

Abstract
Early – intrauterine – environmental factors are linked to the development of cardiovascular disease in later life. Traditionally, these factors are considered to be maternal factors such as maternal under and overnutrition, exposure to toxins, lack of micronutrients, and stress during pregnancy. However, in the recent years, it became obvious that also paternal environmental factors before conception and during sperm development determine the health of the offspring in later life. We will first describe clinical observational studies providing evidence for paternal programming of adulthood diseases in progeny. Next, we describe key animal studies proving this relationship, followed by a detailed analysis of our current understanding of the underlying molecular mechanisms of paternal programming. Alterations of noncoding sperm micro-RNAs, histone acetylation, and targeted as well as global DNA methylation seem to be in particular involved in paternal programming of offspring's diseases in later life.
Keywords: cardiometabolic diseases, epigenetics, offspring, paternal programming, spermatogenesis, transgenerational effects
INTRODUCTION
Currently, the overall population health status of middle and high-income societies is facing an unprecedented challenge because of a rising incidence of the metabolic syndrome, obesity, diabetes mellitus, hypertension, and cardiovascular diseases. It becomes more and more evident that sociocultural (differences in culture, religion, economic status, etc.) and early life environmental factors (insults during pre, peri, and early postnatal periods) are important determinants of human health and disease in adult life [1].
It was suggested that many disadvantageous nutrition and/or environmental factors that occur during critical periods of fetal development may lead to permanent effects on organ morphology, function, and metabolism, which manifest in adulthood. In the late 1960s, Farquhar [2] conducted an epidemiological study, which confirmed a transmission of an impaired maternal phenotype to offspring. This survey demonstrated an increased risk of late childhood overweight in offspring born to diabetic mothers [2]. In the next decade, it has been shown that maternal diabetes leads to an increased risk of impaired glucose tolerance (IGT) and diabetes development in offspring [3]. Dörner et al.[4] showed that type 2 diabetes mellitus (T2DM) is predominantly transmitted over generations via maternal, but not paternal lines, as the incidence of T2DM in maternal great-grandmothers of third-generation offspring with infantile onset diabetes was significantly higher compared with their paternal counterparts. Subsequent epidemiological studies by Barker and his coworkers [5–7] provided evidence that fetal undernutrition is associated with (programs) cardiovascular diseases in later life. This concept is known as fetal programming or fetal origins of adult disease hypothesis. The main assumption behind this theory is that fetuses experiencing suboptimal conditions during prenatal period evolve in a way tending to prepare the growing organism to the similar suboptimal conditions in postnatal life. After its introduction by Barker and Martyn [7], the fetal programming hypothesis was complemented by further studies. In particular, the Dutch Hunger Winter study showed that prenatal famine exposure is linked to physical and mental health in later life [8]. In the mid-2000s, the fetal origins of adult disease hypothesis of Barker and Martyn has been extended by the notion that apart from the most critical intrauterine period, various adverse factors affecting the organism in the later periods of life – childhood, puberty period, and senescence – impact on the resulting health–disease status of a given study participant [9]. This theory is called the Developmental Origins of Health and Disease concept [9,10].
Maternal undernutrition during pregnancy is a well studied factor, which can lead to fetal programming. This was initially recognized in epidemiological studies and subsequently proven in animal experiments [11–14]. Several other mechanisms caused by environmental factors in early life linked to lifelong functional and structural alterations have been reported, including glucocorticoid exposure of the fetus because of 11β-hydroxysteroid dehydrogenase deficiency of the placenta [15,16] or a high-protein diet during pregnancy [17]. An alternative mechanism responsible for programming events during intrauterine life might be related to maternal genes, which influence the fetal phenotype independently of the fetal DNA-based genome. Such an event was reported for the first time for the mutation in wimp gene – a dominant maternal-effect mutation – in Drosophila melanogaster, which leads to the lethal phenotype of the progeny, even when the mutation is not inherited [18]. Our group was the first to translate this to mammalians/humans in 2000 by demonstrating that a genetic variation of a maternal gene most likely involved in the uterine blood flow control is associated with a substantial reduction of offspring's birth weight without being actually transmitted to the offspring [19,20]. Other independent association studies in humans later on, similarly, suggest that certain maternal genes may affect the fetal phenotype even without transmission of that particular gene to the fetus [21,22]. In other words, a gene of a human being may influence the physiology of another study participant without being present in this particular individual. Recently, this hypothesis has finally been proven by experiments showing that a maternal genetic defect can epigenetically alter the progeny phenotype without inheritance of the defect itself [23]. All the abovementioned mechanisms of fetal programming originated from nutritional, environmental, or genetic insults of the mother. There is, however, compelling evidence that also insults to the father may be causal for cardiovascular and mental diseases of the offspring in later life. We will first review studies in humans suggesting that paternal effects may play an important role in the pathogenesis of offspring cardiometabolic diseases in later life, followed by an overview of animal studies aiming to explore the underlying mechanisms of paternal programming.
EPIDEMIOLOGICAL STUDIES SUGGESTING PATERNAL PROGRAMMING
Over the past two decades, many observational studies analyzed the relationship between father's lifestyle-related factors, environmental exposure factors, and offspring's health outcome in early and later life (hypertension, dyslipidemia, hyperglycemia, insulin resistance, obesity, etc.).
Obesity and overnutrition
Overnutrition and obesity – major public health problems in the industrialized countries – are well known lifestyle-related factors. Although mother's influence (lifestyle, diet, etc.) on offspring, adult health has traditionally been considered as a major contributor; potential paternal-mediated effects are less well understood so far. However, there is clear evidence that paternal nutritional factors play an important role. For example, it was shown that children born not only to obese mothers but also to obese fathers tend to be obese [24]. The first study to describe the paternal impact on offspring health in humans was published in 2000 by Figueroa et al.[25], who reported the correlation between paternal absolute and relative amounts of body fat and the same parameters in their daughters aged 4.8–8.9 years. Some efforts have been made to investigate father–offspring and mother–offspring associations with BMI, central obesity, and height. It has been reported that mother–offspring associations are stronger than the ones between father and offspring [26,27], whereas others have reported comparable effects of maternal and paternal obesity on the offspring's phenotype [28,29]. On the other hand, Murrin et al.[26], who conducted a family cohort study, showed that early childhood BMI and height in offspring correlate with only maternal line. As reported by Lawlor et al.[28], who conducted a study in 4091 parent–offspring trios, the amounts of fat in offspring at 9–11 years of age positively correlated with both maternal and paternal BMI, although maternal effect was more pronounced. The results of a large population-based study (29216 parent–offspring trios) suggest that offspring BMI at 3 years of age positively correlated with both maternal and paternal BMI [29]. Contrary to the aforementioned studies in young study participant, Vik et al.[30] conducted a large population-based survey (36528 parent–offspring trios) in adults, and reported that offspring BMI, height, blood pressure, blood lipids, and blood glucose is correlated with both maternal and paternal lines. There are hints that paternal BMI might modulate the offspring phenotype in a sex-dependent manner. Chen et al.[31] in the frames of a family cohort study (899 parent–offspring trios) demonstrated that paternal BMI correlated with birth weight, biparietal diameter, head circumference, abdominal diameter, abdominal circumference, and pectoral diameter in male newborns only. Thus, paternal BMI might be a risk factor for cardiovascular diseases of male progeny in later life [31].
Fathers [32] or grandfathers [33] exposed to either overfeeding or food restriction at 9–12 years of age predetermines male offspring reduced longevity. Similarly, female offspring born to mothers and grandmothers exposed to either overfeeding or food restriction at the age of 9–12 years had a reduced longevity [32]. In addition, the F2 offspring of paternal grandfathers exposed to overnutrition at 9–12 years of age had a four-fold risk of diabetes mortality [34]. Thus, the insults affecting boys in their prepubertal period, when the process of sperm maturation occurs, may be responsible for the aforementioned transgenerational effects.
Taken together, there is clear evidence that paternal nutritional factors not only prior to conception but also as early as in paternal puberty might affect the offspring in a sex-dependent manner. There is also – on the other hand – clear evidence that parental obesity genes – both maternal obesity genes and also paternal obesity genes – are casually linked to obesity and the metabolic syndrome in the offspring [35–39].
Moreover, there is clear evidence that there is an interaction between parental genes and parental environmental factors that causes an effect on the phenotype of the offspring [40]. It was shown, for example, that there is a clear interaction between maternal smoking during pregnancy with maternal genes responsible for smoke toxin metabolism that affect the offspring's birth weight [22].
The gene–environment interaction becomes even more complex, as it is also known that the socioeconomic status of an individual seems to have opposite effects on obesity in poor and rich countries [41].
Diabetes mellitus
The first documented report on correlation between offspring low birth weight and paternal diabetes was provided by Lindsay et al.[42], who conducted their study in 1608 Pima, Native American children of the Gila River Indian Community (Arizona, USA). Offspring birth weight positively correlates with maternal diabetes and negatively correlates with paternal diabetes [42,43]. In addition, nondiabetic children born to fathers with early-onset T2DM had lower body weight compared with offspring born to mothers with early-onset T2DM or both healthy parents [44].
Adverse dietary habits
Paternal unhealthy dietary behaviors were proven to be a detrimental factor not only for fathers themselves but also to their offspring. Chewing of betel nuts (the fruits of Areca catechu palm tree) is known to be a risk factor in the development of T2DM [45], and a dose-dependent positive correlation between betel nuts’ consumption by fathers and the incidence of metabolic syndrome in their offspring has been shown [46]. This observation is in accordance with findings from an earlier animal study, which demonstrated transgenerational diabetogenic effects in F1 and F2 progenies of CD1 mice fed with betel nuts [47].
Exposure to toxic substances
The effects of paternal tobacco smoking have also been shown to be transmitted across generations. The Överkalix study documented a negative correlation between BMI of sons, but not daughters, with the age of smoking onset of their fathers [48]. The analysis of umbilical cord blood cells derived from 39 newborns showed that DNA damage is associated with father's smoking before conception but not with mother's passive smoking during pregnancy [49]. A small study in humans (n = 13) reported that paternal cigarette smoking alters sperm microRNA expression pattern compared with nonsmokers [50]. Such an alteration might cause an impairment of molecular pathways responsible for a normal function of sperm cells and embryo development [50]. These data are in line with animals studies, which showed that the exposure of mature male mice to both firsthand [51] and secondhand cigarette smoke [52] elevated sperm DNA fragmentation rates. Thus, tobacco smoke is a male germ cell mutagen.
Exposure to ionizing radiation
Although ionizing radiation is a well known mutagenic agent in somatic and germ cells, the current knowledge of epigenetic inheritance of ionizing radiation effects through the male germ line remains limited. The recent research focus has been shifted toward investigation of ionizing radiation nontargeted effects, such as bystander effect and genome instability.
In 1990, Gardner et al.[53] reported a three-fold higher incidence of leukemia and non-Hodgkin's lymphoma in children born to fathers occupationally exposed to low-dose ionizing radiation because of their work at Sellafield nuclear power plant (UK). Notably, this study also showed that such association was dose dependent and correlated with father's exposure to ionizing radiation in a period around conception [53]. Another large population-based study reported an increased stillbirth rate in newborns from fathers occupationally exposed to low-dose ionizing radiation in Sellafield nuclear power plant (n = 9078 for live births and n = 130 for stillbirths in the period 1950–1989) compared with the rest of population, in which none of the parents received radiation exposure [54]. A small study in Chernobyl nuclear power plant accident cleanup workers (n = 18) reported abnormal head shape of spermatozoa and impaired sperm motility compared with a control group [55]. Moreover, the children whose fathers were irradiated with low doses while working as liquidators of Chernobyl disaster had an increased rate of chromosomal aberrations [56].
