

Abstract
In physiology, reactive oxygen species (ROS) are produced by most cells for normal function and as a defense mechanism against foreign particles, microbes and viruses. Hepatic macrophages (Kupffer cells), sinusoidal endothelial cells, hepatocytes and hepatic stellate cells (HSCs) are all capable of generating ROS in physiology and pathology. ROS are also produced by infiltrating inflammatory cells during acute and chronic liver injury. Increased levels of ROS have been implicated in apoptotic/necrotic death of hepatocytes, and liver failure. In contrast to causing injury to hepatocytes, ROS and lipid peroxidation products induce transdifferentiation of the quiescent HSCs into an activated highly proliferative myofibroblast-like phenotype. ROS and lipid peroxidation products also stimulate the synthesis of extracellular matrix (ECM) by activated HSCs. Deposition of excessive amounts of ECM is the primary mechanism of fibrosis and cirrhosis of the liver, and interactions between ROS and HSCs appear to play a major role in this pathology. Although these findings suggest that HSCs are resistant to the injurious actions of ROS, there is compelling evidence demonstrating ROS-induced death of activated HSCs. Detailed mechanistic understanding of such paradoxical interactions between ROS and HSCs will be critical for developing therapies for chronic fibrotic liver disease.
Keywords: stellate cells, liver, free radicals, fibrosis, apoptosis
Introduction
Hepatic stellate cells (HSCs), which constitute nearly 10% of the liver cell population, play critical roles in liver physiology and pathology. Physiologically, HSCs are the major cell type to store body’s retinoids, regulate liver blood flow and architecture, and influence hepatic metabolism and function by producing various growth mediators, cytokines and chemokines. During liver injury HSCs are activated by transdifferentiation into a myofibroblastic phenotype (known as activated HSCs). Activated HSCs are highly fibrogenic and contractile, and thus play a major role in hepatic fibrosis and sinusoidal component of portal hypertension. Therefore, selective elimination of activated HSCs has been a topic of intense investigation. Reactive oxygen species (ROS) play an important role in activation and proliferation of HSCs. However, there is compelling in vivo and in vitro evidence for ROS-induced apoptosis of activated HSCs. In this article, I review the work from several laboratories that has provided molecular mechanisms of ROS-induced activation/proliferation and apoptosis of HSCs. A critical examination of these mechanisms may be useful in development of strategies to eliminate activated HSCs as a therapy for liver fibrosis.
The liver cell types
The liver comprises of 4 major cell types: Parenchymal cells (hepatocytes) that account for nearly 80% of the liver volume, and nonparenchymal cells (Kupffer cells, sinusoidal endothelial cells and hepatic stellate cells). Hepatocytes synthesize and store glycogen, secrete glucose via glycogen catabolism, metabolize and produce various proteins (e.g., albumin, clotting factors), bile acids and bile salts, and synthesize and process various lipids. Kupffer cells (the resident macrophages) eliminate invading microbes and their products, viruses and various toxins by phagocytosis and endocytosis. The specialized fenestrated sinusoidal endothelial cells allow free exchange between the substances in plasma and those produced by hepatocytes. Finally, hepatic stellate cells (HSCs), which are located in the space of Disse, regulate hepatic architecture and blood flow by synthesis/degradation of extracellular matrix and contractility respectively [1–4]. All of these hepatic cell types possess antigen-presenting and co-stimulatory capability, and thus perform numerous important functions including innate and adaptive immunological functions. Kupffer cells, sinusoidal endothelial cells and HSCs produce various cytokines, chemokines, and growth-modulating factors that influence cells of the immune system, as well as the survival and growth of hepatocyte (2–4). In addition, the liver also contains natural killer (NK) cells, NKT cells, dendritic cells, and CD4+ and CD8+ T cells [5]. Together, these cell types provide a coordinated system that fights infections (bacterial, viral and parasitic) and destroy toxic substances. Additionally, the coordinated system also determines the course of acceptance or rejection of the liver allograft [6], and regulates hepatic regeneration following various types of organ injury [7].
Hepatic stellate cells (HSCs)
HSCs account for 8–12% of hepatic cell population. They store the major portion (about 80%) of body’s retinoids including vitamin A, and express interstitial filamentous proteins, desmin and glial-fibrillary acidic protein (GFAP) [8]. Vitamin A autofluorescence, desmin and GFAP are used as markers to identify HSCs in vivo, and to ascertain their purity in vitro during cell culture. HSCs exhibit several cytoplasmic processes that can extend over 2–3 hepatocytes, penetrate endothelial fenestrations and contact sinusoidal cells, and can also traverse hepatocyte plates to contact HSCs in the neighboring sinusoid (Figure 1). With this physical characteristic and the capability to produce various cytokines, chemokines, growth factors, and biologically active components of extracellular matrix (ECM), HSCs can influence the functions of hepatocytes on one hand and sinusoidal cells on the other via paracrine and juxtracrine mechanisms [2, 8–10].
Figure 1. Localization of hepatic stellate cell.
