43. (a) representative example of time-lapse image sequence of Cx43 plaque internalization. (b) Quantification of plaque stability in control and meth-treated cultures over 12 h. (*p < 0.05 as compared to control conditions).