However, to date, the vast majority of large epidemiological studies in this field [57–62] failed to demonstrate a direct link between paternal ionizing radiation exposure and offspring long-term health.
Effects of paternal birth weight on offspring birth weight
A positive correlation between maternal low birth weight and offspring low birth weight is well documented. However, to date, little is known about the influence of paternal line birth weight effects. Offspring birth weight was shown to be significantly correlated with their fathers’ birth size and BMI in adulthood [63]. In addition, as shown by Veena et al.[64], who conducted their study in 506 offspring in India, both maternal and paternal low birth weight negatively correlated with the incidence of metabolic syndrome in their children.
Epidemiological studies on programming effects of paternal exposure to various risk factors are summarized in Table 1.
TABLE 1.
List of epidemiological studies on programming effects of paternal exposure to various risk factors
| Risk factor | Source country | Group size (N) | The main findings of the study | Reference |
| Overnutrition | Sweden | 1626 (F0 and F1) 271 (F2) | Longevity of male descendants of paternal grandfathers with overnutrition during SGP was reduced | [32] |
| Overnutrition | Sweden | 239 (F2) | Cardiovascular and diabetes mortality in offspring of paternal grandfather with overnutrition during SGP was increased | [34] |
| Height | United Kingdom | 226 parent–child trios | Height is transmitted to an offspring by both parents | [27] |
| BMI and height | Ireland | 669 families and 529 children | Early childhood BMI in offspring is correlated only with maternal line. Offspring height is correlated with both maternal and paternal lines | [26] |
| BMI | United Kingdom | 4091 parent–child trios | The amount of fat in offspring at 9–11 years of age is positively correlated with both maternal and paternal BMI, although maternal effect was more pronounced | [28] |
| BMI | Norway | 29 216 parent–child trios | Offspring BMI at 3 years of age is positively correlated with both maternal and paternal BMI | [29] |
| BMI | China | 899 parent–child trios | Paternal BMI is correlated with birth parameters of male offspring only: birth weight, biparietal diameter, head circumference, abdominal diameter, abdominal circumference, and pectoral diameter | [31] |
| Body fat | United Kingdom | 39 mothers, 36 fathers, and 47 daughters | Paternal body fat predicts the changes in body fat of premenarcheal daughters | [25] |
| Cardiovascular risk factors | Norway | 36 528 parent–child trios | Parent–offspring associations of anthropometric factors, blood pressure, blood lipids, glucose, and resting heart rate are largely similar between fathers and mothers | [30] |
| Betel quid chewing | China | 5037 parent–child trios | Exposure to paternal betel quid chewing increases the risk of early manifestation of metabolic syndrome in human offspring in a dose-dependent manner | [46] |
| Smoking | Sweden | 1818 (F0 and F1) and 303 F2 | Early paternal smoking is associated with greater BMI at 9 years of age in sons, but not in daughters | [48] |
| Age | New Zealand | 196 fathers and 277 children | Increasing paternal age at childbirth is associated with a more favorable phenotype in their children (taller and slimmer, with better insulin sensitivity in girls) but with a less favorable lipid profile | [65] |
| Type 2 diabetes | United States | 1608 offspring | The offspring of diabetic fathers were lighter than the offspring of nondiabetic fathers and had an increased risk of diabetes in later life | [42] |
| Type 2 diabetes | United States | 36 parent–child trios | Offspring of fathers with early-onset diabetes (age <35 years) were leaner and had lower early insulin secretion | [44] |
| Type 2 diabetes | United Kingdom | 8133 (F0), 6328 (F1), and 2173 (F2) | The offspring of the fathers with diabetes had decreased birth weight | [43] |
| Low birth weight | Denmark | 1097 mothers, 1063 fathers, and 2042 children | Paternal birth weight is positively associated with children's birth weight | [63] |
| Low birth weight | India | 193 mothers, 144 fathers, and 506 children | Paternal birth weight was inversely related to offspring metabolic syndrome | [64] |
F0, grandparents, F1, parents; F2, offspring; SGP, slow growth period.
ANIMAL STUDIES SUGGESTING PATERNAL PROGRAMMING
In general, potential adverse factors influencing the father's organism can be classified into three subgroups based on the time of father's exposure: paternal factors during father's embryonic development, paternal factors during prepubertal and spermatogenesis periods, and preconception paternal factors.
Paternal factors during embryonic development
Transmission via father born to paternal grandmother exposed to gestational diabetes mellitus
It is widely accepted that maternal gestational diabetes mellitus (GDM) is a risk factor for diabetes development in progeny. Ding et al.[66], who investigated transgenerational effects in the F1 and F2 generations of offspring mice born to grandmothers with streptozotocin-induced GDM, revealed that the F2 male offspring of F1-GDM males at the age of 3 and 8 weeks revealed significantly increased fasting insulin and fed insulin plasma levels compared with male littermates born to F1-control males. In contrast, in F2 female offspring of F1-GDM males only fed insulin was elevated, whereas fasting insulin was decreased compared with F2 female littermates derived from the F1-control males. Interestingly, at the age of 8 weeks, male F2 offspring of F1-GDM males had a significant two-fold increase in fed insulin levels compared with their female counterparts [66]. Both F1 and F2 offspring of GDM parents were characterized by pancreatic β-cell-specific downregulation of IGF2 and H19 gene expression levels because of hypermethylation of the differentially DNA-methylated regions (DMRs) of these genes, which was proposed as a potential explanation for an altered islet morphology and function. Independently of the presence or absence of IGT in the phenotype of adult F1-GDM males, their sperm cells exhibited a compromised IGF2 and H19 gene expression [66]. Thus, a paternal line-specific inheritance mode was suggested to be a mechanism of the epigenetic effects in this model.
Transmission via father born to paternal grandmother exposed to food deprivation
To assess the metabolic phenotypes in the F1 and the F2 generation offspring even in the absence of food deprivation, Jimenez-Chillaron et al.[67] used the murine model of low birth weight induced by maternal undernutrition from day 12 of pregnancy until delivery. It has been found that in-utero undernutrition results in lowering of birth weight, IGT, and obesity in both F1 and F2 offspring generations. Decreased birth weight was found to be programmed via paternal line. IGT in both F1 and F2 generations was partially explained by altered regulation of β-cell-specific, ATP-dependent K+ channel Sur1 gene expression.
Transmission via father born to paternal grandmother exposed to the absence of a specific nutrient
Brun et al.[68], in their study on birds, demonstrated that an impaired phenotype of F0 Muscovy ducks grandmothers, induced by a methionine-deficient diet, can be transmitted into the F2 generation via F1 male lineage, which was not even exposed to low-methionine diet. It has been shown that F2 duck progeny fed standard diet exhibited low body weight and impaired lipid metabolism [68].
Transmission via father born to paternal grandmother exposed to a high-fat diet
Dunn et al.[69] detected that in-utero exposure to maternal obesity induced by high-fat diet (HFD) in the period from 4 weeks before mating until the end of weaning in mice results in an increased body length and decreased insulin sensitivity in offspring, which can be inherited for at least two generations. These programming effects were transmitted over the generations via both maternal and paternal lines. The underlying mechanisms of observed programming events include epigenetic effects on growth hormone/insulin-like growth factor 1 axis exerted via decreased methylation status of growth hormone secretagogue receptor gene (GHSR) promoter in hypothalamic arcuate nucleus of the F2 offspring of both sexes, which in turns results in an upregulation of GHSR [69]. GHSR, also known as ghrelin receptor, is responsible for a mediation of numerous biologic effects of ghrelin – one of key regulators of carbohydrate and lipid homeostasis [70]. In the same settings, only female offspring of up to three successive generations had a larger body size (increased body weight and length), and interestingly, this trait was found to be inherited via paternal line [71].
Transmission via father born to paternal grandmother exposed to toxic agents
During the period of gonad development, the exposure to numerous toxic substances can be particularly detrimental. For instance, gestational exposure of rat dams to vapor of JP-8 – a kerosene-based hydrocarbon jet fuel – leads to a higher rate of renal disorders in offspring of both sexes and higher incidence of reproductive system insults, such as disorders of pubertal onset timing and prostate histomorphology in males and abnormal ovarian development and polycystic ovarian syndrome in females [72]. The analysis of sperm cells derived from the F3 offspring detected 33 gene promoters with significantly changed (both increased and decreased) DNA methylation status were detected.
Differential DNA methylation was suggested to be a key mechanism explaining the programming effects of an antiandrogenic endocrine disruptor vinclozolin, which was intraperitoneally given to pregnant mice. In this experiment, vinclozolin's transgenerational effects manifested in the F3 generation in the form of impaired spermatogenesis, testes and prostate histopathology in males, polycystic ovarian syndrome in females, and renal disorders in both sexes [73].
Manikkam et al.[74] investigated the phenotypes of F1, F2, and F3 generations of rat dams subjected to intraperitoneal injections of plastic or low-dose plastic compounds mixture (bisphenol-A, bis(2-ethylhexyl)phthalate, and dibutyl phthalate) from embryonic day 8 till embryonic day 14 of pregnancy – a critical period of gonadal sex determination. Pubertal abnormalities, testicular germ cell apoptosis, obesity, polycystic ovary syndrome, and premature ovarian failure were increased in the F3 generation animals.
Transmission via father born to paternal grandfather exposed to adverse stimuli
Fullston et al.[75] reported that grandfather's obesity can be inherited until at least F2 generation (via both maternal F1 and paternal F1 lineages, and that transmission via maternal line is more pronounced compared with paternal one). Along these lines, Braunschweig et al.[76] showed that F2 offspring of F0 grandfathers fed a high-methionine diet had an increased fat tissue deposition in the shoulders and were leaned compared with the control F2 littermates.
Animal studies on programming effects of paternal exposure to various risk factors during embryonic development are summarized in Table 2.
TABLE 2.