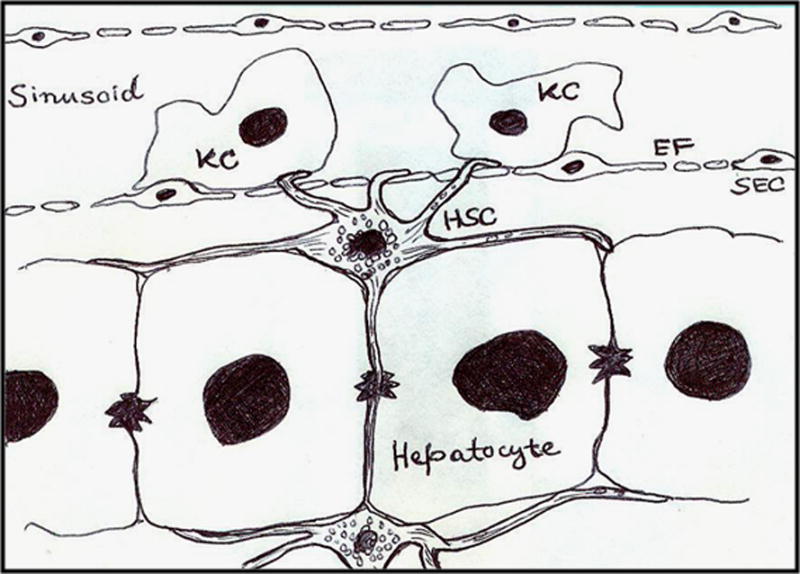
The cartoon depicts localization of a hepatic stellate cell (HSC) in the space of Disse, adjacent to hepatocytes. Note that the cytosolic extensions cover hepatocytes on either side, penetrate the fenestrations (EF) in sinusoidal endothelial cells (SEC), and also traverse the hepatocyte plate and contact HSC in the neighboring perisinusoidal space. KC, Kupffer cell.
Hepatic stellate cells and Fibrosis/Cirrhosis
Physiologically, HSCs are quiescent, but during liver injury, they release retinoids and undergo transdifferentiation into myofibroblast-like phenotype, commonly referred to as activated HSCs. Activation of HSCs is associated with loss of GFAP and acquisition of alpha-smooth muscle actin (α-sma) expression [8,9]. This in vivo activation process is recapitulated significantly when HSCs from normal liver are placed in cell culture. Activated HSCs express receptor for platelet-derived growth factor (PDGF), and proliferate aggressively in response to PDGF and other mitogens produced by infiltrating blood cells, hepatocytes, Kupffer cells, and by HSCs themselves [2–4, 11, 12]. Activated HSCs respond to fibrogenic stimuli such as transforming growth factor-β (TGF-β), and synthesize and deposit excessive amounts of abnormal ECM components. Deposition of ECM is aided by increased expression of tissue inhibitors of matrix metalloproteinases (TIMPs) and reduced or unaltered expression of matrix metalloproteinases (MMPs) by HSCs [13–15]. Thus, activated HSCs contribute profoundly to progressive and excessive fibrosis, the common consequence of chronic liver disease of various etiologies. Therefore, strategies for selective removal of activated HSCs or suppression of their activation, as therapeutic avenues, have been the major topics of intense investigation for a number of years [16–18].
Reactive oxygen species
Reactive oxygen species (ROS) (superoxide radical , hydrogen peroxide [H2O2] and hydroxyl ion [OH−]) are generated as by-products of cellular metabolism [19] by all cell types, with mitochondria and microsomes being the major organelles of their production (Equation 1).
| Equation 1 |
Electron transport associated with the inner mitochondrial membrane is the primary process that generates [20, 21]. Microsomal cytochrome P450 enzyme also generates during oxidation of NADPH [22–24]. Phagocytosis of foreign particles, including microorganisms and derivatives thereof, or senescent/dead cells by macrophages (e.g., Kupffer cells) causes increased O2 uptake and production of via activation of a membrane-bound oxidase. Xanthine oxidase, a widely distributed enzyme that acts on xanthine or hypoxanthine is also a source for the generation of [25,26].
The generation of (half life 10−6 sec) is followed readily by its dismutation to H2O2 by superoxide dismutase (SOD) (Equation 1B). Isomers of SOD are present in mitochondria (Mn-SOD), cytosol (Cu-Zn-SOD) and in extracellular environment (Cu-Zn-SOD) [19]. H2O2 by itself is not a highly reactive molecule, but it is an important biological oxidant that can diffuse through the hydrophobic membranes and generate highly reactive hydroxyl radicals by reacting with redox-active transitional metals (Equation 1C). It has been postulated that activated phagocytic cells generate substantial amounts of H2O2, which is mainly responsible for the cytotoxicity observed at the sites of tissue inflammation [27, 28]. In addition to SOD, the detrimental actions of the ROS, generated on a continuous basis physiologically, are neutralized efficiently by glutathione peroxidase and catalase. Glutathione peroxidase and catalase are also present both in intra- and extracellular compartments [19]. Furthermore, glutathione, vitamins A, C and E, uric acid and ceruloplasmin can act as antioxidants in preventing ROS-induced cell injury.
Moderately increased ROS activate signaling pathways required for physiological functions of the cell. However, at higher concentrations, ROS and their lipid peroxidation products can cause injury to the cells by damaging macromolecules including genomic DNA [19], and induce death of several cell types including hepatocytes [29–32]. Relatively high levels of ROS, generated by Kupffer cells and infiltrating leukocytes during hepatic inflammation [33–35], have been implicated in the initiation and progression of liver pathologies [36, 37].
ROS and activation of hepatic stellate cells
Factors released by apoptotic/necrotic hepatocytes [38, 39], and expression of cell adhesion molecules as well as chemokines by Kupffer cells [38, 40, 41] and HSCs [42] cause infiltration of blood cells such as neutrophils and monocytes in the injured liver. The infiltrated blood cells and Kupffer cells produce inflammatory cytokines and ROS, which were postulated to induce activation and proliferation of HSCs, a hallmark of fibrogenesis in chronic liver disease [9].
