

Abstract
A 16-year-old boy of Caucasian ethnicity was evaluated for recurrent febrile episodes occurring during most of his life without establishment of any microbial aetiology. During febrile episodes he developed extensive splenomegaly, lymphadenopathy, anaemia, severe abdominal pain and general malaise. Lymph node biopsies demonstrated inflammation and sinus histiocytosis but no malignancy or granuloma. The patient underwent seroconversion for Epstein-Barr virus (EBV) infection during the hospitalisation. Genetic testing identified a hemizygous frameshift mutation in the X linked inhibitor of apoptosis (XIAP)-gene as well as variants in the MEFV gene indicating Familial Mediterranean Fever (FMF). XIAP expression was markedly reduced in the patient, while a functional assay assessing tumour necrosis factor (TNF)α production of monocytes in response to NOD2 stimulation displayed reduced activity. We suggest that the heterozygous MEFV variants and the hemizygous XIAP variant in combination triggered the prolonged and pathological inflammatory response to EBV infection.
Background
The distinction between infectious, inflammatory, autoimmune and malignant disease in patients with fever, lymphadenopathy and elevated inflammatory markers in blood often pose a difficult challenge. In the setting of recurrent fever with repeatedly sterile cultures during evaluation for an infectious aetiology, rare autoinflammatory periodic fever syndromes sometimes end up as the final diagnosis. These autoinflammatory fever syndromes are due to excessive production of tumour necrosis factor (TNF)α or interleukin (IL)-1 caused by mutations in molecules involved in cell homoeostasis, TNFα signalling or inflammasome activation and production of biologically active IL-1.
Familial Mediterranean Fever (FMF) is a systemic autoinflammatory fever syndrome characterised by recurrent self-limiting attacks of fever and accompanied by varying degrees of serositis, arthralgias, myalgia, exanthema and abdominal pain, and with the risk of developing complications, most notably amyloidosis and renal insufficiency.1 2 The syndrome is caused by mutations in the MEFV gene located on chromosome 16. MEFV encodes pyrin, which is a ubiquitin (Ub) ligase involved in regulation of inflammasome activation through interaction with caspase-1-dependent processing of pro-IL-1 to biologically active IL-1.3 Although FMF is considered to be inherited in an autosomal recessive manner, many patients have characteristic symptoms despite being heterozygous for mutations in the MEFV gene, and for around 30% of patients no disease-causing mutations are identified.4–6
Mutations in XIAP were first described in 2006 to cause X linked lymphoproliferative syndrome.7 However, more recent studies have revealed that patients with XIAP deficiency may present with a wide clinical spectrum, including periodic fevers, splenomegaly hypogammaglobulinaemia and inflammatory bowel disease, and carriers may be asymptomatic.8 XIAP mediates its effects by limiting apoptosis through inhibition of apoptotic caspases and is also involved in innate immune signalling pathways downstream of the cytosolic pattern recognition receptors NOD1 and NOD2.9–12 Structurally, XIAP consists of three regulatory baculovirus inhibitor of apoptosis protein repeat (BIR) domains, a ubiquitin-associated (UBA) domain and finally a C-terminal RING domain with Ub ligase activity.10 NOD2 activation by the peptidoglycan degradation product muramyl dipeptide (MDP) leads to recruitment of receptor interacting protein kinase 2 and the Ub ligases XIAP, cellular inhibitor of apoptosis 1 and 2.10 13 This event triggers ubiquitination of RIPK2 by XIAP and subsequent downstream activation of mitogen-activated protein kinases as well as the TAK1-TAB1/2/3 and downstream IκB kinase (IKK) complex.14 15 NF-κB activation ultimately results in synthesis of proinflammatory cytokines and chemokines.16
The curative treatment for XIAP deficiency is haematopoietic stem cell transplantation (HSCT). Although the outcome of HSCT has been poor17 recent advances in the treatment regime by reduced conditioning have improved clinical outcome and survival.8 18 19
Case presentation
A 16-year-old boy of Caucasian Danish ethnicity was admitted to the department of infectious diseases, Aarhus University Hospital, for evaluation of recurrent febrile episodes during most of his life but with increasing severity and duration over the past year. The patient was born to healthy non-consanguineous parents and had one healthy brother. His medical history included pituitary insufficiency treated with eltroxin and growth hormone for several years. He had been admitted to other hospitals several times during the past 10 years with episodes of elevated temperatures and evaluated for meningitis, urinary tract infection, appendicitis other viral and bacterial infections without any specific diagnosis and with no positive microbial cultures, serology or PCR indicating infection. During admission to our department, he had a particularly severe and long-lasting inflammatory febrile episode. During this febrile episode lasting almost 2 months, he developed extensive splenomegaly, lymphadenopathy, anaemia, severe intermittent abdominal and thoracic pain (requiring morphine administration), general malaise, fatigue and weight loss.
