

Abstract
Purpose of the review
This review examines therole of FGF-23 in mineral metabolism, innate immunity and adverse cardiovascular outcomes.
Recent findings
FGF-23, produced by osteocytes in bone, activates FGFR/α-Klotho complexes in the kidney. The resulting bone-kidney axis coordinates renal phosphate reabsorption with bone mineralization, and creates a counter-regulatory feedback loop to prevent vitamin D toxicity. FGF-23 acts to counter-regulate the effects of Vitamin D on innate immunity and cardiovascular responses. FGF-23 is ectopically expressed along with α-Klotho in activated macrophages, creating a pro-inflammatory paracrine signaling pathway that counters the anti-inflammatory actions of vitamin D. FGF-23 also inhibits ACE2 expression and increases sodium reabsorption in the kidney, leading to hypertension and left ventricular hypertrophy. Finally, FGF-23 is purported to cause adverse cardiac and impair neutrophil responses through activation of FGFRs in the absence of α-Klotho. While secreted forms of α-Klotho have FGF-23- independent effects, the possibility of α-Klotho-independent effects of FGF-23 is controversial and requires additional experimental validation.
Summary
FGF-23 participates in a bone-kidney axis regulating mineral homeostasis, proinflammatory paracrine macrophage signaling pathways, and in a bone-cardio-renal axis regulating hemodynamics that counteract the effects of Vitamin D.
Keywords: Innate immunity, vitamin D metabolism, chronic kidney disease, inflammation, hypophosphatemia, a-Klotho, renin angiotensin system
Introduction
Fibroblast growth factor 23 (FGF-23) has emerged as an important hormone in regulating mineral homeostasis in chronic kidney disease, and has been implicated in adverse cardiovascular and infectious clinical outcomes. This review outlines what we know and don’t know about the mechanisms underlying FGF-23 regulation and functions.
Hormonal FGFs signal through FGFR/Klotho complexes to create novel endocrine networks
FGF-23, FGF-19, and FGF-21 belong to the subfamily of hormonal FGFs [1]. FGF-23 is a ~32 kDa protein with an N-terminal FGF-homology domain and a 71 amino acid C-terminus separated by a RXXR proprotein convertase cleavage site [2–4] that is principally produced and secreted by osteocytes and osteoblasts in bone [2, 5–8]. FGF-23’s major function is to participate in a bone-kidney axis that coordinates bone mineralization with renal phosphate handling and that counteracts the actions of 1,25(OH)2D [5]. Venous sinusoids of the bone marrow and thymus and in the lateral thalamic nuclei in the brain, express FGF-23, but its functions are unknown [9–12]. FGF-19 is secreted by the ileum and targets the liver to regulate bile acid biosynthesis and cholesterol metabolism; whereas, FGF-21, produced by hepatocytes in the liver, targets adipoctyes to regulate fat and energy metabolism [13, 14].
Hormonal FGF actions are imparted by a C-terminal domain that binds to α-Klotho or β-Klotho, type I membrane, ß-glycosidases that function as obligate co-receptors for FGF-23 binding to FGFRs [1]. α-Klotho (α-Kl) forms a trimeric complex with FGFRs (FGFR1c, 3c or 4, but not FGFR2) and the C-terminus of FGF-23 [15–18] to create a heparin independent FGF-23 signaling complex in kidney tubules, choroid plexus, and parathyroid glands. α-Kl can be ectopically expressed in other tissues under pathological conditions; in addition, recent studies suggest that α-Kl is much more widely expressed than originally described [19, 20]. Nevertheless, FGF-23 and α-Kl have co-dependent effects, as illustrated by in vivo studies showing that FGF-23−/− and α-Kl−/− mice are exact phenocopies [21, 22] and that α-Kl−/− mice are refractory to FGF-23 regulation of phosphate reabsorption and vitamin D metabolism [23].
The kidney is the physiologically most important tissue for FGF-23 activation of FGFRs and α-Kl binary complexes. FGF-23 targets FGFRs in the proximal of the kidney to regulate vitamin D metabolism, phosphate reabsorption and ACE2 expression and in the distal tubule to regulate sodium and calcium transport and α-Klotho expression [24]. Similarly, FGFR/β-Klotho binary complexes constitute the FGF-19 and FGF-21 receptor [23, 25].
There are also circulating isoforms of α-Kl, but not β-Kl, that have FGF-23 independent functions [26]. Transmembrane α-Kl undergoes ectodomain shedding from the kidney by ADAM 10 and 17 to release a processed ~ 130 kDa α-Kl (pKL) into the circulation [27]. An alternative spliced transcript generates a ~70 kDa soluble klotho (sKl). pKl and sKl are purported to have antiaging effects through inhibition of IGF1, Wnt, and TGF-β signaling [26].
Hereditary diseases caused by FGF-23
Autosomal dominant hypophosphatemic rickets (ADHR) is caused by mutations (R176Q and R179W) in the RXXR furin-like cleavage domain of FGF23 that impairs its proteolytic inactivation [28]. In contrast, loss-of-function mutations in polypeptide N-acetylgalactos-aminyltransferase 3 (GalNAc-T3) that prevents O-glycosylation of Thr178 within the RXXR cleavage site causes tumoral calcinosis by increasing FGF-23 proteolysis. In vitro, PC1/3, PC2 and PC5/6, but not furin, cleaves FGF-23 [29]. Understanding the role of posttranslational regulation of FGF-23 in health and disease requires additional study.
Transcriptional regulation of FGF-23 is clinical and pathologically most important. FGF-23 transcription in bone-derived osteoblasts and osteocytes is regulated by both local and systemic factors detail [5–7, 28, 30–32] (Figure 1).
Figure 1. Integrative model of systemic and local factors and signaling pathways that directly regulate FGF-23 gene transcription.
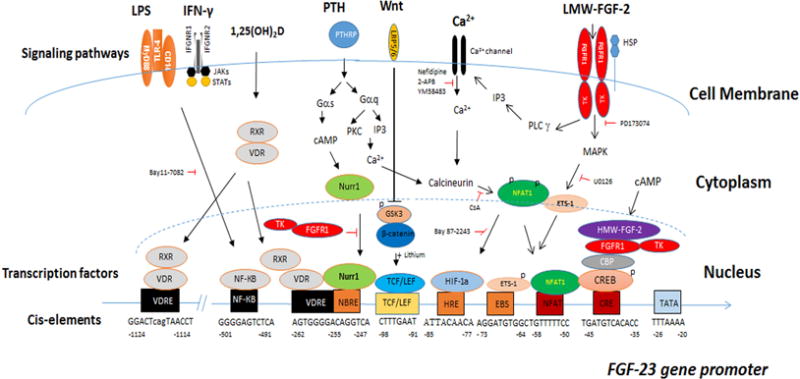
Diverse receptors, including G-protein receptors, nuclear calcium channels, receptor tyrosine kinases, toll-like and cytokine receptors, have the capacity to activate FGF-23 gene transcription through both distinct and overlapping pathways. This diversity permits FGF-23 to participate in regulation by calcitrophic hormones involved in mineral metabolism, cytokine pathways involved in regulating innate immune responses, and oxidative stress, sympathetic nervous system and renin angiontensin system activation that lead to adverse cardiovascular outcomes.
