

Abstract
Each year in the United States, thousands of cases of sudden and unexpected deaths of infants, children, and young adults are assigned an undetermined cause of death after postmortem investigation and autopsy. Heritable genetic variants have been suggested as the cause of up to a third of sudden death (SD) cases. Elucidation of the genetic variants involved in SD cases is important to not only help establish cause and manner of death of these individuals, but to also aid in determining whether familial genetic testing should be considered. Previously, these types of postmortem screenings have not been a feasible option for most county medical examiners’ and coroners’ offices. We sequenced full exons of 64 genes associated with SD in the largest known cohort (351) of infant and young SD decedents using massively parallel sequencing at <$600 per sample. Genetic variants were assessed through literature review and clinical evaluation by a multidisciplinary consortium of experts. Thirteen individuals (3.7%), eight infants (2.8% of those <1 yr of age) and five children/young adults (7.0% of those >1 yr of age), were found to have a reportable genetic variant contributing to SD. These percentages represent an estimate lower than those previously reported. Overall yields and results likely vary between studies due to differences in evaluation techniques and reporting. Additionally, we recommend ongoing assessment of data, including nonreported novel variants, as technology and literature continually advance. This study demonstrates a strategy to implement molecular autopsies in medicolegal investigations of young SD decedents.
Each year in the United States, thousands of infants, children, and young adults die suddenly and unexpectedly with no identifiable cause of death. After extensive medicolegal investigation, including autopsy, death certificates variably list cause of death as undetermined, Sudden Infant Death Syndrome (SIDS) (<1 yr old), or Sudden Unexplained Death (SUD) (from 1 to <40 yr old) (Liberthson 1996; Matthews and MacDorman 2013). The biological mechanisms leading to sudden death (SD) of the young are often unclear. Several exogenous and intrinsic risk factors have been suggested in the pathophysiology of SIDS and SUD (Shephard and Semsarian 2009; Trachtenberg et al. 2012); however, research suggests specific genetic variations underlie the susceptibility of at least some of these individuals to SD (Van Norstrand and Ackerman 2010).
For young people, sudden unexpected death is most often suspected to be of cardiac origin, or sudden cardiac death (SCD). SCD can be described as a natural death caused by sudden arrhythmia often occurring without previous warning or symptoms (Shephard and Semsarian 2009). In many of these cases, structural abnormalities of the heart, cardiomyopathies, are evident at autopsy. Cardiomyopathies, such as hypertrophic cardiomyopathy and dilated cardiomyopathy, have been associated with variants in genes that encode cardiac structural proteins. In cases of SD in which no structural cardiovascular or other anatomical abnormalities are observed or are equivocal at autopsy, a fatal arrhythmogenic disorder may be suspected because of clinical, historical, or circumstantial information; however, due to a negative autopsy exam, the cause of death is left as undetermined (Shephard and Semsarian 2009).
Arrhythmogenic disorders such as Long QT Syndrome (LQTS), Short QT Syndrome (SQTS), Brugada Syndrome, and Catecholoaminergic Polymorphic Ventricular Tachycardia are characterized by electrical disturbances in heart function, unaccompanied by anatomic evidence. These disorders, also known as channelopathies, are linked to putative pathogenic variants in the genes that encode for cardiac ion channels or ion channel–associated proteins (Tester and Ackerman 2009). Up to 15% of SIDS and 35% of SUD cases have been estimated to be due to genetic variants in cardiac channel–associated genes (Ackerman 2005; Ackerman et al. 2011; Tester et al. 2012).
In many case reports and clinical evaluations in which genetic variants or inherited cardiac disease have been associated with a sudden death, some victims are speculated to have de novo pathological variants with SD as the sentinel event (Klaver et al. 2011; Tester et al. 2012; Giudici et al. 2014). However, a variety of reports indicate that many of the known genetic variants underlying SCD due to either cardiomyopathy or channelopathy are autosomal dominant and have a 50% chance of inheritance (Shim et al. 2005; Shephard and Semsarian 2009). Therefore, elucidation of the genetic variants involved in SD cases is important to not only help establish cause and manner of death of these individuals, but to also aid in determining whether familial genetic testing should be considered. Postmortem genetic screening has been recommended as a new best practice standard in some autopsy-negative cases. However, postmortem genetic screening has not been cost-effective due to low yields (Skinner et al. 2008; Basso et al. 2010; Ackerman et al. 2011). An international heart rhythm expert panel further recommended mutation-specific genetic testing and clinical screenings for family members of some sudden death decedents (Ackerman et al. 2011). Due to technological and financial challenges, postmortem genetic screening tools are not currently a feasible option in most medical examiners’ and coroners’ offices as a standard autopsy procedure.