Investigations
The patient was extensively evaluated, including blood tests, histopathology on enlarged lymph nodes, liver and bone marrow, as well as genetic testing by whole exome sequencing (WES). Blood tests revealed elevated C reactive protein (CRP) to 400 mg/L in the setting of a largely normal leucocyte count, although with fluctuating lymphopenia normalising between fever episodes). Sedimentation rate was normal. Liver and renal tests were normal. A profound normocytic anaemia developed with haemoglobin levels as low as 4.2 mmol/L. During some periods, he experienced elevated ferritin levels but triglycerides and IL-2 R remained normal. Computerised axial tomography scan demonstrated hepatosplenomegaly with multiple enlarged lymph nodes in relation to the great vessels in the abdomen. Pathology from an enlarged lymph node demonstrated inflammation and extensive sinus histiocytosis (Langerhans histiocytosis was ruled out by histological stain for CD207 and CD1a, which were negative) and no malignancy or granuloma was found. Biopsy from the liver demonstrated lymphocytic infiltration but no malignancy. A bone marrow aspirate was found to be without malignancy, myelodysplastic syndrome, haemophagocytosis or granuloma. Microbiological testing was negative for HIV, hepatitis A, B and C, cytomegalovirus, Epstein-Barr virus (EBV), influenza virus, toxoplasmosis, Mycobacterium tuberculosis, (negative interferon γ release assay), and there were no positive cultures of common bacteria, mycobacteria or other pathogens in the bone marrow or blood. Of note, the patient underwent seroconversion for EBV during his admission and severe disease. An immunological evaluation was largely normal, including normal levels of immunoglobulins and complement activation. The CD4 count was reduced to 310 cells/μL during the period with general lymphopenia but normalised during disease-free periods. Flow cytometry revealed general lymphopenia (607 cells/μL) but with normal distribution of lymphocytes, including B cells, T cells and natural killer (NK) cells. Notably, no increase in the fraction of double-negative T cells was observed. Finally, degranulation of NK cells was normal.
Since no diagnosis was reached despite this careful evaluation, we decided to perform WES, primarily to search for mutations in genes related to autoinflammatory syndromes (see online supplementary table S1), lymphoproliferative conditions and EBV-related disease (see online supplementary table S2).
Genetic testing by conventional Sanger sequencing of the MEFV gene identified a variant (c.1105C>T, p.P369S) pointing towards an FMF diagnosis, and WES revealed another variant in the MEFV gene. The two variants in MEFV cause amino acid changes P369S and R408Q, of which the P369S variant is predicted to belong to the 10% most damaging variants with a combined annotation dependent depletion (CADD) score of 15.6. However, the variants are frequent (>1%, table 1) and both were present in the mother, but not in the father or healthy brother.
Table 1.
Variants and genotypes detected
| EXaC frequency | Genotype |
||||
|---|---|---|---|---|---|
| Gene/variant | Patient | Mother | Father | Healthy brother | |
| MEFV/c.1105C>T p.P369S |
0.01423 | c.1105C;c.1105T | c.1105C;c.1105T | c.1105C | c.1105C |
| MEFV/c.1223G>A p.R408Q |
0.0128 | c.1223G;c.1223A | c.1223G;c.1223A | c.1223G | c.1223G |
| XIAP/c.1026delT p.I342fs |
– | c.1026delT | c.1026T;c.1026delT | c.1026T | c.1026delT |
| XIAP/c.1027C>A p.H343N |
– | c.1027C>A | c.1027C;c.1027A | c.1027C | c.1027C>A |
| XIAP/c.1031T>A p.L344 |
– | c.1031T>A | c.1031T;c.1031A | c.1031T | c.1031T>A |
EXaC, The Exome Aggregation Consortium; MEFV, NM_000243.2; XIAP, NM_001167.3.