List of animal studies on programming effects of paternal exposure to various risk factors during embryonic development
| Risk factor | Animal | Exposure time | Result | Mechanism | Reference |
| Intrauterine hyperglycemia | Female ICR mice | Intraperitoneal injection of streptozotocin in pregnancy day 1 | Intrauterine hyperglycemia induced IGT and abnormal insulin levels in F1 and F2 offspring, and the IGT of male was obvious than that of female which showed sex-specific characteristics | Intrauterine glycaemia-induced abnormal IGF2/H19 methylation may cause ultrastructural alterations of the pancreatic islets in the F1 and F2 generation. Altered IGF2 and H19 gene expression was also found in sperm of adult F1-GDM offspring | [77] |
| HFD (45% of lipids) | Female C57Bl/6 : 129 hybrid mice | 4 weeks before pregnancy until weaning week 4 | Increased F1 and F2 body length and insulin insensitivity via both maternal and paternal lines. But only increased F3 females body length and body weight via the paternal lineage | Alterations in the GHSR, the GHSR transcriptional repressor AF5q31, plasma IGF-I concentrations, and IGF-binding protein-3 gene expression level suggest a contribution of the growth hormone axis. These studies provide evidence that the heritability of body length and glucose homeostasis are modulated by maternal diet across multiple generations, providing a mechanism where length can increase rapidly in concert with caloric availability. HFD effects seem to be preferentially transmitted via the paternal line even to the F3 generation | [69,71] |
| Maternal undernutrition (50% caloric restriction) | Female ICR mice | From pregnancy day 12.5 until delivery | Reduced birth weight and impaired glucose intolerance progresses to F2 offspring through the paternal line | IGT in both F1 and F2 caused by impaired β-cell function because of dysregulation of β-cell ATP-dependent K+ channel Sur1 gene expression | [67] |
| Methionine deficiency | Muscovy duck | During pregnancy | F2 duck progeny of F0 paternal grandmothers fed a methionine-deficient diet exhibited low body weight and impaired lipid metabolism | The mechanisms were not proposed by the authors | [68] |
| Endocrine disruptor compounds (BPA, DEHP, DBP) | Female Sprague–Dawley rats | From pregnancy days 8–14 | Kidney and prostate disease were observed in the direct fetally exposed F1 generation. Pubertal abnormalities, testis abnormalities, obesity, and ovarian disease (primary ovarian insufficiency and polycystic ovaries) were increased in the F3 generation animals | Analysis of the sperm epigenome identified 197 differential DMRs in gene promoters, including DMR in five known obesity-related genes – TNFRSF12A, ESRRA, FGF19, WNT10B, and GDNF | [74] |
| Endocrine disruptor (vinclozolin) | Female Sprague–Dawley rats | From pregnancy days 8–14 | Transgenerational adult-onset disease in the F3 generation (male and female), including spermatogenic cell defects, testicular abnormalities, prostate abnormalities, kidney abnormalities, and polycystic ovarian disease | 16 differentially methylated gene promoters were identified in F3 generation sperm epigenome | [73] |
| Hydrocarbon mixture involving jet fuel (JP-8) | Female Sprague–Dawley rats | From pregnancy days 8–14 | Increased incidence of primordial follicle loss and polycystic ovarian disease in females, and obesity in both females and males | Analysis of the jet fuel lineage F3 generation sperm epigenome identified 33 differential DMRs. The affected genes are involved in cellular processes such as cell signaling, energy metabolism, and regulation of transcription | [72] |
DMR,7 DNA methylation regions; F0, grandparents, F1, parents; F2, offspring; GDM, gestational diabetes mellitus; GHSR, growth hormone secretagogue receptor; HFD, high-fat diet; IGT, impaired glucose tolerance.
Paternal factors during prepubertal and spermatogenesis periods
In the past 5–7 years, an increasing number of reports suggesting that fathers exposed to environmental adverse factors during their prepubertal and spermatogenesis periods can influence the development of traits in their offspring have been published.
Low-protein diet
At the age of 3 weeks, the livers of F1 progeny of male mice (F0) fed a low-protein diet from weaning until 9–12 weeks of age, in contrast to their counterparts born to fathers fed a normal-protein diet, were characterized by an upregulation of numerous genes, regulating lipid, and cholesterol metabolism [78]. In addition, 3-week-old male F1 offspring of fathers fed a low-protein diet had more than two-fold reduction in cholesterol and cholesterol ester levels in the liver [78]. Interestingly, in liver tissue of F1 offspring born to low-protein diet-treated fathers, many genes exhibited changes in cytosine methylation, most notably of an intergenic CpG island located upstream of PPARA gene – an important regulator of fatty acid metabolism [78].
Watkins et al.[79], who conducted their study in mice, reported transgenerational effects of paternal exposure to low-protein diet. It has been shown that 24-week-old male F1 offspring had an increased birth weight compared with their littermates born to control fathers; however, in later life, offspring body weight did not differ between the groups [79]. Next, a lowering of male to female ratio was detected in progeny of fathers exposed to low-protein diet for 7 weeks before mating. In addition, such offspring of both sexes exhibited vascular dysfunction and lowered glucose tolerance, whereas only the male offspring had mild hypotension and elevation of heart rate [79]. At the age of 24 weeks, male offspring of low-protein diet fathers exhibited an excessive accumulation of fat tissue, lowered relative heart weight, and increased TNFα plasma concentration. Moreover, in offspring of both sexes of paternally programmed F0 generation, in heart and liver tissues, the genes involved in calcium homeostasis [adenylate cyclase 5 (ADCY5), phospholipase C β1(PLCB1), and protein kinase C β (PRKCB)] and FTO – a gene responsible for energy homeostasis, in particular for cardiovascular function and glucose metabolism regulation – revealed a significant downregulation in transcript expression levels [79].
High-fat diet
Numerous reports describing consequences of paternal exposure to HFD before mating on progeny phenotype have recently been published. The models of paternal programming induced by high-fat intake can be classified into the ones with manifested diabetic conditions [80–82] or with normal status of glucose homeostasis in male founders [75,83–85].
The F1 offspring of fathers fed a HFD for 11 weeks before mating, even when fed a normal-fat diet, were reported to have higher body weight, IGT, and excessive fat tissue accumulation compared with control littermates. In addition, female F1 generation progeny of fathers exposed to HFD had an elevated insulin production, decreased mass, and function of pancreatic β cells. Moreover, in female offspring programmed via paternal HFD, numerous genes involved in calcium, MAPK, and Wnt signaling pathways, apoptosis, and the cell cycle showed significant differences in expression levels and methylation status [80,81].
As reported by another study, both males and females offspring born to fathers exposed to HFD exhibited elevated fasting plasma glucose, IGT, and liver steatosis. In addition, such offspring had an excessive fat tissue synthesis, although the status of mitochondrial β-oxidation remained unaffected [82].
The compromised phenotypes (obesity in the absence of diabetes) of F1 offspring born to F0-HFD founders were reported to be inherited to the F2 generation via both maternal and paternal F1 lineages [83]. Noteworthy, the transmission of pathological traits (an impairment of glucose metabolism) in the F2 generation was more pronounced via maternal F1 line compared with the paternal one [83].
It is known that the exposure to the same detrimental factor of the F0 parental generation and the offspring itself leads to an aggravation of the offspring's phenotype. Such a phenomenon is called a ‘second-hit’ mechanism. In the context of HFD-induced paternal programming, such findings have been demonstrated by Fullston et al.[85], who reported that male HFD-treated offspring of F0 fathers exposed to HFD had an excess adiposity, IGT and insulin sensitivity, and male reproductive system derangements. Contrary to the previous reports on paternally transmitted HFD-induced effects, in the study performed by Fullston et al.[85], HFD programming on its own, without HFD exposure in the F1 generation, did not induce significant worsening of phenotype. This fact might be explained by a lower percentage of lipids of the HFD used in this study compared with the others.
High-carbohydrate diet
In the model of D. melanogaster, an exposure of male founders to high-sucrose diet during 2 days resulted in body weight gain, obesity, and an increased food consumption [86]. The F1 generation offspring born to such fathers developed obesity [86]. In this study, the authors reported microarchitecture changes in chromatin landscape in Drosophila sperm, possibly via epigenetic modification of histone mark H3K27me3 – a well known gene repressor [87], which is in turn influencing expression profiles of genes regulating metabolism [86].
Food deprivation
Miersch et al.[88] induced food deprivation in Caenorhabditis elegans – a free-living nematode – by reducing the amount of bactopeptone – a growth media for bacteria, which in turn serves as major food supply for this organism. A paternal C. elegans exposure to low amounts of bactopeptone resulted in an inverted U-shaped relationship between bacterial growth media restriction regime and body fat percentage in the F1 generation offspring of both sexes [88].
The over generational study conducted in Large White pigs revealed that the F2 progeny of F0 grandfathers exposed to a methionine-enriched diet before mating had a fatty infiltration of the shoulder and were leaner compared with their counterparts born to F0 control boars [76]. Numerous genes involved in lipid metabolism were detected to be differently expressed in muscles, liver, and kidney of the F2 offspring born to methionine-treated grandfathers when compared with control F2 counterparts [76]. As methionine is known to be one of the sources of methyl group [89], the transgenerational effects of excessive methionine intake lead to differences in methylation status in promoters of genes (in particular, IYD gene), which showed regulation patterns between F2 offspring with programming and their control counterparts [76].
Animal studies on programming effects of paternal exposure to various risk factors during prepuberty and spermatogenesis period are summarized in Table 3.
TABLE 3.
List of animal studies on programming effects of paternal exposure to various risk factors during prepuberty and spermatogenesis period
| Risk factor | Animal | Exposure time | Result | Mechanism | Reference |
| Low-protein diet (11% of protein) | Male C57/Bl6 mice | From age 3 week to age 9–12 weeks before mating | F1 male offsprings of 3 weeks exhibited elevated hepatic expression of many genes involved in lipid and cholesterol biosynthesis and decreased levels of cholesterol esters | Epigenomic profiling of offspring livers revealed numerous modest (20%) changes in cytosine methylation depending on paternal diet, including reproducible changes in methylation over a likely enhancer for the key lipid regulator PPARA | [78] |
| Low-protein diet (11% of protein) | Male C57BL/6 mice | From age 11 week to age 18 week, 7 weeks before mating | Adult offspring display significantly impaired cardiovascular and metabolic homeostasis. Male offspring birth weight increased. Adult male offspring developed relative hypotension and elevated heart rate | Male offspring had elevated adiposity, reduced heart to body weight ratio, and elevated circulating TNFα levels. Analysis of genes involved in calcium signaling revealed significantly decreased expression of adenylate cyclase 5 (ADCY5), phospholipase C β1(PLCB1), and protein kinase C β (PRKCB) in low-protein diet offspring heart tissue. Cardiovascular function and blood glucose homeostasis-associated gene FTO was also reduced in low-protein diet offspring liver tissue | [79] |
| HFD (43% of lipids) | Male Sprague–Dawley rats | From age 4 weeks to age 13 weeks, 9 weeks before mating | Female offspring of male Sprague–Dawley rats fed with HFD had an early onset of impaired insulin secretion and glucose tolerance that worsened with time | Changes in metabolic health in F1 female offspring were concomitant with altered pancreatic and adipose function, with an increased insulin section, a reduced pancreatic islet cell size, and alterations to methylation and gene profiles | [80,81] |
| HFD (49% of lipids) | Male C57BL/6 mice | From age 4 weeks to age 12 weeks, 8 weeks before mating | Offspring presented high fasting glucose, decreased glucose tolerance, and liver steatosis | SREBP-1c and FASN (hepatic lipogenesis gene) were upregulated in HFD offspring liver tissue | [82] |
| HFD (22% of lipids) | Male C57BL/6 mice | From age 5 weeks to age 15 weeks, 9–10 weeks before mating | HFD causes paternal obesity fertility disturbance and initiate the transmission of obesity and IGT and insulin sensitivity to F1 and F2 generations. Moreover, the metabolic and fertility disturbances in male offspring sired by HFD fathers are exacerbated by a ‘second-hit’ of exposure to the same obesogenic environment postnatally | HFD diminishes the reproductive health of F0, F1, and F2 males (reduced sperm motility, sperm ROS increased, DNA damage increased) and F1 and F2 female (reduced meiotic competence of oocytes and altered mitochondrial MMP in all regions of the oocytes). HFD-induced paternal obesity modulates sperm microRNA content and germ cell methylation status, which are potential signals that program offspring health and initiate the transmission of obesity and impaired metabolic health to future generations | [75,83–85] |
| HFD (22% of lipids) | Male C57BL/6 mice | From age 5 weeks to age 15 weeks, 9–10 weeks before mating | HFD delayed offspring embryo growth and placenta growth | HFD reduced F1 blastocyst cell numbers, increased rates of glycolysis impaired mitochondrial function, alterations to active and repressive embryonic chromatin marks (H3K27me3), and resulting in aberrant placental gene expression | [91,92] |
| HFD (60% of lipids) | Male C57BL/6 mice | From age 6 weeks, 10 weeks before mating | Liver gene expression of male offspring and chromatin of paternal spermatozoa were different | Differences in liver mRNA expression of five genes, namely, metallothionein-1 and 2 (MT1, MT2), fatty acid synthase (FASN), P450 cytochrome oxidoreductase (POR), and acetyl-CoA carboxylase-α (ACACA) at the age of 24 weeks versus control diet F0 male founders. HFD-fed male founders exerted increased histone H3 occupancy in the promoters of genes responsible for embryo development. The rate of H3K4me1 histone modification retention in genes responsible for embryogenesis regulation in spermatozoa derived from HFD-fed male founders was significantly higher compared with the control-fed fathers | [90] |
| High methylating micronutrient diet | Large Swiss white pig | From age 35 days, 5 month before mating | F2 offspring had a higher percentage of shoulder fat and were leaner | Microarray gene expression profiling showed that liver and muscle respective pathways of lipid metabolism and metabolic pathway were overrepresented for the differentially expressed genes between groups. The IYD gene methylation is different | [76] |
| Acute high-sugar diet | Male w1118 Drosophila melanogaster strain | 2 days before mating | Acute paternal dietary sugar increased F1 body weight, obesity, food intake, and changed metabolic control | Paternal diet-induced obesity has been shown to alter epigenetic chromatin marks (H3K27me3) in Drosophila sperm | [86] |
| Dietary restriction of bactopeptone – a growth media for bacteria, which in turn serves as major food supply for the nematode | Nematode Caenorhabditis elegans | Worms were dietary restricted for 48 h and then starved for 24 h or vice versa | Increasing the extent of paternal dietary restriction led to a successive increase in fat content of progeny until reaching a maximum of about 160% of control. Further reduction of paternal food to very low levels decreased fat content of offspring to those levels found in the control experiment | The mechanisms were not proposed by the authors | [88] |
HFD, high-fat diet.