Commercial genotyping tests are available to identify several genetic variants putatively linked to SD. Labor-intensive Sanger sequencing methods for the identification of disease-specific genetic variants can cost $5400 or more per case to sequence fewer than 20 genes (Bai et al. 2009). Faster and more comprehensive whole-genome or exon-specific analyses using next-generation sequencing (NGS) techniques are 20-fold less expensive than Sanger sequencing methods; however, they are not yet economically feasible for routine use by public service agencies (Loporcaro et al. 2014; https://www.genome.gov/27541954/dna-sequencing-costs-data/).
The purpose of this study was to implement a cost-effective molecular autopsy at the Harris County Institute of Forensic Sciences (Houston, Texas), the county medical examiner's office for Harris County, Texas, to investigate sudden unexplained deaths of infants and young people. A cohort of 351 young individuals who died of sudden and mostly unexplained causes was tested for determination of cause of death. Using a custom gene target exon capture array, 64 genes associated with SD were assessed by NGS technology (Table 1; Supplemental Table S1). A crucially important and unique aspect of this approach was the development of a multidisciplinary and multi-institutional panel of experts with expertise in clinical and basic science cardiology, genetics, and pathology to review each case for putatively significant genetic variants. The panel assessed each variant in light of the existing literature for biochemical and functional mutational analysis, known allele frequency, and case history to determine the possible contribution of each sequence variant in SD. Genetic counselors were also enlisted to review the variants thus identified and to be available for dissemination of results to surviving family members in an ethical and accurate manner.
Table 1.
Targeted genes

To our knowledge, this study is the largest cohort to date screened by NGS targeted to genes known to underlie SD and performed as part of medicolegal investigations of sudden unexplained death cases. The use of a multidisciplinary consortium to assess likelihood of relevance for each variant can serve as a model for other death investigators considering adopting molecular testing. At ∼$600 per case, this low-cost SD molecular autopsy has the potential to aid in determination of cause and manner of death in many unexplained cases in Harris County, Texas, each year. Moreover, this approach can also be adopted by other medical examiner offices as a tool to initiate molecular autopsy programs as NGS becomes more widely available and the field continually evolves.
Results
Cohort demographics
Harris County, Texas, is the third most populous county in the United States (United States Census Bureau) (http://quickfacts.census.gov/qfd/states/48/48201.html). As of the 2010 national census, the primary racial/ethnic groups within the county include Hispanic or Latino (41.6%), non-Hispanic or Latino White (31.9%), non-Hispanic Black or African American (19.5%), and Asian (6.8%). In comparison, the demographic characteristics of the Harris County Institute of Forensic Sciences SIDS/SUD cohort selected for this study are described in Table 2. The majority of decedents in the cohort were under 1 yr of age (80.7 %). Within this group, the average age was 2.8 mo old (±2.2 mo) (Fig. 1) with 1.5- and 1.8-fold more black infants than Hispanic and White infants, respectively (Table 2). SUD decedents ranged in age from 1 to 37 yr with an average age of 17.6 yr old (±12.1 yr) (Fig. 1). Ethnicity of the older age group was similar for the three primary racial/ethnic groups of Harris County, Texas. The incidence of SD in males was 1.5 times greater than females in both age groups.
Table 2.