Carbon tetrachloride (CCl4)-induced liver injury in rodents has been employed extensively as a model to investigate the mechanisms of HSC activation and liver fibrosis. CCl4 is metabolized by cytochrome P450 enzymes, primarily CYP2E1, in hepatocytes resulting in the formation of a potent free radical CCl3. CCl3 causes widespread metabolic disturbances, including injury due to peroxidative degeneration of membrane lipids [43]. Since CCl4 is a hepatocyte toxin and does not affect HSCs, substances released by the injured or dying hepatocytes were postulated to be associated with activation of HSCs [44]. Among these, lipid peroxidation products produced by the damaged hepatocytes are particularly important in driving HSC activation. Indeed, transient production of , observed in the pericentral necrotic areas within 24 hours after acute CCl4 treatment, was followed by appearance of HSCs and collagen expression; higher levels of with dramatic and sustained activation of HSCs continued to occur in the necrotic areas during chronic CCl4 treatment of rats [45]. Furthermore, activation of HSCs was observed to correlate closely with hepatic oxidative stress rather than the tissue levels of cytokines [46]. Greater quantities of ROS produced by Kupffer cells from CCl4-treated rats with necrotic injury than those from control rats [47], migration of Kupffer cells to the necrotic area [48], and abrogation of HSC activation by blocking Kupffer cell function [49] during chronic CCl4 treatment of rats, provide additional support for the role of ROS in activation and fibrogenic activity of HSCs.
HSCs cultured on uncoated or collagen I-coated plates in serum-supplemented medium undergo spontaneous activation, but resist activation when cultured in matrigel [50, 51]. These properties of HSCs have been exploited to investigate the mechanisms of their activation. Several cytokines such as tumor necrosis factor-α (TNF-α), transforming growth factor-β (TGF-β), TGF-α, fibroblast growth factor (FGF) and platelet-derived growth factor (PDGF) produced in the hepatic microenvironment during injury were found to facilitate activation and proliferation of HSCs [11, 12]. These observations and activation of HSCs on matrigel by free radicals generated from ascorbate/FeSO4 as well as a lipid peroxidation product, malondialdehyde [52] suggest that complimentary actions of growth mediators and ROS drive both activation and proliferation of HSCs.
Malondialdehyde [CH2(CHO)2] is formed during degradation of polyunsaturated lipids by ROS [53]. Cellular toxicity of this reactive aldehyde, a marker of oxidative stress, is due to its property to form covalent protein adducts known as advanced lipoxidation end products [54]. Malondialdehyde also reacts with deoxyadenosine and deoxyguonosine in DNA forming mutagenic DNA adducts [55]. However, in contrast to its injurious effects on various cell types, malondialdehyde induced activation of NFκB in HSCs [52]. NFκB is a nuclear transcription factor associated with cell survival and proliferation [56]. Malondialdehyde treatment of HSCs also increased protein expression of a transcription factor c-Myb [52], which regulates differentiation and proliferation of many cell types including hematopoietic cells, stem cells and epithelial cells [57]. While blockade of NFκB activation prevented c-Myb expression, treatment of the cells with an antisense oligonucleotide for c-Myb mRNA blocked malondialdehyde-, collagen I- and TGF-α-induced activation of HSCs [52]. These data suggested that NFκB and subsequent c-Myb signaling play a critical role in oxidative stress-induced activation of HSCs. Increased c-Myb expression was also observed in HSCs activated in vivo during CCl4-induced fibrosis [52] and in human fibrotic liver [58]. Since increased expression of c-Myb is associated with activation of anti-apoptotic (Bcl2 and Bcl-xL) genes [59], it is apparent that this transcription factor is required for the survival of HSCs during oxidative stress.
The above-described data indicated that “exogenously” produced ROS or lipid peroxidation products cause activation of HSCs. However, abrogation of TGF-α- or collagen I-induced activation and proliferation of HSCs by antioxidants α-tocopherol or butylated hydoxytoluene [52] suggested a prominent role for “intracellular” oxidative stress as well. HSCs isolated from mouse deficient in NADPH oxidase, a major intracellular producer of ROS, generate less ROS and demonstrate lower level of activation during chronic liver injury than the cells from control mouse [60]. Furthermore, greater level of HSC activation and magnitude of fibrosis was observed in CCl4-treated mice transgenic for Rac (gene that encodes a small GTP-binding protein of Rho family) under the control of α-sma promoter. In vitro, HSCs from Rac-transgenic mice produced greater levels of ROS, expression of activation markers and collagen than control HSCs. Together, these findings suggest a critical role for both extracellular and intracellular oxidative stress in HSC activation and proliferation.
ROS and fibrogenesis
While activation of HSCs is an important step in the initiation of fibrogenic response, continued production and deposition of ECM by retinoid-deficient activated HSCs in chronic liver injury leads to irreversible cirrhosis. In the inflammatory environment of chronic liver injury, ROS can be critical mediators of ECM production by activated HSCs. Since the gene expression as well as the magnitude of the synthesis and deposition of type I collagen is greatly increased in activated HSCs in comparison to their quiescent counterparts that produce very small amounts of this protein [50, 51, 61], expression level of collagen type I is used as a reliable indicator of fibrogenic activity.
Using hypoxanthine (1 mM) and xanthine oxidase (3 mU/ml) as superoxide-generating system, Shina et al [62] demonstrated enhanced collagen synthesis by culture-activated HSCs. Several investigators confirmed these initial findings. Activated neutrophil-derived superoxide [63], superoxide generated from 0.4 mM xanthine and 75 mU/ml xanthine oxidase [63], pro-oxidant ethanol metabolite acetaldehyde [64], and ascorbate/iron [65] were all reported to cause membrane lipid peroxidation and induce collagen I synthesis in activated human HSCs. Furthermore, exogenously added aldehydic lipid peroxidation product 4-hydroxynonenol [65] also induced collagen I production by activated human HSCs.
