

Abstract
Metformin is a commonly prescribed diabetes medication whose mechanism of action is poorly understood. In this study we utilized EHR-linked biobank data to elucidate the impact of genomic variation on glycemic response to metformin. Our study found significant gene- and SNP-level associations within the beta-2 subunit of the heterotrimeric adenosine monophosphate-activated protein kinase complex. Using EHR phenotypes where were able to add additional clarity to ongoing metformin pharmacogenomic dialogue.
Keywords: Electronic Health Records, Biobank, Pharmacogenomics, Metformin, Type 2 Diabetes Mellitus
Introduction
Metformin is recommended as a first-line therapy for type 2 diabetes mellitus (T2DM)[1] and is believed to be the most prescribed drug worldwide[2]. Metformin is primarily utilized to regain glycemic control in diabetic or pre-diabetic patients. While metformin is a relatively safe antidiabetic therapy, serious adverse reactions can occur. Evidence is also accumulating that highlights the potential repurposing of metformin for cancer prevention and treatment[3]. However, the details underlying the molecular mechanism of action for metformin are not fully understood[2]. While the pharmacokinetics (PK) of metformin, the transportation throughout the body, are moderately understood, the pharmacodynamics (PD) of metformin, the physiological and biochemical impact of metformin in the body, are not clearly understood[4]. Further, there is considerable variation in glycemic response to metformin[4], only part of which genetic factors currently explain[2].
Our study aims to add further clarity to metformin pharmacogenomics by using EHR-linked biobank data to understand the impact of common variants in metformin candidate genes (n=16) on altered glycemic response in a clinical population. Candidate genes selected for inclusion in this study are suspected metformin PK or PD determinants as highlighted in systematic reviews of metformin pharmacogenomics[2,4,5]. A series of gene- and SNP-level analyses were performed in this study to elucidate the impact of pharmacogenomic variation.
1. Methods
All genomic data utilized in this study was a part of a local biobank with linked longitudinal electronic health record data[6]. Patients with existing GWAS data, T2DM, known metformin exposure, and longitudinal glycemic indicators were included in this study. Patients without known metformin exposure ≥ 6 months, without A1c measures ≥6 months apart prior to and concurrent with the metformin exposure, and without genomic data with ≥95% call rate within candidate genes were excluded from this study. The study design (Figure 1) and description of cohort of Caucasian patients (n=258) with known metformin exposure, T2DM, longitudinal A1c measures, and genomic data with ≥ 95% call rate utilized in this study can be found in Table 1.
Figure 1.
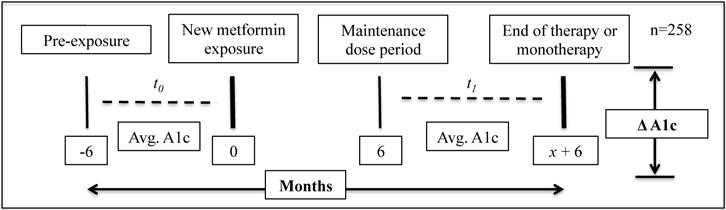
Study Design
Table 1.
Cohort Demographics
| n (%) | |||
|---|---|---|---|
| BMI < 30 | 64 (24.8) | ||
| BMI ≥ 30 to < 35 | 100 (38.8) | ||
| BMI ≥35 | 94 (36.4) | ||
| Median A1c > 7.0 | 101 (39.1) | ||
| Median | Range | ||
| Change in A1c | 0.0733 | −6.45 | 3.51 |
| Age | 64 | 30 | 84 |
Clinical phenotypes were developed using EHR-based algorithms and data, which enabled us to study metformin pharmacogenomics in a naturally occurring population. Attribution of a T2DM phenotype was performed using modified methodology developed by eMERGE [7]. Metformin exposure was ascertained using a combination of validated structured[8] and semi-structured[9] EHR data collection methodologies. To compare the genetic modification of glycemic response to metformin, measures of A1c were compared prior to metformin exposure and during the period of metformin exposure following a 6-month period of delay. In this study, A1c was calculated as the difference between the average of A1c measures within 6 months prior to metformin exposure (t0) and the average of A1c measures between ≥ 6 months after exposure to metformin and the end of metformin exposure(t1)(Figure 1). This approach minimizes the impact of any one A1c measure and biases change in A1c measures towards the null. Age, gender, and morbid obesity (BMI ≥ 35), a known modifier of T2DM state[1], were selected for inclusion as covariates in the model. Age and BMI measures were calculated at first recorded exposure to metformin.