Importantly, novel XIAP mutations were detected by WES (figure 1A) and confirmed by Sanger sequencing (figure 1B). The mutations include a frameshift mutation at position 342 (I342IfsX353) along with two other variants (table 1). The XIAP I342IfsX353 mutation is outside the BIR3 domain, located to the helix immediately folowing the BIR domain and results in loss of UBA and RING domains (figure 1C). The pedigree is shown in figure 1D.
Figure 1.

Identification of novel XIAP mutations. (A) Whole exome sequencing was performed by TruSeq DNA sample preparation, targeting of exomes with SeqCap EZ Human Exome Library V.3.0 (Roche), and sequencing was performed on HiSeq, paired end 2×101 bp indexed. Variants were filtered using ingenuity variant analysis, and visualised using Integrative Genomics Viewer software. Variants of interest were selected on the basis of rarity and evaluation of severity by different prediction tools smart information flow technologies (SIFT), PolyPhen-2 and CADD score. (B) Variants were confirmed by Sanger sequencing. (C) Schematic presentation of the XIAP protein, the mutation in XIAP results in a frameshift after the BIR3 domain. (D) Pedigree showing the inheritance of XIAP and MEFV mutations. wt, wild-type; mt, mutant; Pt, index patient.
To evaluate the effect of the XIAP mutation, we tested the protein expression as well as functional consequences by a flow cytometric assay recently developed to evaluate XIAP deficiency.20 XIAP function is assessed by L18-MDP stimulation of monocytes, recognised by the NOD2 receptor, which then recruits XIAP, causing RIPK2 ubiquitination, NF-κB activation and finally TNFα production. This analysis showed reduced XIAP expression in the patient and his brother, a carrier pattern in the mother (figure 2A) and a diminished L18-MDP response in the patient and his brother (figure 2B).
Figure 2.
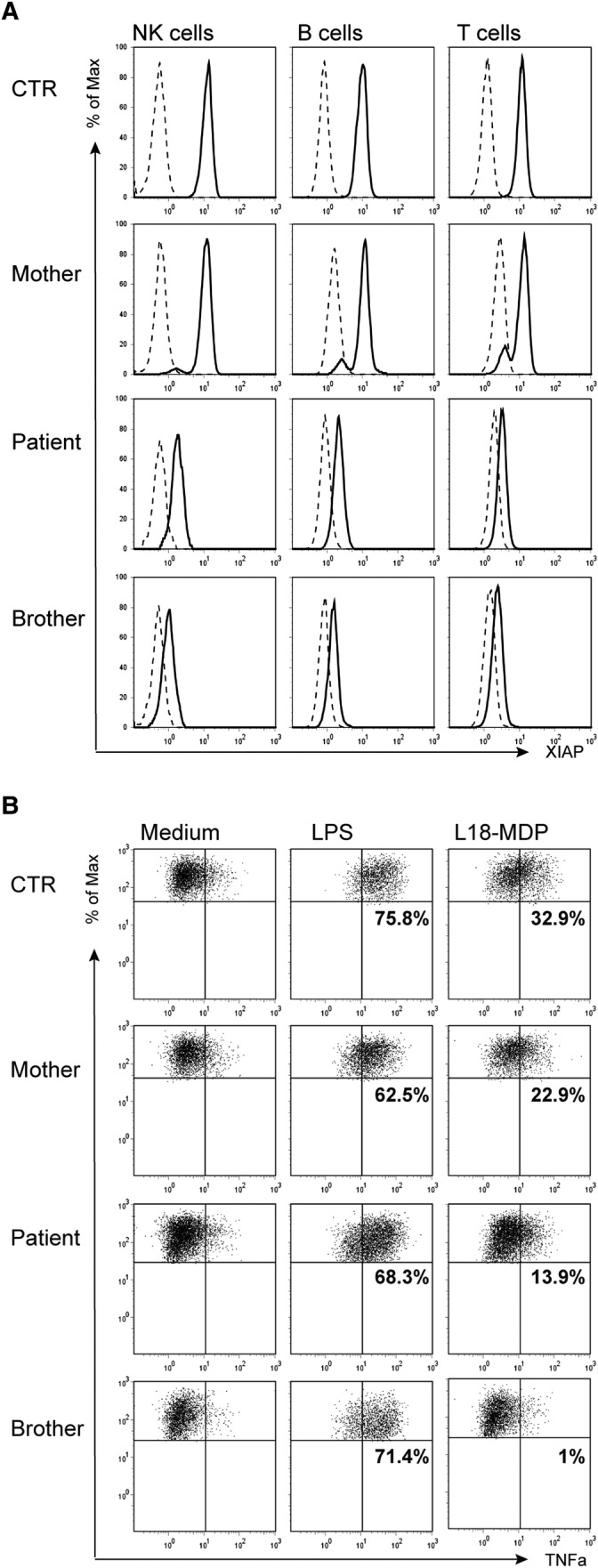
Characterisation of X linked inhibitor of apoptosis (XIAP) presence and function in patient cells. (A) Overlay of staining with anti-XIAP (solid line) and an isotype control antibody (stippled), gated on CD3+ T lymphocytes, CD19+ B lymphocytes and CD56+ NK cells. XIAP patients are shown with their day control. (B) Tumour necrosis factor α (TNFα)-producing monocytes from a healthy donor control (Ctr), the asymptomatic XIAP carrier, the XIAP patient, and his healthy brother (with the same XIAP mutation) cultured in the presence of the medium alone, 200 ng/mL L18-MDP or 200 ng/mL lipopolysaccharide. The percentages indicate the ΔTNFα positive cells of stimulated—medium as a fraction of all human leukocyte antigen-D related (HLA-DR)+CD14 monocytes. BIR, baculovirus inhibitor of apoptosis protein repeat; MDP, muramyl dipeptide; UBA, ubiquitin-associated.
Differential diagnoses
During admission and before the results of genetic analyses demonstrating the presence of mutations in MEFV and XIAP, we considered several differential diagnoses, including other autoinflammatory fever syndromes, lymphoma, acute lymphoproliferative syndrome (ALPS), langerhans cell histiocytosis, Rosai-Dorfman disease, haemophagocytosis, adult Still's disease, porphyria and primary immunodeficiency with mycobacterial or chronic viral infection.
Treatment