Attempts to discover the mechanism of acetaldehyde-induced collagen I production revealed activation of protein kinase C in rat and human activated HSCs [66, 67], and of phosphatidyl-inositol-3-kinase (PI3 kinase) and extracellular signal-regulated mitogen-activated protein kinase (ERK1/2) in human activated HSCs [67]. Activation of ERK1/2 and PI3K is required for proliferation of HSCs [68, 69]. Interestingly, blockade of acetaldehyde-induced protein kinase C activation, but not of ERK1/2 or PI3K activation, abrogated fibrogenic activity of HSCs [67]. These observations indicate that distinct intracellular signaling pathways are responsible for ROS-induced proliferation and fibrogenic activity of HSCs. In contrast to these findings, other investigators [70] did not observe increase in collagen synthesis in culture-activated rat HSCs stimulated by 4-hydroxynonenal or iron/ascorbate. Thus additional studies are required to delineate the biochemical mechanisms involved in ROS-induced fibrogenic activity of HSCs.
Death of activated hepatic stellate cells by ROS- A paradox
The observation that significant number of apoptotic HSCs is present in the actively fibrotic areas of the liver during CCl4 treatment [71] suggested that activated HSCs are vulnerable to apoptotic stimuli generated during chronic liver injury. Sensitivity of activated HSCs to apoptotic stimuli is evident from spontaneous reversibility of CCl4-induced liver fibrosis in rodents upon termination of the toxin treatment [14, 72], and heightened apoptosis of activated HSCs during such reversal [14, 73]. In tissue culture, activated HSCs undergo apoptosis via autocrine Fas/FasL-induced mechanism [74, 75]. However, increase in the number of apoptotic HSCs in chronically CCl4-treated rats upon increasing oxidative stress with tert-butylhydroperoxide (TBHP) administration [76] suggested that ROS can be critical in triggering pro-apoptotic mechanisms in activated HSCs in vivo.
This postulate has been supported strongly by several in vitro studies demonstrating the ability of both exogenous and intracellular oxidative stress to induce death of activated rat and human HSCs [71, 77–80]. Apoptosis of activated HSCs due to superoxide generated from 2 mM hypoxanthine was found to be xanthine oxidase concentration-dependent (0.5–2 mU/ml) [76]. However, during the 24-hour time period of superoxide exposure, about 50% of the cells underwent apoptosis, suggesting differential sensitivity of HSCs in this population to superoxide-induced death. The classical pathway of apoptosis (mitochondrial permeability, cytochrome c release and caspase 3 activation) is instigated in activated HSCs subjected to oxidative stress [71, 79]. Interestingly, superoxide also increased nuclear translocation of a cell survival transcription factor NFkB and up-regulated the expression of anti-apoptotic molecule Bcl-xL in activated HSCs (71). In contrast, quiescent rat HSCs are resistant to superoxide-induced apoptosis in vitro [76]. Even in vivo, oxidative stress induced by TBHP [76] or gliotoxin [78, 81] did not affect survival of quiescent HSCs in the control rats.
The question is why the quiescent and activated HSCs respond in a contrasting manner to apoptosis-inducing effect of ROS? The possibility of altered antioxidant mechanisms should be considered. A major difference between the two phenotypes is the abundance of retinoids, including retinoic acid, in quiescent HSCs and its deficiency in activated HSCs. Retinoic acid has immunomodulatory, anti-inflammatory, and antiapoptotic properties [82,83]. Retinoic acid is also critical in maintaining higher levels of GSH that controls cell’s redox state and glutathione peroxidase activity [19]. In fact, lower GSH content can render cells susceptible to increased oxidative stress and apoptosis [84]. Therefore, high GSH content is likely to contribute to the resistance of quiescent HSCs against damaging effect of ROS, while activated HSCs are vulnerable to ROS-induced death due to relatively lower GSH content [76]. Indeed, retinoic acid was found to protect cultured neurons from oxidative stress by inhibiting GSH depletion [83]. Furthermore, GSH depletion itself was found to induce apoptosis of hepatocytes [85] and to further sensitize them to TNF-α-induced apoptosis [86].
The reported effects of generated by the reaction between hypoxanthine or xanthine and xanthine oxidase on activated human HSCs are somewhat variable. Generation of superoxide by 0.2 mM hypoxanthine + 20 mU/ml xanthine oxidase or 0.05 mM hypoxanthine + 100 mU/ml xanthine oxidase caused apoptosis of activated human HSCs [87]. However, superoxide generated by 0.5 mM xanthine + 2 mU/ml xanthine oxidase caused 6-fold increase in the DNA synthesis [88], while strongly inhibiting PDGF-induced proliferation of activated human HSCs [87]. Up-regulation of anti-apoptotic Bcl2 and down-regulation of pro-apoptotic Bax in activated human HSCs exposed to 0.4 mM hypoxanthine + 2 mU/ml xanthine oxidase may explain their resistance to superoxide-induced apoptosis [89]. Perhaps, PDGF might induce oxidative stress by itself (similar to TGF-α [52]), and this additional intracellular stress along with exogenous oxidative stress could be responsible for inhibition of the observed proliferation of HSCs [87].
Such differential effects of ROS on activated human and rodent HSCs should be evaluated carefully. It is important to consider that, similar to other hepatic cell types, HSCs are a highly heterogeneous population. Characteristics of the hepatic cell types in the periportal region are distinct from those in the pericentral region. Thus in a heterogeneous population of a specific cell type, response to an agent is likely variable. We found that all of the rat HSCs, in a given passage, do not react simultaneously and similarly to a challenge with pro-apoptotic agent such as gliotoxin (unpublished observation) or superoxide [71, 76] (Figure 2). This indicates variability in this so-called homogeneous cell population of activated HSCs. For the investigations involving rodents, the cells were derived from the livers of animals maintained in controlled conditions. On the other hand, human livers used to obtain cells for in vitro studies are generally the ones found unsuitable for transplantation, and have already been subjected to cold ischemic preservation. Alternatively, a portion of the human liver is obtained from the so-called normal part of the organ during elective resection. Even though the human HSCs are used following subcultures, differential reactivity of the subpopulations to ROS, as observed for rodent HSCs, cannot be ruled out. Nevertheless, apoptosis of HSCs during active fibrogenesis in inflammatory liver disease [15, 71] might be an important mechanism in limiting ongoing fibrosis, with ROS playing an integral role. Figure 3 depicts the scheme of ROS-induced effects on HSCs based on the currently available knowledge. Accordingly, several mediators released by Kupffer cells, platelets, infiltrating blood cells and dying hepatocytes induce activation of quiescent HSCs. These mediators continue to act on activated HSCs and promote their proliferation and fibrogenic activity. However, a sub-population of the activated HSCs undergoes apoptosis upon exposure to ROS, which may be a mechanism of limiting the magnitude of chronic liver disease. Continuing investigation of this interesting phenomenon will be necessary to ascertain its clinical relevance, and possible exploitation for treatment of liver fibrosis.