Gene mutations that impair extracellular matrix mineralization identify local factors that regulate FGF-23 gene transcription [5, 7] (Table 1). These include inactivating mutations in phosphate regulating endopeptidase homolog, X-Linked (PHEX), dentin matrix protein 1 (DMP1), ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1), and family with sequence similarity 20, member c (FAM20C)[33]. Mouse genetic studies show that PHEX and DMP1 share a common pathway regulating FGF-23 gene transcription [34]. Inactivating mutations in ENPP1 deplete PPi and local concentrations of phosphate in bone [35], leading to soft tissue calcifications, impaired bone mineralization and increased FGF-23 expression. Inactivation of FAM20C also increases the expression of FGF-23 in bone. Fam20C loss directly phosphorylates FGF-23 on Ser180 within the FGF23 RXXRS180, which inhibits O-glycosylation and degradation of FGF-23 [36]. Many gaps exist in our knowledge of the specific molecular pathways whereby these mutations regulate FGF-23 gene transcription, but all have in common a link between bone mineralization and FGF-23 expression [33]. Impaired mineralization and diminished uptake of phosphate in bone somehow stimulates increased FGF-23 expression and release into the circulation to regulate renal phosphate handing to match the diminished bone phosphate buffering capacity [5, 6].
Table 1.
Mutations causing increased FGF-23 gene transcription in bone.
| Inactivating Mutations | Activating Mutations | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Disorder | ARHR1 | ARHR2 | XLH | RNS ARHR3 | OGD | None | ENS | MAS | HR/HPT | JMC |
| OMIM# | 241520 | 613312 | 30780 | 259775 | 166250 | *134934 163200 *164790 |
174899 | 612089 | 168468 | |
| Mutated Gene | DMP1 | ENPP1 | PHEX | FAM20C | FGFR1 | FGF2-HMW | FGFR3 HRAS NRAS |
GNAS1 | α-Kl | PTH/PTHR |
Abbreviations: ARHR1, autosomal recessive hypophosphatemic rickets 1; ARHR2, autosomal dominant hypophosphatemic rickets 2; HR/HPT, hypophosphatemic rickets and hyperparathyroidism; FGF2-HMW, high-molecular weight FGF2; JMC, Jansen metaphyseal chondrodysplasia; MAS, McCune–Albright syndrome; OGD, osteoglophonic dysplasis; Tg, transgene; XLH, X-linked hypophosphatemic rickets; RNS, Raine’s Syndrome; ENS, Epidermal Nevi Syndrome. PTH, Parathyroid Hormone; PTHR, Parathyroid hormone receptor; DMP1, dentin matrix protein 1; ENPP1, Ecotnucleotide Pyrophosphatase/Phosphodiesterase 1.
Paracrine and intracrine FGFR signaling in bone regulates FGF-23, thereby linking earlier evolved FGF signaling to the later evolved hormonal FGF-23. In this regard, activating mutations in FGFRs, including mutations in FGFR1 in osteoglophonic dysplasia (OGD) and FGFR3 in Epidermal Nevus Syndrome (ENS), lead to increased FGF-23 expression [37]. Selective deletion of FGFR1 in osteocytes of Hyp mice reduces FGF-23 [38]. Low molecular weight (LMW, 18kDa) FGF-2 activates cell surface FGFR signaling and increases FGF-23 gene transcription via PLCγ/calcineurin/NFAT and MAPK pathways and NFAT 1 and pETS-1 binding to the FGF-23 promoter [38–40]. High molecular weight (HMW) FGF-2 isoforms elevates FGF-23 gene transcription via a cAMP-dependent integrative nuclear FGFR1 signaling (INFS) by enhancing FGFR1/CREB binding to a cycle-AMP response element (CRE) in the promoter region of FGF-23 gene [39].
FGF-23 is also the cause of tumor-induced osteomalacia (TIO) and oncogenic osteomalacia paraneoplastic syndromes. Elevated FGF-23 is observed in an adenocarcinoma with an aactivating somatic KRAS mutation (G12V), suggesting that RAS can regulate FGF-23 ectopically in non-osseous tissues. Prostate and colon cancer can also cause hypophosphatemia through ectopic production of FGF-23 [41, 42].
FGF-23 is a counter-regulatory hormone for the pleiotropic actions of Vitamin D
Circulating 1,25(OH)2D is a steroid hormone produced by the kidney proximal tubule that activates VDR/RXR nuclear receptors in multiple tissues to regulate several major biological processes, including bone and mineral metabolism, inflammation and innate immune responses, and cardiovascular system dynamics [43].
FGF-23 counters the effects of vitamin D on bone and mineral metabolism (Figure 2A)
Figure 2. FGF-23 and Vitamin D exhibit counter-regulator effects.
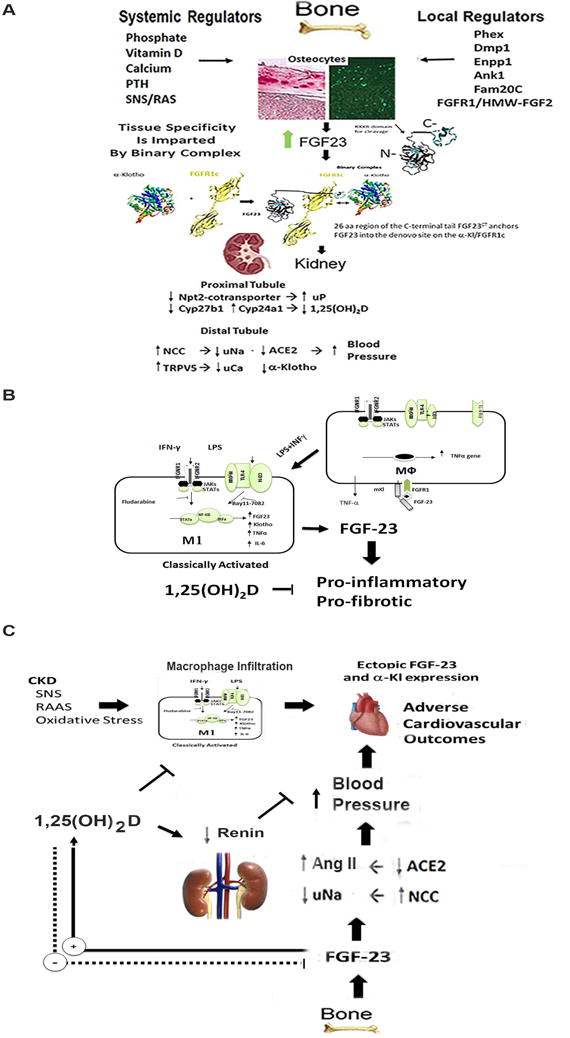
A) Bone-Kidney Endocrine Axis. FGF-23 produced in bone regulates FGFR/α-Klotho complexes in the kidney to inhibit phosphate reabsorption and lower circulating 1,25(OH)2D through suppression of Cyp27b1 synthetic and activation of Cyp24a1 degradative pathways. FGF-23 also suppresses ACE2 and increases distal tubule sodium reabsorption, leading to increased blood pressure. FGF-23 also regulates the expression of α-Klotho, which is released into the circulation by ectodomain shedding and acts as a hormone with FGF-23 independent effects. 1,25(OH)2D stimulates systemic release of FGF-23 from bone that in turn suppresses 1,25(OH)2D release from the kidney to create a negative feedback loop. A positive feedback cardiovascular loop is theoretically created by Ang II and SNS stimulation of FGF-23 expression by bone and effects of circulating FGF-23 to decrease ACE2 and increase renal sodium reabsorption. B) FGF-23 and α-Klotho are also expressed in activated macrophages, which creates a pro-inflammatory paracrine FGF-23 signaling pathway. FGF-23 expression in macrophages and stimulation of TNF-α may offset the anti-inflammatory effects of vitamin D on the innate immune response. In macrophages, 1,25(OH)2D may suppress FGF-23 expression. C) A model of FGF-23 cardiotoxicity derived from FGF-23 effects on the kidney and/or the ectopic expression of FGF-23/α-Klotho in infiltrating macrophages. Stimulation of FGF-23 release into the circulation by RAS, SNS, or oxidative stress or local production of FGF-23 in macrophages could attenuate the beneficial effects of Vitamin D on the cardiovascular and innate immune responses.