The patient was treated with the IL-1R antagonist anakinra (kineret) 200 mg subcutaneous daily together with colchicine 0.5 mg×3 daily, which led to partial resolution of symptoms and CRP decrease to 100 mg/L. However, additional corticosteroid treatment was added, initially in a high dose of solumedrole 100 mg×1 with gradual tapering to the present 7.5 mg daily. Corticosteroid treatment has been continuously required, since the patient starts to experience fever and abdominal and muscular pain whenever prednisolone has been withdrawn. A trial of prophylactic azathioprine 100 mg daily was initiated, but was stopped again after 1 year of treatment without any beneficial effect.
The possibility of performing an HSCT due to the XIAP mutation has been considered and discussed with the patient, but this is not planned given the current excellent state of the patient, although he must be anticipated to require lifelong immunomodulatory treatment and monitoring of immunoglobulin levels once per year.
Outcome and follow-up
The patient has been seen in our immunodeficiency clinic every 4 months and he is currently in a good and stable condition, pursuing his training as a carpenter. His medication include colchicine 500 mg×3, anakinra 100 mg subcutaneously ×1 and prednisolone 7.5 mg daily. Moreover, he keeps morphine tablets in case of rare sudden attacks of fever and serositis with severe abdominal and chest pain. The brother of the patient, who has the same mutation in XIAP but no mutations in MEFV, has been found to be EBV negative. Therefore, he has been encouraged to seek immediate medical advice in case of mononucleosis-like symptoms, since he may have an increased risk of developing a similar severe lymphoproliferative disease during EBV infection. Finally, both brothers have been HLA typed in case of urgent requirement for HSCT.
Discussion
Given the nature and frequency of the MEFV variants identified, we do not consider FMF as the correct and exclusive diagnosis to explain this patient's medical history. The extensive hepatosplenomegaly and lymphadenopathy lasting for more than a month until anakinra and corticosteroids were administered is not typical of FMF, and the continuous requirement for prednisolone together with anakinra to prevent fever and pain is also unusual for this condition. Importantly, WES revealed novel and deleterious mutations in XIAP, which was confirmed by a functional assay demonstrating severely reduced XIAP expression and function. A diagnosis of XIAP deficiency is in agreement with the seroconversion for EBV during the long-lasting severe disease with lymphadenopathy, splenomegaly, fever and biochemical tests resembling, but not fulfilling, the criteria for haemophagocytosis. XIAP deficiency is mostly caused by mutations that either result in severe aberrations in the protein or loss of expression. These mutations (nonsense, frameshifts or deletions) locate throughout XIAP and in general interfere with the C-terminal RING domain,12 18 21 22 which is also the case for the mutations identified in the patient presented here. Furthermore, in the search for causes of idiopathic periodic fever, Ferretti et al23 identified a Q423P polymorphism in XIAP associated with increased risk of periodic fever.