Cohort demographics
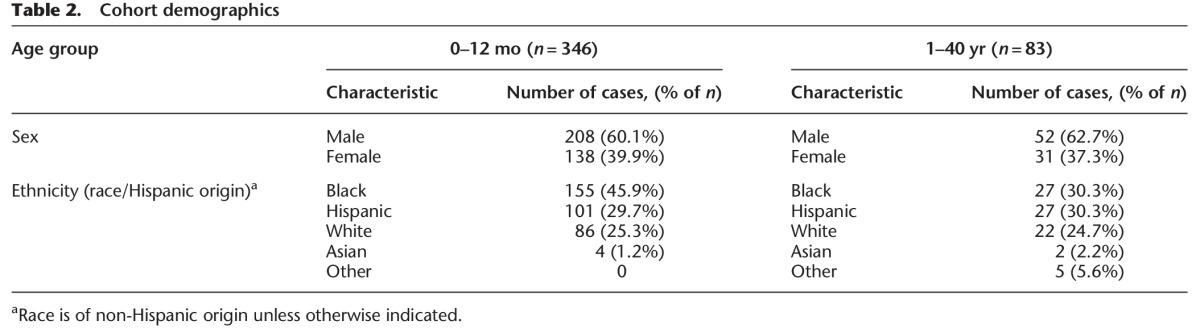
Figure 1.

Sudden death age distribution in the Harris County Institute of Forensic Sciences SIDS/SUD cohort (2004–2012). (A) Age distribution of SIDS victims (0–12 mo, n = 346). The majority of the unexplained death decedents in the cohort were under the age of 1. Distribution of age was unimodal, skewed right, with the highest frequency of death occurring at 2 mo old (95% CI: 2.6–3.0 mo; bin range = 1 mo). (B) Age distribution of SUD victims (1–40 yr, n = 83). The distribution of age was random for unexplained death decedents, with the highest frequency of occurrence in young children between 1 and 2 yr of age (95% CI: 15.0–20.3 yr; bin range = 1 yr). Note the fivefold y-axis scale difference between A and B.
Genetic variants
Of the 429 individuals initially selected for testing (Table 2), 351 (280 infants; 71 > 1 yr of age) had DNA of sufficient quality and quantity for sequencing. After primary quality data analysis, a total of 9318 exonic nonsynonymous or insertion/deletion (indel) variants from the GRCh37 human reference genome were observed in 348 individuals prior to any filtering. Tolerated common polymorphisms or variants with no reference data or literature were filtered out (Fig. 2). Of the remaining 1088 total variants, the same genetic variants occurred in multiple cases (Supplemental Table S2). In total, 77 unique single-nucleotide variants (SNVs) were observed in 29 genes. These variants, plus one indel, were further assessed for clinical review. Thirteen decedents were found to have reportable (defined as likely contributing to the cause of death) pathogenic genetic variants (Tables 3, 4). One individual had two genetic variants likely leading to SD. These cases represent 3.7% of the total cohort that was successfully sequenced. Within the specific age groups, 2.8% of the infants (<1 yr of age) and 7.0% of the children/young adults were found to have pathogenic variants associated with cause of death. The reportable variants were all single-base changes. Additionally, of the 64 genes in the 351 individuals tested, a total of nine genes were found to have pathogenic variants in the 13 reported decedents. Genetic variants in SCN3B and MYL2 were observed twice in these individuals, and variants in SCN5A were observed four times.
Figure 2.
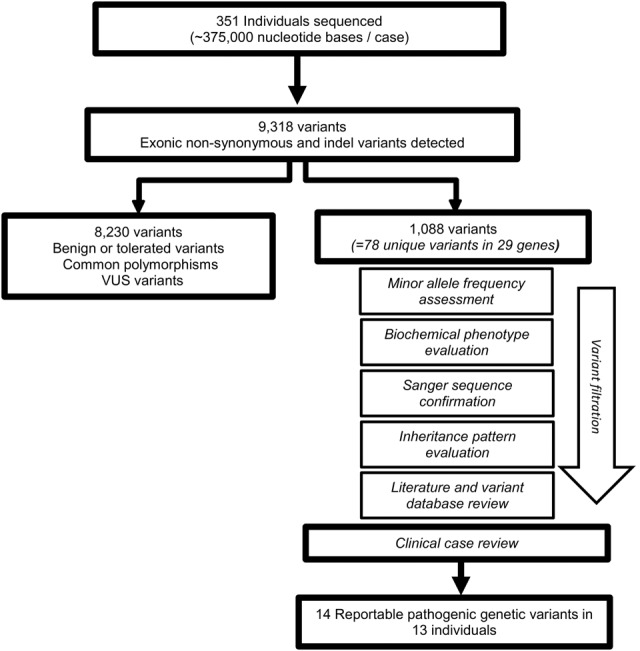
Workflow of genetic variant screening. All quality annotated SNVs and indel variants were filtered in order to classify variants as pathogenic in SD.