Transgenic mouse models overexpressing FGF-23 exhibit hypophosphatemia, suppression of 1,25(OH)2D, increased PTH and rickets/osteomalacia. In contrast, primary deficiency of FGF-23 results in hyperphosphatemia, elevations of 1,25(OH)2D, hypercalcemia, suppression of PTH and soft tissue and vascular calcifications. 1,25(OH)2D stimulates FGF-23 production in bone and FGF-23 suppresses 1,25(OH)2D production by the kidney.
1,25(OH)2D is by far the most important physiological regulator of FGF-23 production (Figures 1 and 2). 1,25(OH)2D stimulation of FGF-23 transcription in bone has been well documented [44–47]. In vivo studies using vitamin D receptor (VDR) knockout mice show that the VDR is necessary for 1,25(OH)2D stimulation of FGF-23 expression in osteoblasts [48], but identification of a VDRE in the FGF-23 promoter remains elusive. While a putative VDR consensus site in the proximal promoter region of mouse FGF-23 has been functionally defined [45], 1,25(OH)2D may stimulate FGF-23 transcription via multiple VDREs outside of the FGF-23 proximal promoter region or indirectly stimulate FGF-23 promoter activity through induction of STAT3 and ETS1 pathways [49].
In the proximal tubule, FGF-23 inhibits sodium-dependent phosphate transport by decreasing the Npt2 transporter expression in the brush border membrane and inhibits 1,25(OH)2D circulating levels, through inhibition of Cyp27b1 mediated 1,25(OH)2D production and stimulation of Cyp24 mediated degradation of 1,25(OH)2D.
PTH has variable actions to directly regulate FGF-23 gene transcription [50] (Figure 1 and Table 1). Activating mutations in GNAS, which encodes the signaling protein Gsα and PTH1R in McCune–Albright syndrome (MAS) and Jansen’s metaphyseal chondrodysplasia (JMC), respectively, increase FGF-23 expression in bone[51]. FGF-23 is activated in mouse models of excess PTH [52]. Parathyroidectomy decreases circulating FGF23 in CKD patients [53]. Calcium-mediated suppression of PTH reduces serum FGF-23 levels in patients with ESRD [54]. PTH also is capable of stimulating FGF-23 indirectly through PTH-mediated increases in 1,25(OH)2D [55]. However, PTH fails to stimulate FGF-23 in some cases of primary hyperparathyroidism [52, 54], in the setting of hypocalcemia and/or 1,25(OH)2D deficiency [56], and after intermittent administration of PTH to mice, which leads to net increments in bone formation [57]. In several studies, PTH did not stimulate production of FGF-23 by osteoblasts in vitro [45]. Bone turnover, the levels of serum calcium and other co-factors likely explain these context-dependent effects of PTH on FGF-23. Indeed, PTH and its downstream effector cAMP, have limited effects to regulate FGF-23 gene transcription in osteoblast cultures. Rather, cAMP-dependent stimulation of FGF-23 promoter activity requires the co-activation of integrative signaling pathways that may involve both nuclear FGFR1 and Nurr1, a member of the NR4A subgroup of orphan nuclear receptors [58]. Interestingly, PTH stimulates Nurr1 expression in osteoblasts prior to FGF-23 elevations and Nurr1 is essential for PTH regulation of FGF-23 gene transcription in UMR-106 osteoblasts [59–61]. VDR is known to activate Nurr1 in T-cells [62], raising the possibility that Nurr1 might also mediate the responses to 1,25(OH)2D to regulate FGF-23 gene transcription [63].
Hypocalcemia is an important negative regulator of FGF-23 production. In elderly humans without CKD serum calcium concentrations, but not phosphate, are positively correlated with serum FGF-23 levels [64]. In mouse genetic models, FGF-23 expression was undetectable in the setting of hypocalcemia, in spite of elevated PTH concentrations. In addition, calcium administration increases FGF-23 levels in the Cyp27b1 null mouse model that lacks 1,25(OH)2D [56]. Extracellular calcium directly upregulates FGF-23 gene expression and promoter activity in osteoblasts in vitro [56, 65] (Figure 2). Calcium regulates FGF-23 through calcium channels. Nifedipine, an L-type calcium channel blocker, inhibits extracellular calcium stimulation of FGF-23 gene transcription [56] and conditional deletion of the Pkd2 (TRPP2) transient receptor potential channel in osteoblasts/osteocytes suppresses FGF-23 expression in bone [66]. Inhibition of the endoplasmic reticulum calcium release-activated calcium channel protein 1 (Orai 1) blocks FGF-23 expression in UMR106 osteoblasts [65]. NFAT transduces the effects of intracellular calcium on FGF-23 promoter activity.
Effects of high phosphate diets on FGF-23 may be to the effects of phosphate to chelate calcium leading to stimulation of PTH and 1,25(OH)2D levels. Dietary phosphate loading (i.e., 1.6 to 2% phosphate diet) in mouse models of CKD result in significant elevations in serum FGF-23 [67], but severe dietary phosphate restriction failed to lower FGF-23 in another model of CKD [68]. In humans, high dietary phosphorus increases and low dietary phosphorus decreases serum FGF-23 [46, 69, 70]; but the changes are small, and there is a delay between phosphate loading and elevations of FGF-23[70, 71]. In the setting of PTH deficiency and hypocalemia, however, high serum phosphorus levels do not stimulate FGF-23 production in bone [56]. Hypophosphatemia caused by primary renal phosphate wasting in SLC34A1 (sodium-phosphate co-transporter 2a) knockout mice is associated with low serum FGF-23, in spite of high serum 1,25(OH)2D and hypercalcemia [72, 73]. Further studies are required to understand how phosphate regulates FGF-23.
FGF-23 counter-regulator effects on vitamin D metabolism are clinically important (Figure EA). In states of vitamin D excess, FGF-23 suppress endogenous 1,25(OH)2D production and increases phosphate excretion by the kidney that offsets the effects of 1,25(OH)2D on the gastrointestinal tract and bone to increase influx of calcium and phosphate into the circulation, and on the parathyroid gland suppresses PTH secretion. On the other hand, in the setting of vitamin D deficiency and low calcium, FGF-23 is suppressed, which along with increments in PTH, maximizes 1,25(OH)2D production, which in turn, increases gastrointestinal calcium and phosphate absorption. In this setting, elevation of FGF-23 may define a threshold for correcting vitamin D deficiency, which occurs a 25(OH)D levels of 20 ng/ml, which is lower than current levels of vitamin D sufficiency [74].
Multiple factors cause elevated FGF-23 levels in CKD. Impaired renal clearance of FGF-23 contributes as evidenced by the increase in circulating FGF-23 before elevations of FGF-23 message expression in bone or elevations in PTH, and the effect of nephrectomy and renal transplantation to respectively rapidly increase and decrease FGF-23 levels. Reductions of 25(OH)D and 1,25(OH)2D were originally thought to respectively represent nutritional deficiencies and loss of renal mass; but FGF-23 mediated suppression of 1,25(OH)2D production and increased degradation of vitamin D are adaptive. In this scenario, suppression of 1,25(OH)2D, by reducing phosphate absorption from the gastrointestinal track and efflux from bone, protects again hyperphosphatemia. In late stages of renal failure, serum FGF-23 levels are markedly elevated and correlate with increased FGF-23 message expression in bone; and at this stage, PTH plays a role in increased FGF-23 expression, since parathyroidectomy leads to reductions in circulating FGF-23 levels. Increases in renin angiotensin and sympathetic nervous systems as well as inflammation in CKD can stimulate FGF-23 expression in bone. Reduced cleavage of FGF-23 into N- and C-terminal fragments may also be impaired in CKD.