1.1. Quality Control
For each candidate gene we selected SNPs 50 kb upstream and downstream of each gene using 1000 genomes project variants and NCBI build 37 as the reference genome. By this mapping rule, a total of 8440 SNPs were mapped to the 16 genes, but only 1065 SNPs were available in the genotype data. For the remaining SNPs, two main quality control filters were applied: (i) SNPs with unacceptable high rates of missing genotype calls (>10%); and (ii) monomorphic SNPs were excluded. The quality control of the genotype data was performed by PLINK v 1.07. The call rate was < 0.90 for 601 SNPs and 1 SNP was monomorphic, leaving 463 SNPs for the single SNP and gene-level analyses. From the total of 285 samples with available genotype, we excluded 27 samples with call rate < 0.95, leaving 258 samples available for analysis in the final cohort. Locus Zoom plots were based on the 1000 Genomes European reference population from March 2012 release.
1.2. Analysis
First, we analyzed the association of each gene with the change in A1c using Van der Waerden rank, or rank based inverse Gaussian, transformed change in A1c. Gene level tests were performed using principal component analysis (PCA). For each gene, principal components (PCs) were created using linear combinations of ordinally scaled SNPs (i.e., 0, 1, 2 copies of minor allele). The smallest set of resulting principal components that explained at least 90% of the SNP variance was included in linear regression models that also controlled for gender, age, and obesity. Instead of including the entire set of SNPs for each gene, the PCA approach reduces the degrees of freedom, avoids model fitting issues due to multi-collinearity of the SNPs from linkage disequilibrium (LD) and potentially improves the statistical power. Finally, to assess overall significance of a gene, we computed the likelihood ratio test by comparing the null model containing only the covariates with the full model containing covariates and the set of resulting principal components. At the gene-level, results of the 16 simultaneous hypothesis tests were utilized for filtering. Second, we tested the association between each SNP and Van der Waerden rank transformed change in A1c using a linear regression model, adjusting for age, gender and morbid obesity. Coefficient estimates were calculated per minor allele, that is, with each minor allele, the A1c level changes by ‘beta’. SNP-level results are not displayed as corrected for multiple testing.
2. Results
The candidate gene PRKAB2, a PD determinant, was identified as being significantly(P-val=0.0194) associated with glycemic response(Table 2). While the estimates for male (Coef=0.04,P-val=0.737), age (Coef=−0.0009,P-val=0.881), morbid obesity (Coef=0.22,P-val=0.083) were not significantly associated with glycemic response at alpha=0.05 significance level for gene- and SNP-level analysis they were included to control for confounding by patient demographic characteristics.
Table 2.