Table 3.
Infant (0–12 mo) cohort with genetic variants contributing to sudden death

Table 4.
Children/young adult (1–40 yr) cohort with genetic variants contributing to sudden death
Discussion
The sudden and unexpected death of an infant or young family member has devastating effects on the family and community (Jind et al. 2010). These effects are compounded by the lack of a definitive cause of death. Medical examiners have the legal responsibility to identify the cause of death. Underlying genetic variants may be suspected; however, advancements in genetic sequencing have only recently opened the door to accessible genetic screenings for victims of SIDS/SUD. No less important is the ability to reduce the risk of potential criminal investigations of those families affected by SDs of infants or young people.
Molecular autopsies in cases of SD are not a new idea (Tester and Ackerman 2006). Genetic screenings performed as part of the epidemiological assessment in SD cases have been recommended by panels of experts for nearly a decade (Skinner et al. 2008; Basso et al. 2010; Ackerman et al. 2011). However, previous studies have been primarily restricted to small cohorts with a limited number of genes tested (Wong and Behr 2014). These types of assessments have also been cost prohibitive for use in medicolegal investigations. The work presented here highlights a transition between research and specific clinical cases to implementation of the molecular autopsy as a cost-effective standard of care in postmortem examinations. By targeting selected exons, cost was kept below $600 per sample (Supplemental Table S3). These costs are substantially lower than other available options and are economically feasible for autopsies.
An unexpected finding of this study is the low percentage of individuals found to have a pathogenic variant compared to previous reports (Ackerman 2005; Ackerman et al. 2011; Tester et al. 2012). Of the total sequenced cohort, <4.0% of unexpected deaths were associated with a pathogenic genetic variant, whereas published reports have estimated up to 15% of SIDS and 35% of SUD deaths were related to specific genetic variants. Overall yield likely varies between studies due to differences in multiple evaluation techniques, such as (1) cohort composition, (2) genetic screening method, or (3) interpretation of genetic testing results.
For this study, a large cohort was selected based on autopsy findings. Cohort demographics were consistent with those observed previously, such as increased incidences of SD reported for males, higher SIDS rates for non-Hispanic black infants compared to Hispanic and non-Hispanic white infants, and the majority of unexpected infant deaths occurred in the youngest of the group (93.5% under 6 mo of age) (Shen et al. 1995; Matthews and MacDorman 2013; Wang et al. 2014). A larger cohort allows for an accurate estimate of the rate of SD of the young in a heterogeneous population that is attributable to pathogenic mutations. Therefore, differences between these findings and previous reports concerning the prevalence of genetic variants contributing to SD is not likely due to intrinsic differences in the cohort evaluated in this study. However, the evaluation of candidate SD cases for genetic screening may differ between medical examiner offices, which would alter the initial size and makeup of cohorts. A more restrictive cohort selection, such as phenotype- or familial history–guided testing may increase the overall likelihood of a positive genetic test result. On the other hand, a less restrictive inclusion criterion with an equally critical genetic evaluation, as utilized in this study, could increase the number of decedents evaluated and decrease the overall percentage of a positive finding. These nuances need to be weighed carefully in discerning the best course of action in development of molecular autopsies for use in death investigations, as they may affect the cost and/or overall yield.