Figure 2. Apoptosis of culture-activated HSCs by superoxide.
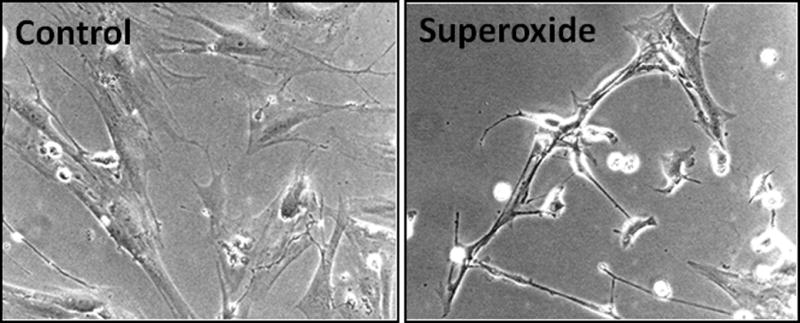
Rat HSCs were cultured and used in passage 3. After the cells had achieved semiconfluence, they were subjected to superoxide challenge (1 mM hypoxanthine + 2 mU/ml xanthine oxidase). Control cells were incubated in the presence of hypoxanthine alone. After 24 hours, photomicrograph was taken. Note that superoxide-challenged cells have shrunk, and several have already undergone apoptosis. Magnification ×100.
Figure 3. A schema showing the effects of ROS and other mediators on HSCs during liver injury.
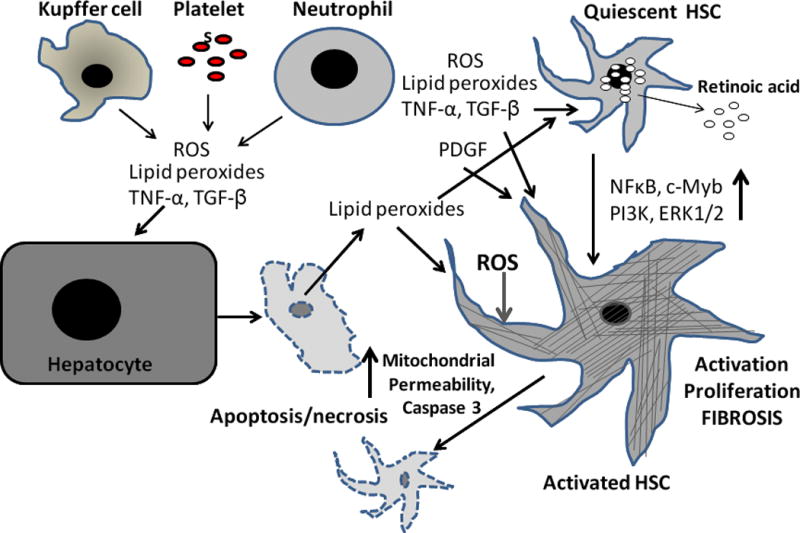
Upon liver injury (e.g., CCl4 administration), Kupffer cells, platelets and infiltrating blood cells such as monocytes and neutrophils produce several mediators including ROS and lipid peroxides. CCl4 also causes hepatocyte apoptosis/necrosis, which release several mediators, including ROS and lipid peroxides. These mediators act on quiescent HSCs, which release retinoids and change their phenotype to myofibroblast-like cells. While these mediators continue to induce HSC activation, and also proliferation of activated HSCs and promote their fibrogenic activity, a subpopulation can undergo apoptosis in response to ROS challenge. NFkB, c-Myb, PI3K and ERK1/2 have been shown to participate in activation/proliferation of HSCs. See text for details.
Acknowledgments
This work was supported by a VA Merit Review Award, and NIH grants DK 54411and PO1A108678
Abbreviations
- HSC
activated hepatic stellate cell
- GSH
glutathione
- ROS
reactive oxygen species
- SOD
superoxide dismutase
Footnotes
Disclosures
The author has no financial conflicts of interest. The content of this work does not represent the views of the Department of Veterans Affairs or the United States Government.