The associations between elevated circulating FGF-23 and adverse outcomes, including increased left ventricular hypertrophy, cardiac mortality, and mortality to due to infections in chronic kidney disease, suggest that FGF-23 has effects beyond those related to mineral metabolism and highlight other functions of FGF-23 that are counter to 1,25(OH)2D beneficial effects.
FGF-23 counters the effects of Vitamin D on inflammation and innate immune responses (Figure 2B)
1,25(OH)2D has immunoregulatory and anti-inflammatory effects. VDR, the receptor for 1,25(OH)2D is expressed in a variety of immune cells, and 1,25(OH)2D ligand is produced locally by monocytes and dendritic cells that express Cp27b1. In dendritic cells, 1,25(OH)2D activation of VDR inhibit the expression of IL-12, IL-23, IL-6, TNFα, INF-γ, whereas IL-10 and IL-8 expression are enhanced [75]. Moreover, vitamin D deficiency is associated with adverse outcomes linked to inflammation, whereas vitamin D administration has anti-inflammatory and pro-survival effects from infections [76, 77].
In contrast, elevations of FGF-23 are associated with enhanced inflammation in a variety of clinical settings [78–82]. FGF-23’s role in adverse outcomes in CKD may be mediated by inflammatory effects and infections[83]. Administration of FGF-23 neutralizing antibody corrected the impaired leukocyte recruitment and host defense in a murine model of CKD [84]. Pro-inflammatory stimuli, such as lipopolysaccharides (LPS), increase FGF-23 expression in bone [85]. Mice injected with LPS have high levels of serum FGF-23 and increased FGF-23 release from activated dendritic cells and macrophages[81]. Furthermore, LPS/IFN-γ activation of M1 macrophages leads to ectopic expression of FGF-23 through activation of a NF-KB-dependent pathway [86]. Po-inflammatory macrophages also express FGFR1 and α-KL transcripts, thereby producing both the ligand and receptor complexes necessary for paracrine FGF-23 signaling in local inflammatory milieu [81]. Recombinant FGF-23 has direct actions to stimulate TNF-α expression in unstimulated macrophages, and to suppress Arg-1 expression in M2 alternatively activated macrophages, actions that are counteracted by 1,25(OH)2D. Interesting, 1,25(OH)2D treatment of M1 macrophages, unlike its effect in osteoblasts, inhibits FGF-23 expression. Thus, FGF-23 and 1,25(OH)2D function together in a paracrine manner to differentially regulate pro-inflammatory and anti-inflammatory macrophage functions in vitro [86].
FGF-23 counteracts the salutary effects of Vitamin D on the cardiovascular system (Figure 2C)
1,25(OH)2D has important effects on the cardiovascular system in patients with normal and impaired renal function. Vitamin D deficiency is associated with hypertension and increased cardiovascular mortality, whereas treatment with vitamin D is associated with improved cardiovascular outcomes and survival in humans [76, 87, 88]. In animal models, disruption of vitamin D signaling promotes hypertension, cardiac hypertrophy, and atherosclerosis [87]. In CKD, the deficiency of 1,25(OH)2D has been implicated in left ventricular hypertrophy and increased cardiovascular mortality, and treatment with calcitriol is associated with reduced mortality in retrospective observational studies [89]. The beneficial effects of vitamin D on cardiovascular system are purported to be mediated by activation of VDR in multiple tissues, leading to suppression of the RAS [90], as well as other salutary direct effects on vascular smooth muscle, endothelial cells and cardiac tissue [91, 92].
While multiple animal studies show that treatment with vitamin D improves LVH, prospective randomized trials of active vitamin D treatment to reduce the risk for cardiovascular disease have been inconclusive [93]. Interestingly, both low and high 25(OH)D levels are associated with cardiovascular mortality, producing a reverse J-shaped association between serum 25(OH)D and cardiovascular disease mortality [94]. Mortality of critically ill patients is also higher in the presence of supra-physiological levels of 25(OH)D (i.e., exceeding 100 ng/ml) [95]. Since FGF-23 is stimulated by vitamin D, could the loss of survival benefits of high dose vitamin D treatment be mediated by offsetting adverse cardiovascular effects of FGF-23?
There are several potential mechanisms underlying FGF-23 adverse effects on the cardiovascular system. One mechanism involves a possible bone-renal-cardiac axis that controls systemic hemodynamic responses through cross-talk between RAS, vitamin D and FGF-23/α-Kl signaling. FGF-23 effects on the kidney to regulate sodium reabsorption and Ang II metabolism is a component of this novel network. In this regard, FGF-23 regulates the expression of renal genes, including ACE2 and the sodium-chloride cotransporter NCC (SLC12A3) that affect blood pressure. Excess FGF-23 elevates blood pressure and causes LVH in both animal models and the clinical setting [96]. For example, in patients with XLH, elevated FGF-23 levels result in increased diastolic blood pressure and increased left ventricular mass [97]. In a mouse model, FGF-23 administration induced hypertension and LVH through stimulation of distal tubule sodium transport [96]. This response required α-Kl, involved SLC12A3, and treatment with chlorothiazide attenuated this effect. Teleologically, these unexpected hemodynamic effects of FGF-23 may represent an adaptive response to vasodilator effects of inflammation, which also stimulates FGF-23.
This network also involves effects of vitamin D to suppress renin and effects of angiotensin II (Ang II) to suppress renal α-Kl expression [98], as well as signaling pathways, including effects of Ang II, the sympathetic nervous system and oxidative stress to stimulate FGF-23 production in osteoblasts in bone. FGF-23 also regulates the expression α-Klotho in the kidney. Reductions is sKl and consequent reductions in TRPC6 expression in the heart is another possible mechanism for inducing LHV in CKD [99, 100]. Consistent with this schema, angiotensin-converting enzyme inhibition has greater effects in patients with chronic systolic heart failure and elevated circulating FGF-23 levels [101]. In this proposed feed-forward bone-cardio-renal endocrine axis, Ang II, SNS and oxidative stress stimulate FGF-23 production in bone and circulating FGF-23 targets the kidney to suppress ACE2 to augment Ang II responses, to increase sodium absorption and blood pressure, to inhibit s-Kl and its circulating cardioprotective effects, and to suppresses 1,25(OH)2D VDR dependent responses. Collectively these kidney effects accounting for FGF-23’s adverse cardiovascular outcomes.
The second mechanism involves local effects of FGF-23 in heart disease. As noted above, FGF-23 is produced by activated macrophages and has proinflammatory effects on macrophages. In addition, macrophage infiltration of the heart has been shown to induce the ectopic expression of FGF-23 and α-Klotho the cardiomyocytes [102]. FGF-23 expression in human cardiomyocytes can be stimulated by oncostatin M (OSM) released from macrophages [103]. Thus, pro-inflammatory macrophages infiltrating the heart by reconstitute the FGF-23/α-Kl local to create a paracrine signaling pathway is responsible for LVH and cardiac fibrosis.
The third and final mechanism proposed to explain FGF-23 cardiotoxic effects and adverse infectious outcomes is a direct, α-Kl independent activation of FGFRs in cardiomyocytes and neutrophils [14, 19]. For example, FGF-23 is purported to target the heart in the absence of α-Kl through direct activation of FGFR4 FGFR4/PLC-dependent signaling [71]. FGF-23 is also purported to activate FGFR2 in the absence of α-Kl in polymorphonuclear neutrophils[84]. These “non-canonical” signaling pathways are controversial, because α-Kl is not expressed in the heart [15] and FGF-23 does not activate FGF2, even in the presence of α-Kl[15].