Paternal factors during preconception stage
Apart from the embryonic, prepubertal, and spermatogenesis periods, paternal factors might also impact during the immediate preconception and zygote period. During the zygote stage, paternal factors may influence the zygote development and play an indirect role in programming offspring disease.
Anderson et al.[93] reported that paternal fasting during 24 h with free access to water before mating resulted in a considerable decreased plasma glucose levels in offspring of both sexes.
Lane et al.[94] investigated the effects of sperm cell exposure to reactive oxygen species on progeny outcome in mice. It has been reported that F1 offspring conceived by the usage of hydrogen peroxide-treated semen had a delayed embryonic development, decreased cell differentiation rate at the blastocyst stage (as proven by a lowering of a ratio of inner cell mass cells – mass of cells inside the primordial embryo – to total cell number in blastocyst), embryo implantation rate lowering, and a decreased fetal length at embryonic day 18 [94]. In adult life, female but not male F1 offspring sired by hydrogen peroxide-treated sperm showed body weight lowering, although they had an increased fat tissue deposition and glucose intolerance [94].
Recently, Chen et al.[95] documented that 30–34 nucleotide-sized transfer RNA-derived small RNAs (tsRNAs) extracted from the sperm cells produced by mouse males exposed to HFD (60% fat) are responsible for transgenerational transmission of paternal influences via sperm. Insertion of such tsRNAs, isolated from sperm cells into zygotes formed by two gametes, both of which derived from normal-fat diet-treated parents, resulted in metabolic disturbances in F1 offspring. Moreover, in early embryos and pancreatic islets of such F1 progeny, impaired expression profiles of the genes associated with metabolism have been observed [95].
Bromfield et al.[96] demonstrated that seminal fluid deficiency induced by surgical removal of seminal vesicles in F0 male mouse founders results in lower chance of pregnancy, but if the pregnancy does occur, F1 generation offspring, predominantly males, exhibit embryonic and postembryonic developmental delay and metabolic disorders (increased adiposity, IGT, and hypertension). In this study, the adverse transgenerational effects of seminal fluid deficiency in F0 founders on F1 progeny health were suggested to be because of both sperm cells damage and to a consequence of a lack of seminal fluid on female reproductive tract tissues. Moreover, oviduct tissues of F0 females made pregnant by semen with a reduced seminal fluid content, genes responsible for synthesis of embryotrophic factors LIF, CSF2, IL-6, and EGF were downregulated, whereas proapoptotic gene TRAIL showed upregulation at gestation day 0.5 [96].
Animal studies on programming effects of paternal exposure to various risk factors during periconceptional and zygote period are summarized in Table 4.
TABLE 4.
List of animal studies on programming effects of paternal exposure to various risk factors during periconceptional and zygote period
| Risk factor | Animal | Exposure time | Result | Mechanism | Reference |
| Hydrogen peroxide | C57Bl6 male mice sperm | Sperm was exposed to hydrogen peroxide, 1500 mmol/l, 1 h before fertilization | Female offspring manifesting as altered body composition and glucose regulation | Intracellular ROS levels, mitochondrial ROS, and lipid peroxidation of hydrogen peroxide-treated sperm increased. Treatment of sperm with hydrogen peroxide resulted in poorer embryo development and reduced fetal growth | [94] |
| Water deprivation | Swiss male mice | One or six times fasting 1–4 weeks before mating | Offspring decrease in average serum glucose | Corticosterone and insulin-like growth factor-1 were changed | [93] |
| RNAs | CD-1 mice | Injection of sperm heads, total RNAs, and tsRNA fractions from HFD male into normal zygotes | Generated metabolic disorders in the F1 offspring | Altered gene expression of metabolic pathways in early embryos and islets of F1 offspring | [95] |
tsRNA, transfer RNA-derived small RNA.
The mentioned animal studies, apart from complimenting the available human epidemiological trials, provide a wide range of possible mechanisms explaining the transmission of paternal insults over the generations via male germ line. Such paternal insults may results in disorders of fertility, sperm cell motility and increased incidence of metabolic, cardiovascular, and other diseases in offspring later live.
The main risk factors, which may potentially cause paternal programming, are summarized in Fig. 1.
FIGURE 1.
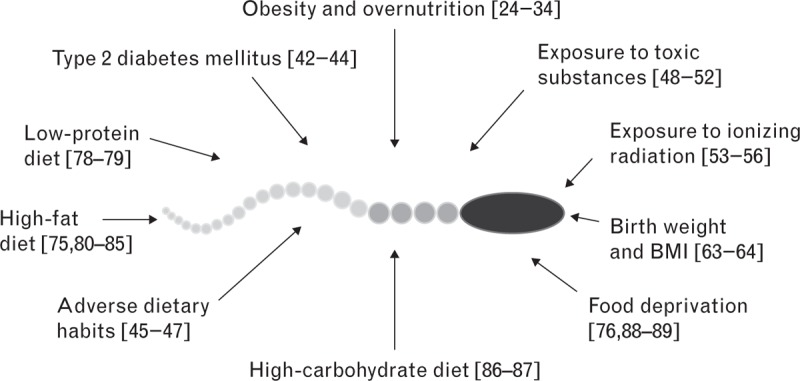
Paternal factors affecting spermatogenesis and causing transgenerational effects.
MECHANISMS OF PATERNAL PROGRAMMING
The molecular mechanism/pathophysiology of paternal programming is currently a hot research topic. The key questions are: how detrimental environmental factors act on sperm cell maturation process in the organism?; Which stage/stages of sperm cell maturation are susceptible to environmental factors?; and how are these alterations in sperm development able to be transferred into the subsequent generations and alter the phenotype of these generations?
In a recent book dedicated to epigenetics, Armstrong [97] defined epigenetic inheritance as ‘the study of changes in gene function that are mitotically and/or meiotically heritable and that do not entail a change in the sequence of DNA’. To date, the main molecular mechanisms of epigenetic inheritance, in particular paternal programming, include methylation of DNA, histone variants modification, and control of gene expression by noncoding RNAs.
The mechanisms of paternal phenotype inheritance to the progeny can generally be classified into: processes influencing paternal sperm cell development and complex interactions between paternal and maternal impacts on formation of offspring health.
Paternal germline epigenetic effects
Sperm DNA methylation
It is widely accepted that DNA hypomethylation (i.e. low levels of DNA 5-methylcytosine) is a sign of gene activation, whereas DNA hypermethylation is associated with the silencing of gene expression. However, there are also studies reporting that methylation of a gene silencer actually activates gene expression because the silencer became silent. Most of newly established germ cells – primordial germ cells – undergo a complete erasure of DNA methylation at CpG sites, that is, global epigenetic reprogramming, during their migration into the genital ridges. Nevertheless, certain DNA sequences in primordial germ cells, such as imprinted clusters and retrotransposons, remain resistant to the process of demethylation during reprogramming events [98–100]. The most well known regions in the genome, resistant to erasure of DNA methylation, are the intracisternal A-particles and long terminal repeat endogenous retroviral sequence 1 elements [99,101]. A human study (n = 298) reported a correlation between the level of global DNA methylation in the genome of fathers and their offspring [102]. An exposure of F0 generation rodent male founders to such toxic agents as plastics mixture (bisphenol-A, bis(2-ethylhexyl)phthalate, and dibutyl phthalate), endocrine disruptor vinclozolin, and hydrocarbon mixture (jet fuel) resulted in changes in methylation status of differential DMR in gene promoters [72–74]. An exposure of F0 rat males to plastic-derived endocrine disruptors led to obesity and other aforementioned abnormalities (see chapter entitled, ‘Transmission via father born to paternal grandmother exposed to a high-fat diet’) in the F3 offspring, which correlated with DMR in gene promoters of five known obesity-related genes, TNFRSF12A, ESRRA, FGF19, WNT10B, and GDNF[74].
Sperm RNAs
Although mature spermatozoa are transcriptionally inactive cells, they still contain diverse RNA species, both coding (ribosomal RNA and mRNA) and noncoding RNAs [microRNAs, double-stranded RNA, derived small RNAs (tsRNAs), piRNAs, transposable elements, annotated long noncoding RNAs, intronic retained elements, exonic elements, chromatin-associated RNAs, small nuclear ILF3/NF30-associated RNAs, quiescent RNAs, mse-tRNAs, and YRNAs] [103].