Results
| Pgx | Gene | Top SNP (by gene) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Name | Chromosome | nSNPs | nPCs | P-value | SNP | MA | Beta | P-value | |
| PK | SLC22A1 | 6 | 38 | 12 | 0.8517 | rs2619268 | T | 0.2117 | 0.0822 |
| SLC22A2 | 6 | 31 | 8 | 0.8993 | rs3103352 | A | 0.2394 | 0.0764 | |
| SLC22A3 | 6 | 33 | 6 | 0.6076 | rs3127602 | T | 0.2307 | 0.0147 | |
| SLC29A4 | 7 | 13 | 9 | 0.1035 | rs10234709 | A | −0.1987 | 0.0318 | |
| SLC47A1 | 17 | 19 | 7 | 0.2030 | rs2120274 | A | 0.1938 | 0.0310 | |
| SLC47A2 | 17 | 20 | 5 | 0.3245 | rs4621031 | C | −0.1908 | 0.0410 | |
| PD | PRKAA1 | 5 | 19 | 6 | 0.5070 | rs11749180 | A | −0.2080 | 0.0464 |
| PRKAA2 | 1 | 24 | 12 | 0.6128 | rs1832899 | C | −0.2711 | 0.0690 | |
| PRKAB1 | 12 | 13 | 4 | 0.7672 | rs7297487 | G | 0.3711 | 0.2770 | |
| PRKAB2 | 1 | 35 | 5 | 0.0194 | rs7541245 | A | −0.7885 | 0.0019 | |
| PRKAG1 | 12 | 8 | 4 | 0.1567 | rs10875914 | G | −0.1498 | 0.0805 | |
| PRKAG2 | 7 | 132 | 46 | 0.8201 | rs4725434 | T | 0.2211 | 0.0136 | |
| PRKAG3 | 2 | 9 | 6 | 0.8242 | rs13013510 | A | −0.1340 | 0.1320 | |
| STK11 | 19 | 10 | 8 | 0.4561 | rs2301759 | C | 0.2203 | 0.0345 | |
| ATM | 11 | 13 | 5 | 0.5447 | rs1800058 | T | 0.4812 | 0.1546 | |
| GCKR | 2 | 14 | 4 | 0.2264 | rs1260326 | T | −0.1488 | 0.0858 | |
Pgx=pharmacogenomic implication, PK=pharmacokinetic, PD=pharmacodynamics, MA=minor allele, Beta=beta coefficient
AMPK is a heterotrimeric enzyme composed of alpha, beta, and gamma subunits, encoded by 5 genes, each of which uniquely determines protein stability and activity. AMPK acts as a metabolic master switch regulating several intracellular systems and plays an important role in cellular energy homeostasis, the maintenance of cellular ATP levels[10]. Metformin is a known AMPK activator, with genetic variations in AMPK suspected to impact the response to metformin[5]. PRKAB2, is the beta subunit 2 of the AMPK complex, represented by 5 PCs of 35 SNPs was marginally significantly associated (p=0.0194) after adjustment for multiple testing with glycemic response (Table 2). The Locus Zoom plot for PRKAB2 (Supplementary Figure 1) placed rs7541245, the most significant (p=0.0019) SNP, as outside the gene boundaries. 5 SNPs (rs6665580, rs6659191, rs6678588, rs7541245, rs10494243) in high LD within PRKAB2, were found to be significantly associated with a decrease in glycemic response after metformin exposure. Only the most significant SNP within each gene was displayed in the results (Table 2). In summary, variation in PRKAB2, the gene encoding the beta subunit 2 of adenosine monophosphate-activated protein kinase complex, appears to be associated with decreases in glycemic response after exposure to metformin, with rs7541245 having the strongest SNP association.
3. Discussion
In this study, we leverage EHR-linked biobank data and EHR-based phenotyping methods to study variants associated with metformin PK and PD determinants. While our cohort is modestly powered (N=258), we posit that utilizing a clinical endpoint that is sensitive to PK and PD determinants strengthens our study. Due to the wide distribution and systemic impact of metformin the distinction between PK and PD determinants may not necessarily be distinct at the clinical level. It is important to note that our study did not replicate PK findings in similar studies[11,12]. However, this could be due to competing directional effects between PCs within candidate genes as several candidates contained SNPs that were significant before bonferoni correction.
While our study is strengthened by its use of robust and sensitive clinical indicators as outcome phenotypes paired with genomic data and ability to replicate findings in an existing clinical population, several limitations exist. The genetic contribution to variation in metformin renal clearance in the kidneys is estimated to be approximately 90%[13], bioavailability and concentration of metformin likely will very widely based on renal function, topics that will be addressed in our future work. Not all SNPs within candidate genes were available for analysis due to GWAS sequencing being originally performed for other studies. The secondary nature of the GWAS data had potential to bias findings either due to original patient selection criteria or sequencing criteria, but was eliminated as a potential effect in sensitivity analysis (not shown). Finally, while an agnostic training step (i.e. GWAS) was not utilized to identify candidate genes, our selection of candidate genes draws strength from the large body of literature and corresponding expertise on which it is based.