In regard to the genetic screening method, we tested a panel of 64 genes associated with multiple diseases linked to SD both from cardiac and noncardiac causes. Others have reported sequence results for disease-specific genetic panels with smaller cohorts or full exome analysis of specific individuals (Ackerman et al. 2004; Tester et al. 2005, 2012; Papadakis et al. 2013; Brion et al. 2014). In this study, we incorporated many genes previously included in separate disease-specific assays into one comprehensive genetic screening panel. A drawback to targeting specific genes is that we are limited in the breadth of exome coverage. By not examining the full exome or genome, we likely miss potentially deleterious genetic variants in nontargeted coding or regulatory intronic regions. Additionally, capture probes included target exons and 35–50 bases of flanking UTRs. These UTRs can exhibit a decrease in selective pressure causing a potential decrease in coverage and variant discovery. These limitations can be further complicated in forensic cases in which there are instances of nonpristine samples. However, this study yielded a moderate to high coverage, and by using a gene targeting approach, we are able to generate a more cost-effective molecular autopsy with relatively high coverage without the burden of massive data sets.
Variations in total yields of pathogenic genetic variants may also be attributed to differences in variant classification. The decision to include or exclude a variant as reportable as contributing to cause of death was based on strength of literature reports, functional studies, and population frequency data at the time of review. How this data is weighed and the overarching goal of a study may differ among laboratories, resulting in variability of the total yield of genetic variants. This total yield may increase using a different variant classification system depending on investigative criteria. Because the goal of this study was to describe a feasible manner in which to identify pathogenic variants as part of death investigation, a drawback to this approach is the limitation on the discovery of novel variants. The identification of novel variants in postmortem genetic screening of large heterogeneous cohorts has been described elsewhere (Wang et al. 2014), and although necessary in the sudden death field, it was beyond the scope of this study. However, it is also important to note that evaluation of putative pathogenic genetic variants reflects a snapshot of current literature and data. Variability in the assignment of pathogenicity has been observed in multiple laboratories and is a limitation of genetic screening studies (Van Driest et al. 2016). As technology, literature, and screening methods evolve, data should be regularly reassessed as novel variants, “variants of unknown significance” (VUS), or variants with conflicting data may become reportable, depending on investigative criteria.
In this report, putative lethal variants were reviewed by certified molecular geneticists using ACMG standards to determine clinical significance on a case-by-case basis. A VUS, in which there is insufficient evidence to support deleteriousness, was not reported as likely pathogenic or pathogenic. Most of the genetic variants that are known to be involved in SD have incomplete penetrance or variable expressivity (Ackerman et al. 2011). As a result of a conservative screening approach, it is likely that novel or VUS genetic variants that were possibly contributing to SD were not identified as pathogenic due to a lack of data at the time of review. Overall, these challenges emphasize the need for a critical evaluation of genetic defects by expert medical geneticists in order to help establish proper evidentiary thresholds for reporting as contributing factors to cause of death.
Multiple factors are considered when determining the potential pathogenicity of a genetic variant. Cosegregation within a family can provide evidence of a heritable genetic defect, whereas incomplete penetrance in unaffected relatives of a proband warrants critical evaluation to identify a true pathogenic variant. De novo variants may also provide strong evidence of a genetic contribution to cause of death, depending on the rarity in the general population. Previous reports have suggested more severe disease expression and earlier onset in de novo carriers (Giudici et al. 2014). However, with an extremely rare variant, a lack of further in vitro/in vivo functional evidence and clinical reports may lead to a categorization of a putative pathogenic variant as a VUS (Dorschner et al. 2013; Tang et al. 2014). Final determinations of cause of death associated with specific genetic variants were concluded by an expert panel based on genetic screening results, autopsy findings, sentinel events, and personal and family medical histories. The cases with a specified cause of death of cardiomyopathy were confirmed, and the associated genetic defect was identified. However, other decedents with negative or ambiguous autopsies also had variants in heritable cardiomyopathy-associated genes as not all cardiomyopathies are unequivocal at autopsy, thus emphasizing the necessity of diagnostic postmortem genetic screening.