As shown by Ostermeier et al.[104], unique sperm cell-derived mRNA repertoires are also present in the tissues of an early embryo, whereas such mRNA species are absent in nonfertilized oocytes. Among entire spermatozoa RNA population, a subgroup of sperm cell-derived microRNAs – single-stranded RNA molecules composed of 20–23 nucleotides – plays a particularly important role in the transmission of paternal epigenetic influences via fertilized oocytes to the subsequent generations [105,106]. Overall, in zygote and subsequent earliest embryo stages, sperm-derived microRNAs exert a fast adaptive feedback to the changes in environmental conditions in terms of gene expression regulation [107]. However, individual microRNAs, implicated in this process, are just starting to be identified. In a mouse model, Rodgers et al.[107] detected certain microRNAs species, which showed elevated levels in spermatozoa of F0 male founders exposed to stress during 42 days before breeding. The F1 progeny of such F0 fathers developed impairment of hypothalamic–pituitary–adrenal axis regulation associated with increased levels of miR-29c, miR-30a, miR-30c, miR-32, miR-193–5p, miR-204, miR-375, miR-5323p, and miR-698 in paternal spermatozoa [107]. Interestingly, the genes responsible for the synthesis of DNA methyltransferase 3a (DNMT3a) – a known DNA-methylating agent, and trinucleotide repeat containing 6b (TNRC6B) and metadherin (MTDH) – two peptides required for microRNA-mediated gene silencing – are reported to be molecular targets for four out of nine sperm microRNAs, which were detected to be elevated in F0 founders. Moreover, in the postfertilization stages, nine sperm microRNAs repertoires sensitive to paternal stress exposure, mentioned above, were reported to translationally repress two maternal mRNA transcripts – sirtuin 1 and ubiquitin protein ligase E3a, both of which are responsible for chromatin remodeling [108]. Fullston et al.[83] conducted a mouse study in which testes and sperm cells of male F0 founders made obese by HFD intake revealed alterations in expression of numerous microRNA species, associated with over generational transmission of obese paternal phenotype via sperm cells. The identified microRNA sequences were found to mediate molecular pathways regulating differentiation of pluripotent stem cells during embryogenesis, lipid and carbohydrate metabolism, inflammatory response, transcriptional regulation, and RNA posttranslational modification [83]. Moreover, in F1 offspring, an impaired microRNAs profile was observed not only in embryonic cells but also in cells of an adult organism [83]. Recently, Chen et al.[95] demonstrated that an injection of a subpopulation (predominantly 5′ tRNA halves of 30–34 nucleotides in length) of tsRNAs isolated from the sperm cells produced by F0 mouse male founders exposed to HFD into the normal zygote results in glucose intolerance and insulin resistance in the F1 offspring. In contrast, the transfer of sperm whole RNA population produced by HFD-treated F0 male founders into the normal zygote in adult F1 offspring caused only glucose tolerance lowering, whereas insulin tolerance remained unaffected [95]. The detrimental effects of HFD-fed male founder sperm tsRNAs caused dysregulation of genes, involved in metabolic pathways (ketone, carbohydrate, and monosaccharide metabolisms) in islet cells of F1 progeny [95]. Thus, a transmission of paternal sperm-derived tsRNAs to the offspring cells and caused by them silencing of metabolism-associated genes is one of the molecular mechanisms explaining the inheritance of paternal phenotype in adult offspring. Interestingly, in the nematode C. elegans, the double-stranded RNA-derived mobile RNAs, produced by neurons – highly differentiated somatic cells – were proven to enter germ cells of the same organism [109]. In turn, neuron cell-derived mobile RNAs cause silencing of certain target genes in germ line cells. Notably, in germ cells, but not in somatic cells, the target gene repression induced through mobile RNAs can be inherited to the progeny for more than 25 generations in the absence of mobile RNAs production by neurons [109].
Histone modifications in the sperm
In eukaryotic cells, each DNA units are surrounded by two molecules of each of the core histone protein H2A, H2B, H3, and H4 forming a histone octamer, called a nucleosome [110]. N-terminal ends of any of four histone protein species can be chemically changed by methylation, acetylation, and phosphorylation [111]. At the end of spermatozoa maturation, haploid sperm chromatin undergoes a process of histone-to-protamine transition [111]. However, not all histones are exchanged to protamines during sperm maturation. In normal human sperm cells, 5–15% of chromatin remains surrounded by histones, which is a higher rate compared with the other mammalians having typically less than 5% of entire DNA packed with histones [112,113]. Nucleosomes, which did not undergo the histone-to-protamine transition (i.e. remain packed by histones), usually predominate at loci, responsible for embryonic development, and include imprinted gene clusters, miRNA clusters, HOX gene clusters, and the promoters of stand-alone developmental transcription and signaling factors [114,115], which play a key role in epigenetic inheritance of paternal traits by the offspring, influence progeny fertility, and embryogenesis.
As shown by Siklenka et al.[116], mice with genetic overexpression of histone H3 lysine 4 (H3K4) demethylase KDM1A had a decreased sperm H3K4 dimethylation, led to developmental disorders, and increased mortality rate in further offspring generations even in the absence of KDM1A overexpression in the sperm cells. Male mouse F0 founders made obese by HFD intake revealed differences in liver mRNA expression of five genes, namely, metallothionein-1 and 2 (MT1, MT2), fatty acid synthase (FASN), P450 cytochrome oxidoreductase (POR), and acetyl-CoA carboxylase-α (ACACA) at the age of 24 weeks versus control diet F0 male founders [90]. Moreover, HFD-fed male founders exerted increased histone H3 occupancy in the promoters of genes responsible for embryo development [90]. In addition, the rate of H3K4me1 histone modification retention in genes responsible for embryogenesis regulation in spermatozoa derived from HFD-fed male founders was significantly higher compared with the control-fed fathers [90]. The sperm cells of D. melanogaster males with high-sugar diet-induced obesity H3K27me3 were reported to be characterized by an epigenetic modification of histone mark H3K27me3 – a well known gene repressor – [87] which is in turn influencing expression profiles of genes regulating metabolism [86].
Thus, the alterations in sperm cells histone profile in the promoters of regulatory genes are one of molecular mechanisms underlying the over generational inheritance of paternal insults induced by obesogenic diet. The schematic representation of the mechanisms influencing father's organisms on different developmental stages, which are related to transgenerational transmission, is presented in Fig. 2.
FIGURE 2.

Epigenetic windows of sensitivity to environmental factors in paternal lineage. The environmental risk factors inducing epigenetic alterations in paternal lineage can be classified into four groups. (a) Factors affecting paternal grandmother and paternal grandfather from embryonic period until conception of a future father. (b) Factors affecting a future father in embryonic period. (c) Factors affecting a future father in prepubertal and spermatogenesis periods. (d) Factors affecting a future father in periconceptional and zygote periods. Epigenetic modifications during the process of spermatogenesis include the following events: primordial germ cells – sperm precursor cells – undergo a genome-wide erasure of epigenetic memory via DNA demethylation and histone demethylation, specifically at lysine positions K4 and K9 of histone H3. Nevertheless, imprinted clusters and retrotransposons remain resistant to demethylation. In addition, on this step, histones H3 and H4 undergo deacetylation and DNA methyltransferases DNMT3A, DNMT3B, and DNMT3L are expressed. In contrast, in spermatogonia – the next sperm maturation step – DNA methylation starts and increases over time. Next, at spermatocyte stage, histone H3 undergoes methylation particularly at lysine positions K4 and K9. Subsequently, at the round spermatid stage, histones, in particular H4, undergo hyperacetylation, which in turn leads to substitution of histones with transition proteins. Sperm chromatin becomes more condensed as a result of the process of histone-to-protamine transition, lysine positions K9 of histone H3 becomes demethylated. Finally, mature spermatozoa possess epigenetic modifications [101,111,117–122].
Imprinted genes in the sperm
In mammalians, each gene in the somatic cell is represented by two alleles inherited from a mother and a father. Although in the vast majority of the genes in the mammalian genome both maternal and paternal alleles are responsible for mRNA production, there is still a minor part of the genes in the genome, in which only one of two alleles is fully transcriptionally functional, whether maternal or paternal one. In other words, when maternally derived gene allele is active, the paternal one is completely or predominantly inactive and vice versa. Such genes are referred to as ‘imprinted’ genes [123]. Mechanistically, gene imprinting is regulated by differential methylation of so called imprinting center regions, also known as imprinting control elements – a relatively short DNA sequences located upstream of the given imprinted genes [124,125]. It has been shown that a deletion of imprinting center regions from the DNA leads to a restoration of normal (biallelic) gene expression [124]. Some of the methylated areas within imprinting center regions of the DNA located, in particular, in the spermatozoa, can be transferred to the progeny in the case if at the zygote stage, such areas do not undergo overall DNA demethylation – a process erasing epigenetic memory of the gametes. In the context of paternal programming, it has been reported that GDM induced in F0 mouse male founders can be transmitted over the generations via paternal lineage, which was mechanistically explained by disturbances in imprinted IGF2 and H19 genes expression in the sperm cells [66]. Interestingly, as suggested by Rondena et al.[126], a prion-like doppel protein, normally present in maturating sperm cells, might serve as an epigenetic agent.
Thus, known so far, players responsible for epigenetic overgenerational transmission of paternal traits include imprinting center region methylation, histone modification, diverse RNA subpopulations, imprinted genes, and doppel protein.
Variations in offspring embryo and placenta development
HFD in male founders causes in the F1 offspring a delay of embryogenesis and placenta growth, decreased blastocyst cell number, and elevated glycolysis rate [91,92]. Moreover, such F1 offspring developed mitochondrial dysfunction and aberrant placental gene expression. This might be because of an impaired ratio of active (H3K4me3) and repressive (H3K27me3) chromatin marks in two-cell embryos [92].
Master et al.[127] reported that the offspring of rat male founders with low birth weight develop alteration in gene expression levels in preimplantation blastocyst (downregulation of growth regulatory genes of the mammalian target of mTOR pathway). Moreover, blastocysts of such offspring had an impaired amino acid metabolism, namely, increased consumption of histidine, methionine, pyruvate, serine, and tyrosine and higher production of aspartate and leucine [127].
Barton et al.[128] showed that the F1 progeny of F0 male rat founders exposed to cyclophosphamide for 4–5 weeks before mating developed an altered chromatin structure in the zygote because of aberrant histone acetylation at lysine 5 of histone H4 and altered DNA methylation during early embryo development.
Variations in offspring organ development and function
Mechanistically, pathways involved in the transmission of paternal adverse insults, particularly cardiovascular disorders, to the progeny, are reported to be localized in many offspring organs, including liver, pancreas, fat, heart, and others. Among them, liver and pancreas are the most well studies organs in terms of father–child transmission mechanisms.
Liver
Paternal low-protein diet results in an upregulation of hepatic mRNA expression of numerous genes responsible for lipid and cholesterol synthesis, a two-fold reduction in liver cholesterol and cholesterol ester levels, and an increase in methylation level of intergenic CpG island located 50 kb upstream of PPARA gene in the liver [78]. Watkins et al.[79] in a trial with an identic study design showed that paternal low-protein diet for 7 weeks before mating led to a downregulation of ADCY, PLCB, and PRKCB liver expression – genes involved in calcium signaling, and FTO downregulation – a gene associated with fat metabolism. The F1 progeny of mouse male F0 founder fed an HFD for 8 weeks before conception exhibited an increase in liver SREBP-1c and FAS – hepatic lipogenesis regulators – gene expression level and protein concentration [82]. Phenotypically, such F1 offspring developed glucose intolerance, excessive lipid synthesis without affecting mitochondrial β-oxidation, and increased hepatic steatosis [82].
Pancreas
Ng et al.[81] demonstrated that the female F1 offspring of male rat founders fed HFD for 9 weeks before mating developed an early appearance of impaired insulin secretion and glucose tolerance lowering that progressed over time. Phenotypically, female F1 offspring exhibited the following alterations in pancreas morphology: reduced relative islet area mostly at the expense of large islets – a sign of an impaired β-cell replication – and an increase in small β cells – a possible compensatory reaction, as well as a tendency toward β cell area reduction and a suppressed insulin granule exocytosis [81]. On the genomic level, the adult female offspring of HFD-fed fathers were characterized by dysregulation in 642 pancreatic islet genes, which could be classified into 13 functional clusters, responsible for cation and ATP binding, cytoskeleton, and intracellular transport regulation [81]. In addition, female progeny of male founders treated with HFD revealed a hypomethylation of IL-13RA2 gene – a player in the Jak–Stat signaling pathway, modulating growth and invasion of various pancreatic cancer cells. Chen et al.[95] reported that in the cells of the pancreatic islets of normal-fat diet-fed F1 offspring born to HFD-fed fathers, downregulated genes (mostly regulating ketone, carbohydrate, and glucose metabolism) have a higher methylation rate in comparison to F1 counterparts born to F0 fathers fed a normal-fat diet.
POTENTIAL INTERVENTIONS TO PREVENT ADVERSE PATERNAL PROGRAMMING
An understanding of the pathophysiology of maternal and paternal programming may result in approaches to avoid adverse programming, and hence reduce the disease burden in the offspring.