The primary purpose of this study was to add clarity to metformin pharmacogenomics by understanding the impact of common variants in metformin PK and PD candidate genes (n=16) on altered glycemic response in a clinical population derived from an EHR-linked biobank. By utilizing EHR phenotypes and biobank data we were able to add additional clarity to ongoing metformin pharmacogenomic dialogue by reinforcing the importance of PRKAB2.
Supplementary Material
References
- 1.Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of Hyperglycemia in Type 2 Diabetes: A Patient-Centered Approach. Diabetes Care. 2012;35:1364–1379. doi: 10.2337/dc12-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Todd JN, Florez JC. An update on the pharmacogenomics of metformin: progress, problems and potential. Pharmacogenomics. 2014;15(4):529–539. doi: 10.2217/pgs.14.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franciosi M, Lucisano G, Lapice E, Strippoli GF, Pellegrini F, Nicolucci A. Metformin therapy and risk of cancer in patients with type 2 diabetes: systematic review. PloS one. 2013;8(8):e71583. doi: 10.1371/journal.pone.0071583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gong L, Goswami S, Giacomini KM, Altman RB, Klein TE. Metformin pathways: pharmacokinetics and pharmacodynamics. Pharmacogenetics and genomics. 2012 Nov;22(11):820–827. doi: 10.1097/FPC.0b013e3283559b22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen S, Zhou J, Xi M, et al. Pharmacogenetic Variation and Metformin Response. Current Drug Metabolism. 2013;14:1070–1082. doi: 10.2174/1389200214666131211153933. [DOI] [PubMed] [Google Scholar]
- 6.Bielinski SJ, Chai HS, Pathak J, et al. Mayo Genome Consortia: A Genotype-Phenotype Resource for Genome-Wide Association Studies With an Application to the Analysis of Circulating Bilirubin Levels. Mayo Clinic proceedings. 2011;86(7):606–614. doi: 10.4065/mcp.2011.0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kho AN, Hayes MG, Rasmussen-Torvik L, et al. Use of diverse electronic medical record systems to identify genetic risk for type 2 diabetes within a genome-wide association study. Journal of the American Medical Informatics Association: JAMIA. 2012 Mar-Apr;19(2):212–218. doi: 10.1136/amiajnl-2011-000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chute CG, Beck SA, Fisk TB, Mohr DN. The Enterprise Data Trust at Mayo Clinic: a semantically integrated warehouse of biomedical data. Journal of the American Medical Informatics Association: JAMIA. 2010 Mar-Apr;17(2):131–135. doi: 10.1136/jamia.2009.002691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pathak J, Murphy SP, Willaert BN, et al. Using RxNorm and NDF-RT to Classify Medication Data Extracted from Electronic Health Records: Experiences from the Rochester Epidemiology Project. American Medical Informatics Association Annual Symposium. 2011:1089–1098. [PMC free article] [PubMed] [Google Scholar]
- 10.Winder WW, Hardie DG. AMP-activated protein kinase, a metabolic master switch: possible roles in Type 2 diabetes. American Journal of Physiology. 1999;277(1) doi: 10.1152/ajpendo.1999.277.1.E1. [DOI] [PubMed] [Google Scholar]
- 11.Christensen MM, Brasch-Andersen C, Green H, et al. The pharmacogenetics of metformin and its impact on plasma metformin steady-state levels and glycosylated hemoglobin A1c. Pharmacogenetics and genomics. 2011 Dec;21(12):837–850. doi: 10.1097/FPC.0b013e32834c0010. [DOI] [PubMed] [Google Scholar]
- 12.Pawlyk AC, Giacomini KM, McKeon C, Shuldiner AR, Florez JC. Metformin Phramacogenomics: Current Status and Future Directions. Diabetes. 2014;63:2590–2599. doi: 10.2337/db13-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leabman MK, Giacomini KM. Estimating the contribution of genes and environment to variation in renal drug clearance. Pharmacogenetics. 2003 Sep;13(9):581–584. doi: 10.1097/00008571-200309000-00007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.