This case report clearly demonstrates some of the challenges in distinguishing between infection, inflammation and malignancy in patients with recurrent or long-lasting fever, haematological abnormalities and elevated inflammatory parameters. The clinical presentation of this young man, including fever and serositis typical of FMF (but without the typical ethnicity) combined with lymphoproliferation, hepatosplenomegaly, lymphopenia and anaemia more characteristic of XIAP deficiency, ALPS or lymphoma, could initially not be easily explained by just one disease entity. A narrow genetic approach employing Sanger sequencing identified the MEFV variant pointing towards FMF; however, the inheritance in FMF is normally autosomal recessive, and the clinical picture did not correlate well with a frequent, heterozygous variant in MEFV. A broadened genetic approach consisting of WES subsequently led to the identification of hemizygous, deleterious mutations in the XIAP gene, indicating XIAP deficiency as the correct diagnosis. However, a combined effect of these mutations may contribute to the clinical phenotype and possibly explain why the brother is asymptomatic. It could be speculated that the broad spectrum of symptoms seen in XIAP-deficient patients is due to the existence of modifying variants in other genes (such as MEFV).
Several questions remain, including the long-term risk of developing malignant lymphoproliferative disease, the optimal treatment strategy in case of deterioration and eventually the need for HSCT. Moreover, the healthy, so far EBV-naïve, brother shared the XIAP mutation but not the MEFV variant; thus, it will be of uttermost importance to follow the course of an EBV infection in this individual. We believe that it is essential for clinicians, clinical immunologists and geneticists to be aware of the existence of such heterogenous clinical conditions with a mixed genetic background, which may require extensive evaluation. Finally, this case illustrates the need for individually tailored treatment and control of patients with such rare autoinflammatory and lymphoproliferative diseases.
Learning points.
Autoinflammatory fever syndromes and lymphoproliferative diseases may have a wide spectrum of presentations and be difficult to differentiate from malignancy, particularly lymphoma.
The clinical presentation of XIAP deficiency is highly variable and may be modified in the presence of additional genetic defects.
A clinical picture may present in an atypical way if two different entities, that is, here XIAP deficiency and FMF coexist. This scenario may be an increasingly frequent challenge for clinicians given the use of broad-range genetic tests, such as WES.
The optimal immunomodulatory treatment for autoinflammatory/lymphoproliferative conditions often need to be searched and optimised in each individual patient.
The differentiation between infectious, inflammatory and malignant diseases may be complex and difficult and require extensive analysis of histopathology, functional immunological assay and cell biological assay as well as sequencing of the entire exome or, alternatively, a more targeted sequencing approach.
Acknowledgments
The authors wish to thank the patient and his family for participation and for permission to publication of the medical story.