This provocative mechanism awaits experimental validation, however. Direct in vivo evidence for FGF-23 activation of FGFR4 in a model with cardiac specific deletion of FGFR4 is lacking. In addition, activation of FGFR signaling in the heart appears to be cardioprotecitive, not cardiotoxic. In this regard,FGF2 knockout mice are resistant to myocardial infarction-[25], isoproterenol- [104] and Ang II- induced cardiac hypertrophy[105], suggesting that activation of FGFR signaling is cardioprotective. Moreover, both Fgfr1c and β-Klotho are expressed in cardiomyocytes, and circulating concentrations of FGF-21 are increased in CKD [25]. Fgf-21 knockout mice develop a dilated cardiomyopathy and exhibit increased susceptibility to isoproterenol-mediated cardiac hypertrophy, whereas activation of FGFR signaling through FGF-21 is purported to have cardioprotective effects [106]. Finally, if Klotho or another co-factor were not needed, then circulating FGFs would have generalized, overlapping, and potentially unwanted, effects. Unrestricted activation of FGFRs by hormonal FGFs would mimic gain-of-function mutations of FGFR1, FGFR3 and FGFR4, which result congenital bone diseases [107], disordered bile-acid metabolism [14], and various cancers [108, 109].
Conclusions
FGF-23 was original call “phosphatonin” to reflect its role in the regulation of serum phosphate concentrations. Unlike serum calcium, however, that activates a calcium sensing receptor (CASR) in the parathyroid gland to regulate the release of PTH in response to changes in serum calcium, changes in serum phosphate do not result in rapid changes in circulating FGF-23 [45, 110]. Rather, FGF-23 is stimulated by 1,25(OH)2D, regulated by other systemic and local factors to create unexpected endocrine networks that counteract the biological effects of 1,25(OH)2D. Thus, FGF-23 is the “yin” and vitamin D is the “yang” in complex physiological and pathological networks.
Key points.
FGF-23 is produced by bone and targets FGFR/a-Klotho binary complexes that constitute the FGF-23 receptor in physiologically relevant tissues
FGF-23 coordinates bone mineralization with renal handling of phosphate to maintain phosphate homeostasis in response to alterations in bone remodeling.
FGF-23 counteracts the effects of Vitamin D on mineral metabolism, innate immunity and cardiovascular responses.
The association between FGF-23 and adverse outcomes may be due to a-Klotho-dependent effects of circulating FGF-23 on the kidney to regulate blood pressure or to ectopic expression of FGF-23 in macrophages and diseased tissues to enhance inflammation and tissue fibrosis.
Acknowledgments
None
Financial support and sponsorship
This work was supported by grant R01-AR045955 to LDQ from the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript
Abbreviations
- FGF-23
Fibroblastic growth factor 23
- 1,25(OH)2D
1,25 dihydroxy -vitamin D3
Footnotes
Conflicts of interest
Dr. Quarles has received honoraria from Amgen, Inc. The remaining authors have no conflicts of interest.
References and recommended reading
- 1.Itoh N. Hormone-like (endocrine) Fgfs: their evolutionary history and roles in development, metabolism, and disease. Cell and tissue research. 2010;342:1–11. doi: 10.1007/s00441-010-1024-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun. 2000;277:494–498. doi: 10.1006/bbrc.2000.3696. [DOI] [PubMed] [Google Scholar]
- 3.Itoh N, Ornitz DM. Functional evolutionary history of the mouse Fgf gene family. Dev Dyn. 2008;237:18–27. doi: 10.1002/dvdy.21388. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki M, Uehara Y, Motomura-Matsuzaka K, et al. {beta}Klotho Is Required for Fibroblast Growth Factor (FGF) 21 Signaling through FGF Receptor (FGFR) 1c and FGFR3c. Molecular endocrinology (Baltimore, Md) 2008;22:1006–1014. doi: 10.1210/me.2007-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pi M, Wu Y, Quarles LD. GPRC6A Mediates Responses to Osteocalcin in beta-Cells In Vitro and Pancreas In Vivo. J Bone Miner Res. 2011 doi: 10.1002/jbmr.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quarles LD. FGF23, PHEX, and MEPE regulation of phosphate homeostasis and skeletal mineralization. Am J Physiol Endocrinol Metab. 2003;285:E1–9. doi: 10.1152/ajpendo.00016.2003. [DOI] [PubMed] [Google Scholar]
- 7.Quarles LD. Evidence for a bone-kidney axis regulating phosphate homeostasis. J Clin Invest. 2003;112:642–646. doi: 10.1172/JCI19687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu S, Quarles LD. How fibroblast growth factor 23 works. J Am Soc Nephrol. 2007;18:1637–1647. doi: 10.1681/ASN.2007010068. [DOI] [PubMed] [Google Scholar]
- 9.Andersen IA, Huntley BK, Sandberg SS, et al. Elevation of circulating but not myocardial FGF23 in human acute decompensated heart failure. Nephrol Dial Transplant. 2015 doi: 10.1093/ndt/gfv398. [DOI] [PubMed] [Google Scholar]
- 10.Leifheit-Nestler M, Grosse Siemer R, Flasbart K, et al. Induction of cardiac FGF23/FGFR4 expression is associated with left ventricular hypertrophy in patients with chronic kidney disease. Nephrol Dial Transplant. 2015 doi: 10.1093/ndt/gfv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lang F, Foller M. Enigmatic Cassandra: renal FGF23 formation in polycystic kidney disease. Kidney international. 2014;85:1260–1262. doi: 10.1038/ki.2013.534. [DOI] [PubMed] [Google Scholar]
- 12.Spichtig D, Zhang H, Mohebbi N, et al. Renal expression of FGF23 and peripheral resistance to elevated FGF23 in rodent models of polycystic kidney disease. Kidney international. 2014;85:1340–1350. doi: 10.1038/ki.2013.526. [DOI] [PubMed] [Google Scholar]
- 13.Degirolamo C, Sabba C, Moschetta A. Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nature reviews. Drug discovery. 2016;15:51–69. doi: 10.1038/nrd.2015.9. [DOI] [PubMed] [Google Scholar]
- 14.Owen BM, Mangelsdorf DJ, Kliewer SA. Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends in endocrinology and metabolism: TEM. 2015;26:22–29. doi: 10.1016/j.tem.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Urakawa I, Yamazaki Y, Shimada T, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–774. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 16.Yu X, Ibrahimi OA, Goetz R, et al. Analysis of the biochemical mechanisms for the endocrine actions of fibroblast growth factor-23. Endocrinology. 2005;146:4647–4656. doi: 10.1210/en.2005-0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li SA, Watanabe M, Yamada H, et al. Immunohistochemical localization of Klotho protein in brain, kidney, and reproductive organs of mice. Cell structure and function. 2004;29:91–99. doi: 10.1247/csf.29.91. [DOI] [PubMed] [Google Scholar]
- 18.Yamazaki Y, Tamada T, Kasai N, et al. Anti-FGF23 Neutralizing Antibodies Demonstrate the Physiological Role and Structural Features of FGF23. J Bone Miner Res. 2008 doi: 10.1359/jbmr.080417. [DOI] [PubMed] [Google Scholar]
- 19.Lim K, Groen A, Molostvov G, et al. alpha-Klotho Expression in Human Tissues. J Clin Endocrinol Metab. 2015;100:E1308–1318. doi: 10.1210/jc.2015-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Six I, Okazaki H, Gross P, et al. Direct, acute effects of Klotho and FGF23 on vascular smooth muscle and endothelium. PLoS One. 2014;9:e93423. doi: 10.1371/journal.pone.0093423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuro-o M, Matsumura Y, Aizawa H, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 22.Nakatani T, Sarraj B, Ohnishi M, et al. In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23) -mediated regulation of systemic phosphate homeostasis. FASEB J. 2009;23:433–441. doi: 10.1096/fj.08-114397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomiyama K, Maeda R, Urakawa I, et al. Relevant use of Klotho in FGF19 subfamily signaling system in vivo. Proc Natl Acad Sci U S A. 2010;107:1666–1671. doi: 10.1073/pnas.0913986107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindberg K, Amin R, Moe OW, et al. The kidney is the principal organ mediating klotho effects. J Am Soc Nephrol. 2014;25:2169–2175. doi: 10.1681/ASN.2013111209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.!!! INVALID CITATION !!!