Ma and Hardy [129] summarized all so far know strategies, which can lower the risk of metabolic syndrome in progeny born to mothers with metabolic disorders. They predict that most promising pharmacological and dietary agents in reducing the risk of offspring morbidity because of maternal negative impacts would be approaches targeting transcription factors, for example, nuclear receptors. Dietary supplements, ascorbic acid, folic acid, multiple micronutrients, ω-3 fatty acids, resveratrol, melatonin, exendin-4, and nuclear receptor agonists were reported to be such agents [129].
So far, there is only one study published aiming to interfere with paternally transmitted programming effects [130]. It was demonstrated that female offspring born to obese male founders, whose feeding program was changed from HFD to normal-fat diet for 9 weeks before mating and who were exposed to physical activity had better metabolic health (in terms of increased insulin sensitivity and normalized body fat content) in comparison with their counterparts born to fathers exposed to only HFD and without physical exercises [130]. Mechanistically, such female offspring outcome improvements were associated with normalization in X-linked sperm microRNAs levels implicated in pathways mediating cell cycle arrest, apoptosis, oocyte meiotic resumption, and early embryogenesis [130]. It would be of major interest to show whether or not these relatively simple alterations of paternal diet and physical activity might also be applicable to humans.
CONCLUSION
Epidemiological and animal studies suggest that many factors, including paternal under and overnutrition, exposures to environmental toxins, father's health conditions such as diabetes, and even grandfather's nutritional status can program diseases in the following generations in particular via germ cell-mediated transmission and independent of maternal lineage. Better understanding of mechanisms through which adverse stimuli exert their influences on paternal sperm cells will help to clarify the cause of many diseases and the aspects of their inheritance. Thus, in the future, it will be important to conduct epidemiological studies using large population-based cohorts to assess the exact role of paternal detrimental experiences on the health status of further generations and offer possibilities for clinical intervention. These studies together with animal studies addressing molecular pathways of paternal programming might even be the basis of first human studies aiming to reduce adverse paternal effects on the offspring.
ACKNOWLEDGEMENTS
The present review content has never been published before.
Cited papers from the authors were partially supported by the Deutsche Forschungsgemeinschaft to B.H. and by the National Natural Science Foundation of China (Grant No. 81300557), by Hunan Province Science and Technology Plan (Grant No.2014SK3003), and the Program for Excellent Talents of Hunan Normal University (Grant No. ET14106)" to J.L.
Conflicts of interest
There are no conflicts of interest.
Footnotes
Abbreviations: AF5q31, ALL-1 fused gene on chromosome 4 / fragile XE-associated familial mental retardation protein 2 family member (4AF4/FMR2) family member 4; BPA, bisphenol-A; CpG, cytosine-phosphate-guanine motif; CSF2, colony-stimulating factor 2; DBP, dibutyl phthalate; DEHP, bis(2-ethylhexyl)phthalate; DMR, DNA methylation regions; EGF, epidermal growth factor; ESRRA, estrogen-related receptor alpha; FASN, fatty acid synthase gene; FGF19, fibroblast growth factor 19; GDM, gestational diabetes mellitus; GDNF, glial cell line-derived neurotrophic factor; GHSR, growth hormone secretagogue receptor gene; HFD, high-fat diet; HOX, homeobox genes; H3K27me3, trimethylation of lysine 27 on histone H3; H3K4me1, histone H3 lysine 4 monomethylation; H3K4me3, histone H3 lysine 4 trimethylation; H19, imprinted maternally expressed transcript; ICR, Institute of Cancer Research mice; IGF2, insulin-like growth factor 2; IGT, impaired glucose tolerance; IL-13RA2, interleukin 13 receptor subunit alpha 2; IYD, iodotyrosine deiodinase; JP-8, jet propulsion fuel-8; KDM1A, lysine-specific demethylase 1A; LIF, leukemia inhibitory factor; MMP, mitochondrial membrane potential; mse-tRNAs, mature-sperm-enriched tRNA-derived small RNAs; mTOR, mechanistic target of rapamycin; piRNAs, PIWI-interacting RNAs; PPARA, peroxisome proliferator-activated receptor alpha; ROS, reactive oxygen species; SREBP-1c, sterol-regulatory-element-binding protein 1c; TNFRSF12A, tumor necrosis factor receptor superfamily member 12a; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; T2DM, type 2 diabetes mellitus; tsRNA, transfer RNA-derived small RNA; WNT10B, wingless-type mouse mammary tumor virus integration site family member 10B (Wnt) family member 10B; YRNAs, small cytoplasmic non-coding RNAs which are components of the Ro ribonucleoprotein complexes
REFERENCES
- 1.Skogen JC, Overland S. The fetal origins of adult disease: a narrative review of the epidemiological literature. JRSM Short Rep 2012; 3:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farquhar JW. Prognosis for babies born to diabetic mothers in Edinburgh. Arch Dis Child 1969; 44:36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amendt P, Michaelis D, Hildmann W. Clinical and metabolic studies in children of diabetic-mothers. Endokrinologie 1976; 67:351–361. [PubMed] [Google Scholar]
- 4.Dörner G, Plagemann A, Reinagel H. Familial diabetes aggregation in type I diabetics: gestational diabetes an apparent risk factor for increased diabetes susceptibility in the offspring. Exp Clin Endocrinol 1987; 89:84–90. [DOI] [PubMed] [Google Scholar]
- 5.Barker DJ, Bull AR, Osmond C, Simmonds SJ. Fetal and placental size and risk of hypertension in adult life. BMJ 1990; 301:259–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1986; 1:1077–1081. [DOI] [PubMed] [Google Scholar]
- 7.Barker DJ, Martyn CN. The maternal and fetal origins of cardiovascular disease. J Epidemiol Community Health 1992; 46:8–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kyle UG, Pichard C. The Dutch Famine of 1944-1945: a pathophysiological model of long-term consequences of wasting disease. Curr Opin Clin Nutr Metab Care 2006; 9:388–394. [DOI] [PubMed] [Google Scholar]
- 9.Barouki R, Gluckman PD, Grandjean P, Hanson M, Heindel JJ. Developmental origins of noncommunicable disease: implications for research and public health. Environ Health 2012; 11:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wadhwa PD, Buss C, Entringer S, Swanson JM. Developmental origins of health and disease: brief history of the approach and current focus on epigenetic mechanisms. Semin Reprod Med 2009; 27:358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barker DJ. The developmental origins of adult disease. J Am Coll Nutr 2004; 23 (6 Suppl):588S–595S. [DOI] [PubMed] [Google Scholar]
- 12.Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med 2008; 359:61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ravelli AC, van der Meulen JH, Michels RP, Osmond C, Barker DJ, Hales CN, et al. Glucose tolerance in adults after prenatal exposure to famine. Lancet 1998; 351:173–177. [DOI] [PubMed] [Google Scholar]
- 14.Vehaskari VM, Aviles DH, Manning J. Prenatal programming of adult hypertension in the rat. Kidney Int 2001; 59:238–245. [DOI] [PubMed] [Google Scholar]
- 15.Aufdenblatten M, Baumann M, Raio L, Dick B, Frey BM, Schneider H, et al. Prematurity is related to high placental cortisol in preeclampsia. Pediatric research 2009; 65:198–202. [DOI] [PubMed] [Google Scholar]
- 16.Seckl JR, Cleasby M, Nyirenda MJ. Glucocorticoids, 11beta-hydroxysteroid dehydrogenase, and fetal programming. Kidney Int 2000; 57:1412–1417. [DOI] [PubMed] [Google Scholar]
- 17.Thone-Reineke C, Kalk P, Dorn M, Klaus S, Simon K, Pfab T, et al. High-protein nutrition during pregnancy and lactation programs blood pressure, food efficiency, and body weight of the offspring in a sex-dependent manner. Am J Physiol Regul Integr Comp Physiol 2006; 291:R1025–R1030. [DOI] [PubMed] [Google Scholar]
- 18.Parkhurst SM. Ish-Horowicz D. wimp, a dominant maternal-effect mutation, reduces transcription of a specific subset of segmentation genes in Drosophila. Genes Dev 1991; 5:341–357. [DOI] [PubMed] [Google Scholar]
- 19.Hocher B, Slowinski T, Bauer C, Halle H. The advanced fetal programming hypothesis. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association: European Renal Association 2001; 16:1298–1299. [DOI] [PubMed] [Google Scholar]
- 20.Hocher B, Slowinski T, Stolze T, Pleschka A, Neumayer HH, Halle H. Association of maternal G protein beta3 subunit 825T allele with low birthweight. Lancet 2000; 355:1241–1242. [DOI] [PubMed] [Google Scholar]
- 21.Masuda K, Osada H, Iitsuka Y, Seki K, Sekiya S. Positive association of maternal G protein beta3 subunit 825T allele with reduced head circumference at birth. Pediatr Res 2002; 52:687–691. [DOI] [PubMed] [Google Scholar]
- 22.Wang X, Zuckerman B, Pearson C, Kaufman G, Chen C, Wang G, et al. Maternal cigarette smoking, metabolic gene polymorphism, and infant birth weight. JAMA 2002; 287:195–202. [DOI] [PubMed] [Google Scholar]
- 23.Hocher B, Haumann H, Rahnenführera J, Reichetzeder C, Kalk P, Pfab T, et al. Maternal eNOS deficiency determines a fatty liver phenotype of the offspring in a sex dependent manner. Epigenetics 2016; 13:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whitaker RC, Wright JA, Pepe MS, Seidel KD, Dietz WH. Predicting obesity in young adulthood from childhood and parental obesity. N Engl J Med 1997; 337:869–873. [DOI] [PubMed] [Google Scholar]
- 25.Figueroa CR, Arani RB, Goran MI, Weinsier RL. Paternal body fat is a longitudinal predictor of changes in body fat in premenarcheal girls. Am J Clin Nutr 2000; 71:829–834. [DOI] [PubMed] [Google Scholar]
- 26.Murrin CM, Kelly GE, Tremblay RE, Kelleher CC. Body mass index and height over three generations: evidence from the lifeways cross-generational cohort study. BMC Public Health 2012; 12:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ajala O, Fr Meaux AE, Hosking J, Metcalf BS, Jeffery AN, Voss LD, et al. The relationship of height and body fat to gender-assortative weight gain in children. A longitudinal cohort study (EarlyBird 44). Int J Pediatr Obes 2011; 6:223–228. [DOI] [PubMed] [Google Scholar]
- 28.Lawlor DA, Timpson NJ, Harbord RM, Leary S, Ness A, McCarthy MI, et al. Exploring the developmental overnutrition hypothesis using parental-offspring associations and FTO as an instrumental variable. PLoS Med 2008; 5:e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fleten C, Nystad W, Stigum H, Skjaerven R, Lawlor DA, Davey SG, et al. Parent-offspring body mass index associations in the Norwegian Mother and Child Cohort Study: a family-based approach to studying the role of the intrauterine environment in childhood adiposity. Am J Epidemiol 2012; 176:83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vik KL, Romundstad P, Carslake D, Smith GD, Nilsen TI. Comparison of father-offspring and mother-offspring associations of cardiovascular risk factors: family linkage within the population-based HUNT Study, Norway. Int J Epidemiol 2014; 43:760–771. [DOI] [PubMed] [Google Scholar]