References
- 1.Soto-Gutierrez A, Navarro-Alvarez N, Kobayashi N. In: Molecular Pathology of Liver Diseases. Monga SP, editor. Springer; New York: 2010. p. 17. [Google Scholar]
- 2.Gandhi CR. In: Molecular Pathology of Liver Diseases. Monga SP, editor. Springer; New York: 2010. p. 53. [Google Scholar]
- 3.Gandhi CR. In: Molecular Pathology of Liver Diseases. Monga SP, editor. Springer; New York: 2010. p. 81. [Google Scholar]
- 4.Stolz DB. In: Molecular Pathology of Liver Diseases. Monga SP, editor. Springer; New York: 2010. p. 97. [Google Scholar]
- 5.Crispe IN. Annu Rev Immunol. 2009;27:147. doi: 10.1146/annurev.immunol.021908.132629. [DOI] [PubMed] [Google Scholar]
- 6.Thomson AW, Knolle PA. Nat Rev Immunol. 2010;10:753. doi: 10.1038/nri2858. [DOI] [PubMed] [Google Scholar]
- 7.Michalopoulos GK. J Cell Physiol. 2007;213:286. doi: 10.1002/jcp.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geerts A, DeBleser P, Hautekeete ML, Niki T, Wisse E. In: The Liver: Biology and Pathobiology. Arias IM, Boyer JL, Fasuto N, Jakoby WB, Schachter DL, Shafritz DA, editors. Raven Press, Ltd.; New York: 1994. p. 819. [Google Scholar]
- 9.Friedman SL. J Biol Chem. 2000;275:2247. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 10.Pinzani M, Marra F. Semin Liver Dis. 2001;21:397. doi: 10.1055/s-2001-17554. [DOI] [PubMed] [Google Scholar]
- 11.Pinzani M, Gesualdo L, Sabbah GM, Abboud HE. J Clin Invest. 1989;84:1786. doi: 10.1172/JCI114363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pinzani M, Knauss TC, Pierce GF, Hsieh P, Kenney W, Dubyak GR, Abboud HE. Am J Physiol. 1991;260:C485. doi: 10.1152/ajpcell.1991.260.3.C485. [DOI] [PubMed] [Google Scholar]
- 13.Arthur MJ, Mann DA, Iredale JP. J Gastroenterol Hepatol. 1998;13:S33. [PubMed] [Google Scholar]
- 14.Iredale JP, Benyon RC, Pickering J, McCullen M, Northrop M, Pawley S, Hovell C, Arthur MJ. J Clin Invest. 1998;102:538. doi: 10.1172/JCI1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iredale JP. Semin Liver Dis. 2001;21:427. doi: 10.1055/s-2001-17557. [DOI] [PubMed] [Google Scholar]
- 16.Friedman SL, Bansal MB. Hepatology. 2006;43:S82. doi: 10.1002/hep.20974. [DOI] [PubMed] [Google Scholar]
- 17.Kisseleva T, Brenner DA. J Gastroenterol Hepatol. 2007;22:S73. doi: 10.1111/j.1440-1746.2006.04658.x. [DOI] [PubMed] [Google Scholar]
- 18.Ghiassi-Nejad Z, Friedman SL. Expert Rev Gastroenterol Hepatol. 2008;2:803. doi: 10.1586/17474124.2.6.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu BP. Physiol Rev. 1994;74:139. doi: 10.1152/physrev.1994.74.1.139. [DOI] [PubMed] [Google Scholar]
- 20.McCord JM, Fridovich I. J Biol Chem. 1970;245:1374. [PubMed] [Google Scholar]
- 21.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 3rd. Oxford Press; Oxford: 1999. [Google Scholar]
- 22.Svingen BA, O’Neal FO, Aust SD. Photochem Photobiol. 1978;28:803. doi: 10.1111/j.1751-1097.1978.tb07022.x. [DOI] [PubMed] [Google Scholar]
- 23.Swartz HM. In: Free Radicals, Lipid Peroxidation, and Cancer. McBrien DCH, Slater TF, editors. Academic Press; London: 1982. p. 5. [Google Scholar]
- 24.Thomas CE, Aust SD. Free Radical Biol Med. 1985;1:293. doi: 10.1016/0748-5514(85)90134-5. [DOI] [PubMed] [Google Scholar]
- 25.McCord JM, Fridovich I. J Biol Chem. 1968;243:5753. [PubMed] [Google Scholar]
- 26.McCord JM, Fridovich I. J Biol Chem. 1969;244:6056. [PubMed] [Google Scholar]
- 27.Freeman BD. In: Free Radicals Molecular Biology, Aging, and Disease. Armstrong D, Sohal RS, Cutler RG, Slater TE, editors. Raven; New York: 1984. p. 43. [Google Scholar]
- 28.Heffner JE, Repine JE. Am Rev Respir Dis. 1989;140:531. doi: 10.1164/ajrccm/140.2.531. [DOI] [PubMed] [Google Scholar]
- 29.Li PF, Dietz R, von Harsdorf R. Circulation. 1997;96:3602. doi: 10.1161/01.cir.96.10.3602. [DOI] [PubMed] [Google Scholar]
- 30.Knight TR, Ho YS, Farhood A, Jaeschke H. J Pharmacol Exp Ther. 2002;303:468. doi: 10.1124/jpet.102.038968. [DOI] [PubMed] [Google Scholar]
- 31.Rauen U, Elling B, de Groot H. Free Radic Biol Med. 1997;23:392. doi: 10.1016/s0891-5849(96)00618-1. [DOI] [PubMed] [Google Scholar]
- 32.Rauen U, Polzar B, Stephan H, Mannherz HG, de Groot D. FASEB J. 1999;13:155. doi: 10.1096/fasebj.13.1.155. [DOI] [PubMed] [Google Scholar]