- 26.Doi S, Zou Y, Togao O, et al. Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J Biol Chem. 2011;286:8655–8665. doi: 10.1074/jbc.M110.174037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kurosu H, Yamamoto M, Clark JD, et al. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.White KE, Carn G, Lorenz-Depiereux B, et al. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney international. 2001;60:2079–2086. doi: 10.1046/j.1523-1755.2001.00064.x. [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto H, Ramos-Molina B, Lick AN, et al. Posttranslational processing of FGF23 in osteocytes during the osteoblast to osteocyte transition. Bone. 2016;84:120–130. doi: 10.1016/j.bone.2015.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quarles LD. Skeletal secretion of FGF-23 regulates phosphate and vitamin D metabolism. Nat Rev Endocrinol. 2012 doi: 10.1038/nrendo.2011.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu S, Guo R, Simpson LG, et al. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. The Journal of biological chemistry. 2003;278:37419–37426. doi: 10.1074/jbc.M304544200. [DOI] [PubMed] [Google Scholar]
- 32.Liu S, Zhou J, Tang W, et al. Pathogenic role of Fgf23 in Hyp mice. American journal of physiology. Endocrinology and metabolism. 2006;291:E38–49. doi: 10.1152/ajpendo.00008.2006. [DOI] [PubMed] [Google Scholar]
- 33.Feng JQ, Ward LM, Liu S, et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nature genetics. 2006;38:1310–1315. doi: 10.1038/ng1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin A, Liu S, David V, et al. Bone proteins PHEX and DMP1 regulate fibroblastic growth factor Fgf23 expression in osteocytes through a common pathway involving FGF receptor (FGFR) signaling. Faseb J. 2011 doi: 10.1096/fj.10-177816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harmey D, Hessle L, Narisawa S, et al. Concerted regulation of inorganic pyrophosphate and osteopontin by akp2, enpp1, and ank: an integrated model of the pathogenesis of mineralization disorders. The American journal of pathology. 2004;164:1199–1209. doi: 10.1016/S0002-9440(10)63208-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tagliabracci VS, Engel JL, Wiley SE, et al. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc Natl Acad Sci U S A. 2014;111:5520–5525. doi: 10.1073/pnas.1402218111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zutt M, Strutz F, Happle R, et al. Schimmelpenning-Feuerstein-Mims syndrome with hypophosphatemic rickets. Dermatology. 2003;207:72–76. doi: 10.1159/000070948. [DOI] [PubMed] [Google Scholar]
- 38.Xiao Z, Huang J, Cao L, et al. Osteocyte-specific deletion of Fgfr1 suppresses FGF23. PLoS One. 2014;9:e104154. doi: 10.1371/journal.pone.0104154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han X, Xiao Z, Quarles LD. Membrane and integrative nuclear fibroblastic growth factor receptor (FGFR) regulation of FGF-23. J Biol Chem. 2015;290:20101. doi: 10.1074/jbc.A114.609230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiao L, Esliger A, Hurley MM. Nuclear fibroblast growth factor 2 (FGF2) isoforms inhibit bone marrow stromal cell mineralization through FGF23/FGFR/MAPK in vitro. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2013;28:35–45. doi: 10.1002/jbmr.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leaf DE, Pereira RC, Bazari H, Juppner H. Oncogenic osteomalacia due to FGF23-expressing colon adenocarcinoma. J Clin Endocrinol Metab. 2013;98:887–891. doi: 10.1210/jc.2012-3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee EK, Martinez MC, Blakely K, et al. FGF23: mediator of poor prognosis in a sizeable subgroup of patients with castration-resistant prostate cancer presenting with severe hypophosphatemia? Medical hypotheses. 2014;83:482–487. doi: 10.1016/j.mehy.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 43.Christakos S, Dhawan P, Verstuyf A, et al. Vitamin D: Metabolism, Molecular Mechanism of Action, and Pleiotropic Effects. Physiol Rev. 2016;96:365–408. doi: 10.1152/physrev.00014.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barthel TK, Mathern DR, Whitfield GK, et al. 1,25-Dihydroxyvitamin D3/VDR-mediated induction of FGF23 as well as transcriptional control of other bone anabolic and catabolic genes that orchestrate the regulation of phosphate and calcium mineral metabolism. The Journal of steroid biochemistry and molecular biology. 2007;103:381–388. doi: 10.1016/j.jsbmb.2006.12.054. [DOI] [PubMed] [Google Scholar]
- 45.Liu S, Tang W, Zhou J, et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol. 2006;17:1305–1315. doi: 10.1681/ASN.2005111185. [DOI] [PubMed] [Google Scholar]
- 46.Saito H, Maeda A, Ohtomo S, et al. Circulating FGF-23 is regulated by 1alpha,25-dihydroxyvitamin D3 and phosphorus in vivo. The Journal of biological chemistry. 2005;280:2543–2549. doi: 10.1074/jbc.M408903200. [DOI] [PubMed] [Google Scholar]
- 47.Masuyama R, Stockmans I, Torrekens S, et al. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. The Journal of clinical investigation. 2006;116:3150–3159. doi: 10.1172/JCI29463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu X, Sabbagh Y, Davis SI, et al. Genetic dissection of phosphate- and vitamin D-mediated regulation of circulating Fgf23 concentrations. Bone. 2005;36:971–977. doi: 10.1016/j.bone.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 49.Saini RK, Kaneko I, Jurutka PW, et al. 1,25-dihydroxyvitamin D(3) regulation of fibroblast growth factor-23 expression in bone cells: evidence for primary and secondary mechanisms modulated by leptin and interleukin-6. Calcified tissue international. 2013;92:339–353. doi: 10.1007/s00223-012-9683-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quarles LD. Skeletal secretion of FGF-23 regulates phosphate and vitamin D metabolism. Nat Rev Endocrinol. 2012;8:276–286. doi: 10.1038/nrendo.2011.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown WW, Juppner H, Langman CB, et al. Hypophosphatemia with Elevations in Serum Fibroblast Growth Factor 23 in a Child with Jansen’s Metaphyseal Chondrodysplasia. J Clin Endocrinol Metab. 2009;94:17–20. doi: 10.1210/jc.2008-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawata T, Imanishi Y, Kobayashi K, et al. Parathyroid hormone regulates fibroblast growth factor-23 in a mouse model of primary hyperparathyroidism. J Am Soc Nephrol. 2007;18:2683–2688. doi: 10.1681/ASN.2006070783. [DOI] [PubMed] [Google Scholar]
- 53.Sato T, Tominaga Y, Ueki T, et al. Total parathyroidectomy reduces elevated circulating fibroblast growth factor 23 in advanced secondary hyperparathyroidism. Am J Kidney Dis. 2004;44:481–487. [PubMed] [Google Scholar]
- 54.Tebben PJ, Singh RJ, Clarke BL, Kumar R. Fibroblast growth factor 23, parathyroid hormone, and 1alpha,25-dihydroxyvitamin D in surgically treated primary hyperparathyroidism. Mayo Clin Proc. 2004;79:1508–1513. doi: 10.4065/79.12.1508. [DOI] [PubMed] [Google Scholar]