- 31.Chen YPM, Li XX, Reichetzeder J, Wang C, Hocher ZNB. Paternal body mass index (BMI) is associated with offspring intrauterine growth in a gender dependent manner. PLoS One 2012; 7:e36329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaati G, Bygren LO, Pembrey M, Sjostrom M. Transgenerational response to nutrition, early life circumstances and longevity. Eur J Hum Genet 2007; 15:784–790. [DOI] [PubMed] [Google Scholar]
- 33.Bygren LO, Kaati G, Edvinsson S. Longevity determined by paternal ancestors’ nutrition during their slow growth period. Acta Biotheor 2001; 49:53–59. [DOI] [PubMed] [Google Scholar]
- 34.Kaati G, Bygren LO, Edvinsson S. Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period. Eur J Hum Genet 2002; 10:682–688. [DOI] [PubMed] [Google Scholar]
- 35.Horikoshi M, Yaghootkar H, Mook-Kanamori DO, Sovio U, Taal HR, Hennig BJ, et al. New loci associated with birth weight identify genetic links between intrauterine growth and adult height and metabolism. Nat Genet 2013; 45:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tyrrell J, Richmond RC, Palmer TM, Feenstra B, Rangarajan J, Metrustry S, et al. Genetic evidence for causal relationships between maternal obesity-related traits and birth weight. JAMA 2016; 315:1129–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bellia A, Giardina E, Lauro D, Tesauro M, Di Fede G, Cusumano G, et al. The Linosa Study’: epidemiological and heritability data of the metabolic syndrome in a Caucasian genetic isolate. Nutr Metab Cardiovasc Dis 2009; 19:455–461. [DOI] [PubMed] [Google Scholar]
- 38.Luo BF, Du L, Li JX, Pan BY, Xu JM, Chen J, et al. Heritability of metabolic syndrome traits among healthy younger adults: a population based study in China. J Med Genet 2010; 47:415–420. [DOI] [PubMed] [Google Scholar]
- 39.Henneman P, Aulchenko YS, Frants RR, van Dijk KW, Oostra BA, van Duijn CM. Prevalence and heritability of the metabolic syndrome and its individual components in a Dutch isolate: the Erasmus Rucphen Family study. J Med Genet 2008; 45:572–577. [DOI] [PubMed] [Google Scholar]
- 40.Hocher B. More than genes: the advanced fetal programming hypothesis. J Reprod Immunol 2014; 104-105:8–11. [DOI] [PubMed] [Google Scholar]
- 41.Pampel FC, Denney JT, Krueger PM. Obesity, SES, and economic development: a test of the reversal hypothesis. Soc Sci Med 2012; 74:1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lindsay RS, Dabelea D, Roumain J, Hanson RL, Bennett PH, Knowler WC. Type 2 diabetes and low birth weight: the role of paternal inheritance in the association of low birth weight and diabetes. Diabetes 2000; 49:445–449. [DOI] [PubMed] [Google Scholar]
- 43.Hypponen E, Smith DG, Power C. Parental diabetes and birth weight of offspring: intergenerational cohort study. BMJ 2003; 326:19–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Penesova A, Bunt JC, Bogardus C, Krakoff J. Effect of paternal diabetes on pre-diabetic phenotypes in adult offspring. Diabetes Care 2010; 33:1823–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tung TH, Chiu YH, Chen LS, Wu HM, Boucher BJ, Chen TH. A population-based study of the association between areca nut chewing and type 2 diabetes mellitus in men (Keelung Community-based Integrated Screening programme No. 2). Diabetologia 2004; 47:1776–1781. [DOI] [PubMed] [Google Scholar]
- 46.Chen TH, Chiu YH, Boucher BJ. Transgenerational effects of betel-quid chewing on the development of the metabolic syndrome in the Keelung Community-based Integrated Screening Program. Am J Clin Nutr 2006; 83:688–692. [DOI] [PubMed] [Google Scholar]
- 47.Boucher BJ, Ewen SW, Stowers JM. Betel nut (Areca catechu) consumption and the induction of glucose intolerance in adult CD1 mice and in their F1 and F2 offspring. Diabetologia 1994; 37:49–55. [DOI] [PubMed] [Google Scholar]
- 48.Pembrey ME, Bygren LO, Kaati G, Edvinsson S, Northstone K, Sjöström M, et al. Sex-specific, male-line transgenerational responses in humans. Eur J Hum Genet 2006; 14:159–166. [DOI] [PubMed] [Google Scholar]
- 49.Laubenthal J, Zlobinskaya O, Poterlowicz K, Baumgartner A, Gdula MR, Fthenou E, et al. Cigarette smoke-induced transgenerational alterations in genome stability in cord blood of human F1 offspring. FASEB J 2012; 26:3946–3956. [DOI] [PubMed] [Google Scholar]
- 50.Marczylo EL, Amoako AA, Konje JC, Gant TW, Marczylo TH. Smoking induces differential miRNA expression in human spermatozoa: a potential transgenerational epigenetic concern? Epigenetics 2012; 7:432–439. [DOI] [PubMed] [Google Scholar]
- 51.Yauk CL, Berndt ML, Williams A, Rowan-Carroll A, Douglas GR, Stämpfli MR. Mainstream tobacco smoke causes paternal germ-line DNA mutation. Cancer Res 2007; 67:5103–5106. [DOI] [PubMed] [Google Scholar]
- 52.Marchetti F, Rowan-Carroll A, Williams A, Polyzos A, Berndt-Weis ML, Yauk CL. Sidestream tobacco smoke is a male germ cell mutagen. Proc Natl Acad Sci U S A 2011; 108:12811–12814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gardner MJ, Snee MP, Hall AJ, Powell CA, Downes S, Terrell JD. Results of case-control study of leukaemia and lymphoma among young people near Sellafield nuclear plant in West Cumbria. BMJ 1990; 300:423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Parker L, Pearce MS, Dickinson HO, Aitkin M, Craft AW. Stillbirths among offspring of male radiation workers at Sellafield nuclear reprocessing plant. Lancet 1999; 354:1407–1414. [DOI] [PubMed] [Google Scholar]
- 55.Fischbein A, Zabludovsky N, Eltes F, Grischenko V, Bartoov B. Ultramorphological sperm characteristics in the risk assessment of health effects after radiation exposure among salvage workers in Chernobyl. Environ Health Perspect 1997; 105 (Suppl 6):1445–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aghajanyan A, Kuzmina N, Sipyagyna A, Baleva L, Suskov I. Analysis of genomic instability in the offspring of fathers exposed to low doses of ionizing radiation. Environ Mol Mutagen 2011; 52:538–546. [DOI] [PubMed] [Google Scholar]
- 57.Doyle P, Maconochie N, Roman E, Davies G, Smith PG, Beral V. Fetal death and congenital malformation in babies born to nuclear industry employees: report from the nuclear industry family study. Lancet 2000; 356:1293–1299. [DOI] [PubMed] [Google Scholar]
- 58.Green LM, Dodds L, Miller AB, Tomkins DJ, Li J, Escobar M. Risk of congenital anomalies in children of parents occupationally exposed to low level ionising radiation. Occup Environ Med 1997; 54:629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Laurier D, Jacob S, Bernier MO, Leuraud K, Metz C, Samson E, et al. Epidemiological studies of leukaemia in children and young adults around nuclear facilities: a critical review. Radiat Prot Dosimetry 2008; 132:182–190. [DOI] [PubMed] [Google Scholar]
- 60.Neel.F JV, Schull WJ, McDonald DJ, Morton NE, Kodani M, Takeshima K, et al. The effect of exposure to the atomic bombs on pregnancy termination in Hiroshima and Nagasaki: preliminary report. Science 1953; 118:537–541. [DOI] [PubMed] [Google Scholar]
- 61.Otake M, Schull WJ, Neel JV. Congenital malformations, stillbirths, and early mortality among the children of atomic bomb survivors: a reanalysis. Radiat Res 1990; 122:1–11. [PubMed] [Google Scholar]
- 62.Wakeford R. Childhood leukaemia and radiation exposure of fathers: the end of the road, perhaps? J Radiol Prot 2003; 23:359–362. [DOI] [PubMed] [Google Scholar]
- 63.Klebanoff MA, Mednick BR, Schulsinger C, Secher NJ, Shiono PH. Father's effect on infant birth weight. Am J Obstet Gynecol 1998; 178:1022–1026. [DOI] [PubMed] [Google Scholar]
- 64.Veena SR, Geetha S, Leary SD, Saperia J, Fisher DJ, Kumaran K, et al. Relationships of maternal and paternal birthweights to features of the metabolic syndrome in adult offspring: an inter-generational study in South India. Diabetologia 2007; 50:43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Savage T, Derraik JG, Miles HL, Mouat F, Hofman PL, Cutfield WS. Increasing paternal age at childbirth is associated with taller stature and less favourable lipid profiles in their children. Clin Endocrinol (Oxf) 2014; 80:253–260. [DOI] [PubMed] [Google Scholar]
- 66.Ding GL, Wang FF, Shu J, Tian S, Jiang Y, Zhang D, et al. Transgenerational glucose intolerance with Igf2/H19 epigenetic alterations in mouse islet induced by intrauterine hyperglycemia. Diabetes 2012; 61:1133–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jimenez-Chillaron JC, Isganaitis E, Charalambous M, Gesta S, Pentinat-Pelegrin T, Faucette RR, et al. Intergenerational transmission of glucose intolerance and obesity by in utero undernutrition in mice. Diabetes 2009; 58:460–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brun JM, Bernadet MD, Cornuez A, Leroux S, Bodin L, Basso B, et al. Influence of grand-mother diet on offspring performances through the male line in Muscovy duck. BMC Genet 2015; 16:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dunn GA, Bale TL. Maternal high-fat diet promotes body length increases and insulin insensitivity in second-generation mice. Endocrinology 2009; 150:4999–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yin Y, Li Y, Zhang W. The growth hormone secretagogue receptor: its intracellular signaling and regulation. Int J Mol Sci 2014; 15:4837–4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dunn GA, Bale TL. Maternal high-fat diet effects on third-generation female body size via the paternal lineage. Endocrinology 2011; 152:2228–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tracey R, Manikkam M, Guerrero-Bosagna C, Skinner MK. Hydrocarbons (jet fuel JP-8) induce epigenetic transgenerational inheritance of obesity, reproductive disease and sperm epimutations. Reprod Toxicol 2013; 36:104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guerrero-Bosagna C, Covert TR, Haque MM, Settles M, Nilsson EE, Anway MD, et al. Epigenetic transgenerational inheritance of vinclozolin induced mouse adult onset disease and associated sperm epigenome biomarkers. Reprod Toxicol 2012; 34:694–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Manikkam M, Tracey R, Guerrero-Bosagna C, Skinner MK. Plastics derived endocrine disruptors (BPA, DEHP and DBP) induce epigenetic transgenerational inheritance of obesity, reproductive disease and sperm epimutations. PLoS One 2013; 8:e55387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fullston T, Palmer NO, Owens JA, Mitchell M, Bakos HW, Lane M. Diet-induced paternal obesity in the absence of diabetes diminishes the reproductive health of two subsequent generations of mice. Hum Reprod 2012; 27:1391–1400. [DOI] [PubMed] [Google Scholar]
- 76.Braunschweig M, Jagannathan V, Gutzwiller A, Bee G. Investigations on transgenerational epigenetic response down the male line in F2 pigs. PLoS One 2012; 7:e30583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ding GL, Huang HF. Paternal transgenerational glucose intolerance with epigenetic alterations in second generation offspring of GDM. Asian J Androl 2013; 15:451–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Carone BR, Fauquier L, Habib N, Shea JM, Hart CE, Li R, et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 2010; 143:1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Watkins AJ, Sinclair KD. Paternal low protein diet affects adult offspring cardiovascular and metabolic function in mice. Am J Physiol Heart Circ Physiol 2014; 306:H1444–H1452. [DOI] [PubMed] [Google Scholar]