Footnotes
Contributors: THM identified and cared for the patient. MC performed genetic analysis and prepared the figures. SA performed the XIAP assay. CS provided discussion and advice on the XIAP assay and patient management. THM and MC wrote the manuscript. All authors read and approved the final version of the manuscript.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Shohat M, Halpern GJ. Familial Mediterranean fever—a review. Genet Med 2011;13:487–98. 10.1097/GIM.0b013e3182060456 [DOI] [PubMed] [Google Scholar]
- 2.Lidar M, Yaqubov M, Zaks N et al. The prodrome: a prominent yet overlooked pre-attack manifestation of familial Mediterranean fever. J Rheumatol 2006;33:1089–92. [PubMed] [Google Scholar]
- 3.Manukyan G, Aminov R. Update on pyrin functions and mechanisms of familial Mediterranean fever. Front Microbiol 2016;7:456 10.3389/fmicb.2016.00456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Booty MG, Chae JJ, Masters SL et al. Familial Mediterranean fever with a single MEFV mutation: where is the second hit? Arthritis Rheum 2009;60:1851–61. 10.1002/art.24569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marek-Yagel D, Berkun Y, Padeh S et al. Clinical disease among patients heterozygous for familial Mediterranean fever. Arthritis Rheum 2009;60:1862–6. 10.1002/art.24570 [DOI] [PubMed] [Google Scholar]
- 6.Ben-Zvi I, Herskovizh C, Kukuy O et al. Familial Mediterranean fever without MEFV mutations: a case-control study. Orphanet J Rare Dis 2015;10:34 10.1186/s13023-015-0252-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rigaud S, Fondanèche M-C, Lambert N et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature 2006;444:110–14. 10.1038/nature05257 [DOI] [PubMed] [Google Scholar]
- 8.Speckmann C, Lehmberg K, Albert MH et al. X-linked inhibitor of apoptosis (XIAP) deficiency: the spectrum of presenting manifestations beyond hemophagocytic lymphohistiocytosis. Clin Immunol 2013;149:133–41. 10.1016/j.clim.2013.07.004 [DOI] [PubMed] [Google Scholar]
- 9.Bauler LD, Duckett CS, O'Riordan MXD. XIAP regulates cytosol-specific innate immunity to Listeria infection. PLoS Pathog 2008;4:e1000142 10.1371/journal.ppat.1000142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Damgaard RB, Nachbur U, Yabal M et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell 2012;46:746–58. 10.1016/j.molcel.2012.04.014 [DOI] [PubMed] [Google Scholar]
- 11.Krieg A, Correa RG, Garrison JB et al. XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci USA 2009;106:14524–9. 10.1073/pnas.0907131106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damgaard RB, Fiil BK, Speckmann C et al. Disease-causing mutations in the XIAP BIR2 domain impair NOD2-dependent immune signalling. EMBO Mol Med 2013;5:1278–95. 10.1002/emmm.201303090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertrand MJM, Doiron K, Labbé K et al. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity 2009;30:789–801. 10.1016/j.immuni.2009.04.011 [DOI] [PubMed] [Google Scholar]
- 14.Beug ST, Cheung HH, LaCasse EC et al. Modulation of immune signalling by inhibitors of apoptosis. Trends Immunol 2012;33:535–45. 10.1016/j.it.2012.06.004 [DOI] [PubMed] [Google Scholar]
- 15.Damgaard RB, Gyrd-Hansen M. Inhibitor of apoptosis (IAP) proteins in regulation of inflammation and innate immunity. Discov Med 2011;11:221–31. [PubMed] [Google Scholar]
- 16.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol 2004;25:280–8. 10.1016/j.it.2004.03.008 [DOI] [PubMed] [Google Scholar]
- 17.Marsh RA, Rao K, Satwani P et al. Allogeneic hematopoietic cell transplantation for XIAP deficiency: an international survey reveals poor outcomes. Blood 2013;121:877–83. 10.1182/blood-2012-06-432500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marsh RA, Vaughn G, Kim MO et al. Reduced-intensity conditioning significantly improves survival of patients with hemophagocytic lymphohistiocytosis undergoing allogeneic hematopoietic cell transplantation. Blood 2010;116:5824–31. 10.1182/blood-2010-04-282392 [DOI] [PubMed] [Google Scholar]
- 19.Chellapandian D, Krueger J, Schechter T et al. Successful allogeneic hematopoietic stem cell transplantation in XIAP deficiency using reduced-intensity conditioning. Pediatr Blood Cancer 2016;63:355–7. 10.1002/pbc.25756 [DOI] [PubMed] [Google Scholar]
- 20.Ammann S, Elling R, Gyrd-Hansen M et al. A new functional assay for the diagnosis of X-linked inhibitor of apoptosis (XIAP) deficiency. Clin Exp Immunol 2014;176:394–400. 10.1111/cei.12306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Filipovich AH, Zhang K, Snow AL et al. X-linked lymphoproliferative syndromes: brothers or distant cousins? Blood 2010;116:3398–408. 10.1182/blood-2010-03-275909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pachlopnik Schmid J, Canioni D, Moshous D et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood 2011;117:1522–9. 10.1182/blood-2010-07-298372 [DOI] [PubMed] [Google Scholar]
- 23.Ferretti M, Gattorno M, Chiocchetti A et al. The 423Q polymorphism of the X-linked inhibitor of apoptosis gene influences monocyte function and is associated with periodic fever. Arthritis Rheum 2009;60:3476–84. 10.1002/art.24905 [DOI] [PubMed] [Google Scholar]