- 55.Saji F, Shigematsu T, Sakaguchi T, et al. Fibroblast growth factor 23 production in bone is directly regulated by 1{alpha},25-dihydroxyvitamin D, but not PTH. American journal of physiology. Renal physiology. 2010;299:F1212–1217. doi: 10.1152/ajprenal.00169.2010. [DOI] [PubMed] [Google Scholar]
- 56.David V, Dai B, Martin A, et al. Calcium regulates FGF-23 expression in bone. Endocrinology. 2013;154:4469–4482. doi: 10.1210/en.2013-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Samadfam R, Richard C, Nguyen-Yamamoto L, et al. Bone formation regulates circulating concentrations of fibroblast growth factor 23. Endocrinology. 2009;150:4835–4845. doi: 10.1210/en.2009-0472. [DOI] [PubMed] [Google Scholar]
- 58.Lee YW, Terranova C, Birkaya B, et al. A novel nuclear FGF Receptor-1 partnership with retinoid and Nur receptors during developmental gene programming of embryonic stem cells. J Cell Biochem. 2012;113:2920–2936. doi: 10.1002/jcb.24170. [DOI] [PubMed] [Google Scholar]
- 59.Meir T, Durlacher K, Pan Z, et al. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney international. 2014;86:1106–1115. doi: 10.1038/ki.2014.215. [DOI] [PubMed] [Google Scholar]
- 60.Roforth MM, Liu G, Khosla S, Monroe DG. Examination of nuclear receptor expression in osteoblasts reveals Rorbeta as an important regulator of osteogenesis. J Bone Miner Res. 2012;27:891–901. doi: 10.1002/jbmr.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Imai Y, Youn MY, Inoue K, et al. Nuclear receptors in bone physiology and diseases. Physiol Rev. 2013;93:481–523. doi: 10.1152/physrev.00008.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Achiron A, Feldman A, Gurevich M. Characterization of multiple sclerosis traits: nuclear receptors (NR) impaired apoptosis pathway and the role of 1-alpha 25-dihydroxyvitamin D3. Journal of the neurological sciences. 2011;311:9–14. doi: 10.1016/j.jns.2011.06.038. [DOI] [PubMed] [Google Scholar]
- 63.Kaneko I, Saini RK, Griffin KP, et al. FGF23 gene regulation by 1,25-dihydroxyvitamin D: opposing effects in adipocytes and osteocytes. The Journal of endocrinology. 2015;226:155–166. doi: 10.1530/JOE-15-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schoppet M, Hofbauer LC, Brinskelle-Schmal N, et al. Serum level of the phosphaturic factor FGF23 is associated with abdominal aortic calcification in men: the STRAMBO study. J Clin Endocrinol Metab. 2012;97:E575–583. doi: 10.1210/jc.2011-2836. [DOI] [PubMed] [Google Scholar]
- 65.Zhang B, Yan J, Schmidt S, et al. Lithium- Sensitive Store-Operated Ca2+ Entry in the Regulation of FGF23 Release. Neurosignals. 2015;23:34–48. doi: 10.1159/000442602. [DOI] [PubMed] [Google Scholar]
- 66.Xiao Z, Cao L, Liang Y, et al. Osteoblast-specific deletion of Pkd2 leads to low-turnover osteopenia and reduced bone marrow adiposity. PLoS One. 2014;9:e114198. doi: 10.1371/journal.pone.0114198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arai-Nunota N, Mizobuchi M, Ogata H, et al. Intravenous phosphate loading increases fibroblast growth factor 23 in uremic rats. PLoS One. 2014;9:e91096. doi: 10.1371/journal.pone.0091096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang S, Gillihan R, He N, et al. Dietary phosphate restriction suppresses phosphaturia but does not prevent FGF23 elevation in a mouse model of chronic kidney disease. Kidney international. 2013;84:713–721. doi: 10.1038/ki.2013.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Antoniucci DM, Yamashita T, Portale AA. Dietary phosphorus regulates serum fibroblast growth factor-23 concentrations in healthy men. The Journal of clinical endocrinology and metabolism. 2006;91:3144–3149. doi: 10.1210/jc.2006-0021. [DOI] [PubMed] [Google Scholar]
- 70.Perwad F, Azam N, Zhang MY, et al. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology. 2005;146:5358–5364. doi: 10.1210/en.2005-0777. [DOI] [PubMed] [Google Scholar]
- 71.Grabner A, Amaral AP, Schramm K, et al. Activation of Cardiac Fibroblast Growth Factor Receptor 4 Causes Left Ventricular Hypertrophy. Cell metabolism. 2015;22:1020–1032. doi: 10.1016/j.cmet.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schlingmann KP, Ruminska J, Kaufmann M, et al. Autosomal-Recessive Mutations in SLC34A1 Encoding Sodium-Phosphate Cotransporter 2A Cause Idiopathic Infantile Hypercalcemia. Journal of the American Society of Nephrology : JASN. 2016;27:604–614. doi: 10.1681/ASN.2014101025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Segawa H, Onitsuka A, Furutani J, et al. Npt2a and Npt2c in mice play distinct and synergistic roles in inorganic phosphate metabolism and skeletal development. American journal of physiology. Renal physiology. 2009;297:F671–678. doi: 10.1152/ajprenal.00156.2009. [DOI] [PubMed] [Google Scholar]
- 74.Alshayeb H, Showkat A, Wall BM, et al. Activation of FGF-23 mediated vitamin D degradative pathways by cholecalciferol. J Clin Endocrinol Metab. 2014;99:E1830–1837. doi: 10.1210/jc.2014-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Barragan M, Good M, Kolls JK. Regulation of Dendritic Cell Function by Vitamin D. Nutrients. 2015;7:8127–8151. doi: 10.3390/nu7095383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Autier P, Boniol M, Pizot C, Mullie P. Vitamin D status and ill health: a systematic review. The lancet. Diabetes & endocrinology. 2014;2:76–89. doi: 10.1016/S2213-8587(13)70165-7. [DOI] [PubMed] [Google Scholar]
- 77.Chowdhury R, Kunutsor S, Vitezova A, et al. Vitamin D and risk of cause specific death: systematic review and meta-analysis of observational cohort and randomised intervention studies. Bmj. 2014;348:g1903. doi: 10.1136/bmj.g1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Munoz Mendoza J, Isakova T, Ricardo AC, et al. Fibroblast growth factor 23 and Inflammation in CKD. Clinical journal of the American Society of Nephrology : CJASN. 2012;7:1155–1162. doi: 10.2215/CJN.13281211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mendoza JM, Isakova T, Ricardo AC, et al. Fibroblast Growth Factor 23 and Inflammation in CKD. Clinical journal of the American Society of Nephrology : CJASN. 2012;7:1155–1162. doi: 10.2215/CJN.13281211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kubin T, Poling J, Kostin S, et al. Oncostatin M is a major mediator of cardiomyocyte dedifferentiation and remodeling. Cell stem cell. 2011;9:420–432. doi: 10.1016/j.stem.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 81.Yuki M, Ohta H, Morita Y, et al. Expression of Fgf23 in activated dendritic cells and macrophages in response to immunological stimuli in mice. Biological & pharmaceutical bulletin. 2015 doi: 10.1248/bpb.b14-00276. [DOI] [PubMed] [Google Scholar]