- 80.Ng SF, Lin RC, Maloney CA, Youngson NA, Owens JA, Morris MJ. Paternal high-fat diet consumption induces common changes in the transcriptomes of retroperitoneal adipose and pancreatic islet tissues in female rat offspring. FASEB J 2014; 28:1830–1841. [DOI] [PubMed] [Google Scholar]
- 81.Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature 2010; 467:963–966. [DOI] [PubMed] [Google Scholar]
- 82.Ornellas F, Souza-Mello V, Mandarim-de-Lacerda CA, Aguila MB. Programming of obesity and comorbidities in the progeny: lessons from a model of diet-induced obese parents. PLoS One 2015; 10:e0124737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fullston T, Ohlsson Teague EM, Palmer NO, DeBlasio MJ, Mitchell M, Corbett M, et al. Paternal obesity initiates metabolic disturbances in two generations of mice with incomplete penetrance to the F2 generation and alters the transcriptional profile of testis and sperm microRNA content. FASEB J 2013; 27:4226–4243. [DOI] [PubMed] [Google Scholar]
- 84.McPherson NO, Fullston T, Bakos HW, Setchell BP, Lane M. Obese father's metabolic state, adiposity, and reproductive capacity indicate son's reproductive health. Fertil Steril 2014; 101:865–873. [DOI] [PubMed] [Google Scholar]
- 85.Fullston T, McPherson NO, Owens JA, Kang WX, Sandeman LY, Lane M. Paternal obesity induces metabolic and sperm disturbances in male offspring that are exacerbated by their exposure to an ‘obesogenic’ diet. Physiol Rep 2015; 3:e12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ost A, Lempradl A, Casas E, Weigert M, Tiko T, Deniz M, et al. Paternal diet defines offspring chromatin state and intergenerational obesity. Cell 2014; 159:1352–1364. [DOI] [PubMed] [Google Scholar]
- 87.Kim J, Kim H. Recruitment and biological consequences of histone modification of H3K27me3 and H3K9me3. ILAR J 2012; 53:232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Miersch C, Doring F. Paternal dietary restriction affects progeny fat content in Caenorhabditis elegans. IUBMB Life 2012; 64:644–648. [DOI] [PubMed] [Google Scholar]
- 89.Glier MB, Green TJ, Devlin AM. Methyl nutrients, DNA methylation, and cardiovascular disease. Mol Nutr Food Res 2014; 58:172–182. [DOI] [PubMed] [Google Scholar]
- 90.Terashima M, Barbour S, Ren J, Yu W, Han Y, Muegge K. Effect of high fat diet on paternal sperm histone distribution and male offspring liver gene expression. Epigenetics 2015; 10:861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Binder NK, Hannan NJ, Gardner DK. Paternal diet-induced obesity retards early mouse embryo development, mitochondrial activity and pregnancy health. PLoS One 2012; 7:e52304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McPherson NO, Bell VG, Zander-Fox DL, Fullston T, Wu LL, Robker RL, et al. When two obese parents are worse than one! Impacts on embryo and fetal development. Am J Physiol Endocrinol Metab 2015; 309:E568–E581. [DOI] [PubMed] [Google Scholar]
- 93.Anderson LM, Riffle L, Wilson R, Travlos GS, Lubomirski MS, Alvord WG. Preconceptional fasting of fathers alters serum glucose in offspring of mice. Nutrition 2006; 22:327–331. [DOI] [PubMed] [Google Scholar]
- 94.Lane M, McPherson NO, Fullston T, Spillane M, Sandeman L, Kang WX, et al. Oxidative stress in mouse sperm impairs embryo development, fetal growth and alters adiposity and glucose regulation in female offspring. PLoS One 2014; 9:e100832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen Q, Yan M, Cao Z, Li X, Zhang Y, Shi J, et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 2016; 351:397–400. [DOI] [PubMed] [Google Scholar]
- 96.Bromfield JJ, Schjenken JE, Chin PY, Care AS, Jasper MJ, Robertson SA. Maternal tract factors contribute to paternal seminal fluid impact on metabolic phenotype in offspring. Proc Natl Acad Sci U S A 2014; 111:2200–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Armstrong L. Epigenetics. New York:Front Genet; 2014. [Google Scholar]
- 98.Lane N, Dean W, Erhardt S, Hajkova P, Surani A, Walter J, et al. Resistance of IAPs to methylation reprogramming may provide a mechanism for epigenetic inheritance in the mouse. Genesis 2003; 35:88–93. [DOI] [PubMed] [Google Scholar]
- 99.Guibert S, Forne T, Weber M. Global profiling of DNA methylation erasure in mouse primordial germ cells. Genome Res 2012; 22:633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Abdalla H, Hirabayashi M, Hochi S. Demethylation dynamics of the paternal genome in pronuclear-stage bovine zygotes produced by in vitro fertilization and ooplasmic injection of freeze-thawed or freeze-dried spermatozoa. J Reprod Dev 2009; 55:433–439. [DOI] [PubMed] [Google Scholar]
- 101.Lane N, Dean W, Erhardt S, Hajkova P, Surani A, Walter J, et al. Resistance of IAPs to methylation reprogramming may provide a mechanism for epigenetic inheritance in the mouse. Genesis 2003; 35:88–93. [DOI] [PubMed] [Google Scholar]
- 102.Hillemacher T, Frieling H, Moskau S, Muschler MA, Semmler A, Kornhuber J, et al. Global DNA methylation is influenced by smoking behaviour. Eur Neuropsychopharmacol 2008; 18:295–298. [DOI] [PubMed] [Google Scholar]
- 103.Jodar M, Selvaraju S, Sendler E, Diamond MP, Krawetz SA. The presence, role and clinical use of spermatozoal RNAs. Hum Reprod Update 2013; 19:604–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ostermeier GC, Miller D, Huntriss JD, Diamond MP, Krawetz SA. Reproductive biology: delivering spermatozoan RNA to the oocyte. Nature 2004; 429:154. [DOI] [PubMed] [Google Scholar]
- 105.Gapp K, Jawaid A, Sarkies P, Bohacek J, Pelczar P, Prados J, et al. Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Nat Neurosci 2014; 17:667–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rassoulzadegan M, Grandjean V, Gounon P, Vincent S, Gillot I, Cuzin F. RNA-mediated nonmendelian inheritance of an epigenetic change in the mouse. Nature 2006; 441:469–474. [DOI] [PubMed] [Google Scholar]
- 107.Rodgers AB, Morgan CP, Bronson SL, Revello S, Bale TL. Paternal stress exposure alters sperm microRNA content and reprograms offspring HPA stress axis regulation. J Neurosci 2013; 33:9003–9012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rodgers AB, Morgan CP, Leu NA, Bale TL. Transgenerational epigenetic programming via sperm microRNA recapitulates effects of paternal stress. Proc Natl Acad Sci U S A 2015; 112:13699–13704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Devanapally S, Ravikumar S, Jose AM. Double-stranded RNA made in C. elegans neurons can enter the germline and cause transgenerational gene silencing. Proc Natl Acad Sci U S A 2015; 112:2133–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Natsume-Kitatani Y, Mamitsuka H. Classification of promoters based on the combination of core promoter elements exhibits different histone modification patterns. PLoS One 2016; 11:e0151917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shirakata Y, Hiradate Y, Inoue H, Sato E, Tanemura K. Histone h4 modification during mouse spermatogenesis. J Reprod Dev 2014; 60:383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Erenpreiss J, Spano M, Erenpreisa J, Bungum M, Giwercman A. Sperm chromatin structure and male fertility: biological and clinical aspects. Asian J Androl 2006; 8:11–29. [DOI] [PubMed] [Google Scholar]
- 113.Ioannou D, Miller D, Griffin DK, Tempest HG. Impact of sperm DNA chromatin in the clinic. J Assist Reprod Genet 2016; 33:157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Arpanahi A, Brinkworth M, Iles D, Krawetz SA, Paradowska A, Platts AE, et al. Endonuclease-sensitive regions of human spermatozoal chromatin are highly enriched in promoter and CTCF binding sequences. Genome Res 2009; 19:1338–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hammoud SS, Nix DA, Zhang H, Purwar J, Carrell DT, Cairns BR. Distinctive chromatin in human sperm packages genes for embryo development. Nature 2009; 460:473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Siklenka K, Erkek S, Godmann M, Lambrot R, McGraw S, Lafleur C, et al. Disruption of histone methylation in developing sperm impairs offspring health transgenerationally. Science 2015; 350:aab2006. [DOI] [PubMed] [Google Scholar]
- 117.Smith ZD, Meissner A. The simplest explanation: passive DNA demethylation in PGCs. EMBO J 2013; 32:318–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Boissonnas CC, Jouannet P, Jammes H. Epigenetic disorders and male subfertility. Fertil Steril 2013; 99:624–631. [DOI] [PubMed] [Google Scholar]
- 119.Oakes CC, La Salle S, Smiraglia DJ, Robaire B, Trasler JM. Developmental acquisition of genome-wide DNA methylation occurs prior to meiosis in male germ cells. Dev Biol 2007; 307:368–379. [DOI] [PubMed] [Google Scholar]
- 120.Stuppia L, Franzago M, Ballerini P, Gatta V, Antonucci I. Epigenetics and male reproduction: the consequences of paternal lifestyle on fertility, embryo development, and children lifetime health. Clin Epigenetics 2015; 7:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dada R, Kumar M, Jesudasan R, Fernández JL, Gosálvez J, Agarwal A. Epigenetics and its role in male infertility. J Assist Reprod Genet 2012; 29:213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sonnack V, Failing K, Bergmann M, Steger K. Expression of hyperacetylated histone H4 during normal and impaired human spermatogenesis. Andrologia 2002; 34:384–390. [DOI] [PubMed] [Google Scholar]
- 123.Alberts B. Molecular biology of the cell. 6 edNew York, USA:Garland Science; 2015. [Google Scholar]
- 124.Eroglu A, Layman L C. Role of ART in imprinting disorders. Semin Reprod Med 2012; 30:92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shin JY, Fitzpatrick GV, Higgins MJ. Two distinct mechanisms of silencing by the KvDMR1 imprinting control region. EMBO J 2008; 27:168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rondena M, Ceciliani F, Comazzi S, Pocacqua V, Bazzocchi C, Luvoni C, et al. Identification of bovine doppel protein in testis, ovary and ejaculated spermatozoa. Theriogenology 2005; 63:1195–1206. [DOI] [PubMed] [Google Scholar]
- 127.Master JS, Thouas GA, Harvey AJ, Sheedy JR, Hannan NJ, Gardner DK, et al. Fathers that are born small program alterations in the next-generation preimplantation rat embryos. J Nutr 2015; 145:876–883. [DOI] [PubMed] [Google Scholar]
- 128.Barton TS, Robaire B, Hales BF. Epigenetic programming in the preimplantation rat embryo is disrupted by chronic paternal cyclophosphamide exposure. Proc Natl Acad Sci U S A 2005; 102:7865–7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ma N, Hardy DB. The fetal origins of the metabolic syndrome: can we intervene? J Pregnancy 2012; 2012:482690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.McPherson NO, Owens JA, Fullston T, Lane M. Preconception diet or exercise intervention in obese fathers normalizes sperm microRNA profile and metabolic syndrome in female offspring. Am J Physiol Endocrinol Metab 2015; 308:E805–E821. [DOI] [PubMed] [Google Scholar]