- 33.Drath DB, Karnovsky ML. J Exp Med. 1975;141:257. doi: 10.1084/jem.141.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnston RB, Jr, Godzik CA, Cohn ZA. J Exp Med. 1978;148:115. doi: 10.1084/jem.148.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shiratori Y, Takikawa H, Kawase T, Sugimoto T. Gastroenterol Jpn. 1986;21:135. doi: 10.1007/BF02774831. [DOI] [PubMed] [Google Scholar]
- 36.Klebanoff SL. In: Inflammation: Basic Princliples and Clinical Correlates. Gallin JI, Goldstein IM, Snyderman I, editors. Raven Press; New York: 1988. p. 391. [Google Scholar]
- 37.Pietrangelo A. Sem Liver Dis. 1996;16:13. doi: 10.1055/s-2007-1007215. [DOI] [PubMed] [Google Scholar]
- 38.Jaeschke H, Smith CW, Clemens MG, Ganey PE, Roth RA. Toxicol Appl Pharmacol. 1996;139:213. doi: 10.1006/taap.1996.0160. [DOI] [PubMed] [Google Scholar]
- 39.Ziol M, Tepper M, Lohez M, Arcangeli G, Ganne N, Christidis C, Trinchet JC, Beaugrand M, Guillet JG, Guettier C. J Hepatol. 2001;34:254. doi: 10.1016/s0168-8278(00)00047-7. [DOI] [PubMed] [Google Scholar]
- 40.Jaeschke H. J Gastroenterol Hepatol. 2011;26:S173. [Google Scholar]
- 41.Abu-Amara M, Yang SY, Tapuria N, Fuller B, Davidson B, Seifalian A. Liver Transpl. 2010;16:1016. doi: 10.1002/lt.22117. [DOI] [PubMed] [Google Scholar]
- 42.Knittel T, Dinter C, Kobold D, Neubauer K, Mehde M, Eichhorst S, Ramadori G. Am J Pathol. 1999;154:153. doi: 10.1016/s0002-9440(10)65262-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manibusan MK, Odin M, Eastmond DA. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2007;25:185. doi: 10.1080/10590500701569398. [DOI] [PubMed] [Google Scholar]
- 44.Bedossa P, Houglum K, Trautwein C, Holstege A, Chojkier M. Hepatology. 1994;19:1262. [PubMed] [Google Scholar]
- 45.Montosi G, Garuti C, Iannone A, Pietrangelo A. Am J Pathol. 1998;152:1319. [PMC free article] [PubMed] [Google Scholar]
- 46.Kim KY, Choi I, Kim SS. Mol Cells. 2000;10:289. [PubMed] [Google Scholar]
- 47.Alric L, Orfila C, Carrere N, Beraud M, Carrera G, Lepert JC, Duffaut M, Pipy B, Vinel JP. Inflamm Res. 2000;49:700. doi: 10.1007/s000110050649. [DOI] [PubMed] [Google Scholar]
- 48.Kawashima R, Mochida S, Matsui A, YouLuTuZ Y, Ishikawa K, Toshima K, Yamanobe F, Inao M, Ikeda H, Ohno A, Nagoshi S, Uede T, Fujiwara K. Biochem Biophys Res Commun. 1999;256:527. doi: 10.1006/bbrc.1999.0372. [DOI] [PubMed] [Google Scholar]
- 49.Rivera CA, Bradford BU, Hunt KJ, Adachi Y, Schrum LW, Koop DR, Burchardt ER, Rippe RA, Thurman RG. Am J Physiol Gastrointest Liver Physiol. 2001;281:G200. doi: 10.1152/ajpgi.2001.281.1.G200. [DOI] [PubMed] [Google Scholar]
- 50.Davis BH, Pratt BM, Madri JA. J Biol Chem. 1987;262:10280. [PubMed] [Google Scholar]
- 51.Friedman SL, Roll FJ, Boyles J, Arenson DM, Bissell DM. J Biol Chem. 1989;264:10756. [PubMed] [Google Scholar]
- 52.Lee KS, Buck S, Houglum K, Chojkier M. J Clin Invest. 1995;96:2461. doi: 10.1172/JCI118304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pryor WA, Stanley JP. J Org Chem. 1975;40:3615. doi: 10.1021/jo00912a038. [DOI] [PubMed] [Google Scholar]
- 54.Farmer EE, Davoine C. Curr Opin Plant Biol. 2007;10:380. doi: 10.1016/j.pbi.2007.04.019. [DOI] [PubMed] [Google Scholar]
- 55.Marnett LJ. Mutat Res. 1999;424:83. doi: 10.1016/s0027-5107(99)00010-x. [DOI] [PubMed] [Google Scholar]
- 56.Karin M. Cancer J Sci Am. 1998;4:S92. [PubMed] [Google Scholar]
- 57.Zhou Y, Ness SA. Front Biosci. 2011;16:1109. doi: 10.2741/3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kitada T, Seki S, Nakatani K, Kawada N, Kuroki T, Monna T. Hepatology. 1997;26:1506. doi: 10.1053/jhep.1997.v26.pm0009397991. [DOI] [PubMed] [Google Scholar]
- 59.Ramsay RG, Barton AL, Gonda TJ. Expert Opin Ther Targets. 2003;7:235. doi: 10.1517/14728222.7.2.235. [DOI] [PubMed] [Google Scholar]
- 60.Choi SS, Sicklick JK, Ma Q, Yang L, Huang J, Qi Y, Chen W, Li YX, Goldschmidt-Clermont PJ, Diehl AM. Hepatology. 2006;44:1267. doi: 10.1002/hep.21375. [DOI] [PubMed] [Google Scholar]
- 61.Brenner DA, Chojkier M. J Biol Chem. 1987;262:17690. [PubMed] [Google Scholar]
- 62.Shina S, Shiratori Y, Kawase T, Sugimoto T. In: Cells of the Hepatic sinusoids. Wisse E, Knook DL, Decker K, editors. Vol. 2. Kupffer Cell Foundation; The Netherlands: 1989. p. 52. [Google Scholar]
- 63.Casini A, Ceni E, Salzano R, Biondi P, Parola M, Galli A, Foschi M, Caligiuri A, Pinzani M, Surrenti C. Hepatology. 1997;25:361. doi: 10.1053/jhep.1997.v25.pm0009021948. [DOI] [PubMed] [Google Scholar]