- 82.Dai B, David V, Martin A, et al. A comparative transcriptome analysis identifying FGF23 regulated genes in the kidney of a mouse CKD model. PLoS One. 2012;7:e44161. doi: 10.1371/journal.pone.0044161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chonchol M, Greene T, Zhang Y, et al. Low Vitamin D and High Fibroblast Growth Factor 23 Serum Levels Associate with Infectious and Cardiac Deaths in the HEMO Study. J Am Soc Nephrol. 2016;27:227–237. doi: 10.1681/ASN.2014101009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rossaint J, Oehmichen J, Van Aken H, et al. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J Clin Invest. 2016;126:962–974. doi: 10.1172/JCI83470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ito N, Wijenayaka AR, Prideaux M, et al. Regulation of FGF23 expression in IDG-SW3 osteocytes and human bone by pro-inflammatory stimuli. Molecular and cellular endocrinology. 2015;399:208–218. doi: 10.1016/j.mce.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 86.Han X, Li L, Yang J, et al. Counter-regulatory paracrine actions of FGF-23 and 1,25(OH)2 D in macrophages. FEBS letters. 2016;590:53–67. doi: 10.1002/1873-3468.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang TJ. Vitamin D and Cardiovascular Disease. Annual review of medicine. 2016;67:261–272. doi: 10.1146/annurev-med-051214-025146. [DOI] [PubMed] [Google Scholar]
- 88.Wang TJ, Pencina MJ, Booth SL, et al. Vitamin D deficiency and risk of cardiovascular disease. Circulation. 2008;117:503–511. doi: 10.1161/CIRCULATIONAHA.107.706127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Teng M, Wolf M, Lowrie E, et al. Survival of patients undergoing hemodialysis with paricalcitol or calcitriol therapy. The New England journal of medicine. 2003;349:446–456. doi: 10.1056/NEJMoa022536. [DOI] [PubMed] [Google Scholar]
- 90.Li YC, Kong J, Wei M, et al. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110:229–238. doi: 10.1172/JCI15219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Simpson RU, Hershey SH, Nibbelink KA. Characterization of heart size and blood pressure in the vitamin D receptor knockout mouse. J Steroid Biochem Mol Biol. 2007;103:521–524. doi: 10.1016/j.jsbmb.2006.12.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen S, Law CS, Grigsby CL, et al. Cardiomyocyte-specific deletion of the vitamin D receptor gene results in cardiac hypertrophy. Circulation. 2011;124:1838–1847. doi: 10.1161/CIRCULATIONAHA.111.032680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thadhani R, Appelbaum E, Pritchett Y, et al. Vitamin D therapy and cardiac structure and function in patients with chronic kidney disease: the PRIMO randomized controlled trial. Jama. 2012;307:674–684. doi: 10.1001/jama.2012.120. [DOI] [PubMed] [Google Scholar]
- 94.Durup D, Jorgensen HL, Christensen J, et al. A Reverse J-Shaped Association Between Serum 25-Hydroxyvitamin D and Cardiovascular Disease Mortality: The CopD Study. J Clin Endocrinol Metab. 2015;100:2339–2346. doi: 10.1210/jc.2014-4551. [DOI] [PubMed] [Google Scholar]
- 95.Ralph R, Peter JV, Chrispal A, et al. Supraphysiological 25-hydroxy vitamin D3 level at admission is associated with illness severity and mortality in critically ill patients. Journal of bone and mineral metabolism. 2015;33:239–243. doi: 10.1007/s00774-014-0585-7. [DOI] [PubMed] [Google Scholar]
- 96.Andrukhova O, Slavic S, Smorodchenko A, et al. FGF23 regulates renal sodium handling and blood pressure. EMBO molecular medicine. 2014;6:744–759. doi: 10.1002/emmm.201303716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nehgme R, Fahey JT, Smith C, Carpenter TO. Cardiovascular abnormalities in patients with X-linked hypophosphatemia. J Clin Endocrinol Metab. 1997;82:2450–2454. doi: 10.1210/jcem.82.8.4181. [DOI] [PubMed] [Google Scholar]
- 98.de Borst MH, Vervloet MG, ter Wee PM, Navis G. Cross talk between the renin-angiotensin-aldosterone system and vitamin D-FGF-23-klotho in chronic kidney disease. J Am Soc Nephrol. 2011;22:1603–1609. doi: 10.1681/ASN.2010121251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xie J, Cha SK, An SW, et al. Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nature communications. 2012;3:1238. doi: 10.1038/ncomms2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xie J, Yoon J, An SW, et al. Soluble Klotho Protects against Uremic Cardiomyopathy Independently of Fibroblast Growth Factor 23 and Phosphate. J Am Soc Nephrol. 2015;26:1150–1160. doi: 10.1681/ASN.2014040325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wohlfahrt P, Melenovsky V, Kotrc M, et al. Association of Fibroblast Growth Factor-23 Levels and Angiotensin-Converting Enzyme Inhibition in Chronic Systolic Heart Failure. JACC. Heart failure. 2015;3:829–839. doi: 10.1016/j.jchf.2015.05.012. [DOI] [PubMed] [Google Scholar]
- 102.Poling J, Gajawada P, Richter M, et al. Therapeutic targeting of the oncostatin M receptor-beta prevents inflammatory heart failure. Basic research in cardiology. 2014;109:396. doi: 10.1007/s00395-013-0396-3. [DOI] [PubMed] [Google Scholar]
- 103.Richter M, Lautze HJ, Walther T, et al. The failing heart is a major source of circulating FGF23 via oncostatin M receptor activation. The Journal of heart and lung transplantation : the official publication of the International Society for Heart Transplantation. 2015;34:1211–1214. doi: 10.1016/j.healun.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 104.House SL, House BE, Glascock B, et al. Fibroblast Growth Factor 2 Mediates Isoproterenol-induced Cardiac Hypertrophy through Activation of the Extracellular Regulated Kinase. Molecular and cellular pharmacology. 2010;2:143–154. doi: 10.4255/mcpharmacol.10.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pellieux C, Foletti A, Peduto G, et al. Dilated cardiomyopathy and impaired cardiac hypertrophic response to angiotensin II in mice lacking FGF-2. J Clin Invest. 2001;108:1843–1851. doi: 10.1172/JCI13627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lin Z, Zhou Z, Liu Y, et al. Circulating FGF21 levels are progressively increased from the early to end stages of chronic kidney diseases and are associated with renal function in Chinese. PLoS One. 2011;6:e18398. doi: 10.1371/journal.pone.0018398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Su N, Jin M, Chen L. Role of FGF/FGFR signaling in skeletal development and homeostasis: learning from mouse models. Bone research. 2014;2:14003. doi: 10.1038/boneres.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Turkington RC, Longley DB, Allen WL, et al. Fibroblast growth factor receptor 4 (FGFR4): a targetable regulator of drug resistance in colorectal cancer. Cell death & disease. 2014;5:e1046. doi: 10.1038/cddis.2014.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ahmad I, Iwata T, Leung HY. Mechanisms of FGFR-mediated carcinogenesis. Biochimica et biophysica acta. 2012;1823:850–860. doi: 10.1016/j.bbamcr.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 110.Mirams M, Robinson BG, Mason RS, Nelson AE. Bone as a source of FGF23: regulation by phosphate? Bone. 2004;35:1192–1199. doi: 10.1016/j.bone.2004.06.014. [DOI] [PubMed] [Google Scholar]