- 64.Moshage H, Casini A, Lieber CS. Hepatology. 1990;12:511. doi: 10.1002/hep.1840120311. [DOI] [PubMed] [Google Scholar]
- 65.Parola M, Pinzani M, Casini A, Albano E, Poli G, Gentilini A, Gentilini P, Dianzani MU. Biochem Biophys Res Commun. 1993;194:1044. doi: 10.1006/bbrc.1993.1927. [DOI] [PubMed] [Google Scholar]
- 66.Anania FA, Womack L, Potter JJ, Mezey E. Alcohol Clin Exp Res. 1999;23:279. [PubMed] [Google Scholar]
- 67.Svegliati-Baroni G, Ridolfi F, Di Sario A, Saccomanno S, Bendia E, Benedetti A, Greenwel P. Hepatology. 2001;33:1130. doi: 10.1053/jhep.2001.23788. [DOI] [PubMed] [Google Scholar]
- 68.Marra F, Pinzani M, DeFranco R, Laffi G, Gentilini P. FEBS Lett. 1995;376:141. doi: 10.1016/0014-5793(95)01261-0. [DOI] [PubMed] [Google Scholar]
- 69.Reeves HL, Thompson MG, Dack CL, Burt AD, Day CP. Hepatology. 2000;31:95. doi: 10.1002/hep.510310116. [DOI] [PubMed] [Google Scholar]
- 70.Maher JJ, Neuschwander-Tetri BA. Hepatology. 1997;26:618. doi: 10.1002/hep.510260313. [DOI] [PubMed] [Google Scholar]
- 71.Thirunavukkarasu C, Watkins S, Harvey SAK, Gandhi CR. J Hepatol. 2004;41:567. doi: 10.1016/j.jhep.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 72.Anselmi K, Subbotin VM, Nemoto EM, Gandhi CR. J Gastroenterol Hepatol. 2002;17:589. doi: 10.1046/j.1440-1746.2002.02705.x. [DOI] [PubMed] [Google Scholar]
- 73.Thirunavukkarasu C, Yang Y, Subbotin VM, Harvey SAK, Fung J, Gandhi CR. Gut. 2004;53:1010. doi: 10.1136/gut.2003.026534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gong W, Pecci A, Roth S, Lahme B, Beato M, Gressner AM. Hepatology. 1998;28:492. doi: 10.1002/hep.510280229. [DOI] [PubMed] [Google Scholar]
- 75.Saile B, Knittel T, Matthes N, Schott P, Ramadori G. Am J Pathol. 1997;151:1265. [PMC free article] [PubMed] [Google Scholar]
- 76.Jameel NM, Thirunavukkarasu C, Wu T, Watkins C, Friedman SL, Gandhi CR. J Cell Physiol. 2009;218:157. doi: 10.1002/jcp.21581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li L, Tao J, Davaille J, Feral C, Mallat A, Rieusset J, Vidal H, Lotersztajn S. J Biol Chem. 2001;276:38152. doi: 10.1074/jbc.M101980200. [DOI] [PubMed] [Google Scholar]
- 78.Wright MC, Issa R, Smart DE, Trim N, Murray GI, Primrose JN, Arthur MJ, Iredale JP, Mann DA. Gastroenterology. 2001;121:685. doi: 10.1053/gast.2001.27188. [DOI] [PubMed] [Google Scholar]
- 79.Kweon YO, Paik YH, Schnabl B, Qian T, Lemasters JJ, Brenner DA. J Hepatol. 2003;39:38. doi: 10.1016/s0168-8278(03)00178-8. [DOI] [PubMed] [Google Scholar]
- 80.Montiel-Duarte C, Ansorena E, Lopez-Zabalza MJ, Cenarruzabeitia E, Iraburu MJ. Biochem Pharmacol. 2004;67:1025. doi: 10.1016/j.bcp.2003.10.020. [DOI] [PubMed] [Google Scholar]
- 81.Anselmi K, Nalesnik M, Watkins SC, Beer-Stolz D, Gandhi CR. J Hepatology. 2007;47:103. doi: 10.1016/j.jhep.2007.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fumarulo R, Conese M, Riccardi S, Giordano D, Montemuro P, Golucci M, Semeraro N. Agents Actions. 1991;34:339. doi: 10.1007/BF01988726. [DOI] [PubMed] [Google Scholar]
- 83.Ahlemeyer B, Krieglstein J. Neurochem Int. 2000;36:1. doi: 10.1016/s0197-0186(99)00101-1. [DOI] [PubMed] [Google Scholar]
- 84.Hayes JD, McLellan LI. Free Rad Res. 1999;31:273. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- 85.Rauen U, Reuters I, Fuchs A, de Groot H. Hepatology. 1997;26:351. doi: 10.1002/hep.510260215. [DOI] [PubMed] [Google Scholar]
- 86.Colell A, García-Ruiz C, Miranda M, Ardite E, Marí M, Morales A, Corrales F, Kaplowitz N, Fernández-Checa JC. Gastroenterology. 1998;115:1541. doi: 10.1016/s0016-5085(98)70034-4. [DOI] [PubMed] [Google Scholar]
- 87.Novo E, Marra F, Zamara E, Valfre di Bonzo L, Caligiuri A, Cannito S, Antonaci C, Colombatto S, Pinzani M, Parola M. Gut. 2006;55:90. doi: 10.1136/gut.2005.069633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Galli A, Svegliati-Baroni G, Ceni E, Milani S, Ridolfi F, Salzano R, Tarocchi M, Grappone C, Pellegrini G, Benedetti A, Surrenti C, Casini A. Hepatology. 2005;41:1074. doi: 10.1002/hep.20683. [DOI] [PubMed] [Google Scholar]
- 89.Novo E, Marra F, Zamara E, Valfrè di Bonzo L, Monitillo L, Cannito S, Petrai I, Mazzocca A, Bonacchi A, De Franco RS, Colombatto S, Autelli R, Pinzani M, Parola M. Gut. 2006;55:1174. doi: 10.1136/gut.2005.082701. [DOI] [PMC free article] [PubMed] [Google Scholar]