The wider implementation of molecular autopsy tools, such as the one presented here, can aid in bringing clarity to these ambiguous molecular pathways and help to identify pathogenic variants. This highlights the need for medical examiners to not only maintain archives of blood samples of SIDS/SUD victims, but also detailed case history records and databases of sequencing results for continuous reassessment as new molecular, biochemical, and functional evidence is discovered and gene candidates are further defined. Agencies need to be cognizant of the ethical implications of molecular genetic testing and reporting. In some cases, disclosure of sensitive genetic results may lead to the discovery of unwanted information, increased insurance burdens, stressful unnecessary medical testing, or altered family planning (Ross et al. 2013; Clarke 2014). Therefore, molecular autopsy programs are best served by the inclusion of experts in biomedical ethics and genetic counselors on review boards. For this study, a consortium of clinical and genetic experts from the Harris County Institute of Forensic Sciences and Baylor College of Medicine collaborated with the Baylor College of Medicine Center for Medical Ethics and Health Policy and genetic counselors to establish guidelines on the disclosure of genetic information (McGuire et al. 2016). The results of these molecular autopsies are envisioned to become part of the normal medicolegal death investigation and will, in some instances, result in amendments of autopsy reports and death certificates.
These results demonstrated a strategy for application of molecular autopsies for public medical examiners’ and coroners’ offices. However, with the unexpected low yield of reportable pathogenic variants, it is conceivable that detection of pathogenic variants may still not be cost-effective in all medicolegal investigations with an undetermined cause of death. This study is limited by its retrospective nature, with an additional goal of determining the yield of reportable pathogenic variants using methods presented here. As expected in a large retrospective cohort, some costs would be amortized. However, as costs continue to fall with advancements in massively parallel sequencing and data mining software, the role of genetic variants in SD will be elucidated further, thus possibly increasing the overall benefit of postmortem genetic screening in medicolegal investigations.
In conclusion, the present study represents the largest heterogeneous cohort of SD cases evaluated by a targeted sequencing panel for genes associated with SIDS and SUD. In addition to being diagnostic in nature, these screenings allow for affected families to make appropriate choices regarding medical testing, treatment, and lifestyle. As the dynamic field of molecular diagnostics advances, molecular autopsy tools have begun to be within reach as a new standard of care for autopsy negative cases.
Methods
Cohort
Autopsy reports of young adults (≤40 yr), children, and infants (≤1 yr) from 2004–2012 at the Harris County Institute of Forensic Sciences were reviewed, and cases classified as “undetermined,” “SIDS,” or “undetermined (cosleeping)” were culled for further analysis. Any case with indicators of non-natural etiology, including cases with any suspicion of death by an inflicted mechanism (e.g., suspicion of intentional suffocation), was excluded. An initial heterogeneous cohort of 429 decedents was defined by medical examiners for postmortem genetic screening. Also included in the cohort were a few cases that had nonspecific anatomic cardiac changes, suggesting a manifestation of an underlying genetic defect (i.e., borderline cardiomyopathy), one case with a listed cause of death as hypertrophic cardiomyopathy, and an additional case with previous medical history suggesting LQTS. These cases were included in order to obtain genetic confirmation and/or specific diagnosis of a disorder in which anatomic findings were equivocal. In addition to the genetic screenings, the demographic composition of the selected cohort was compared to age-matched populations in Harris County, Texas, to identify possible at-risk groups.
DNA extraction
Total DNA was extracted from archived blood spots dried on Whatman bloodstain cards from the selected cohort using a QIAsymphony DSP DNA Midi Kit (Qiagen) according to the manufacturer's protocol. Briefly, bloodstain card cuttings (∼3 cm2) were incubated in 1 mL Buffer ATL (Qiagen) containing 1 mg/mL Proteinase K (Qiagen) at 56°C with shaking at 900 RPM for 8 h. Genomic DNA was isolated from the lysates on a QIAsymphony SP instrument and eluted at 100 µL per sample.
Capture array design and validation
Multiple genes associated with SD due to arrhythmogenic channelopathies and other non-channelopathy disorders were chosen based on literature review and database entries. The 64 selected genes included 22 associated with known cardiac channelopathies, 29 associated with cardiomyopathies, and 13 genes linked to SD without a reported association with a cardiac condition (Table 1). A library of capture array probes (NimbleGen SeqCap EZ Choice Library; Roche) was designed across 94% of the targeted nucleotides (337 kb). The applicable genome coordinates from the human genome build hg19 were obtained from the UCSC Genome Browser (http://genome.ucsc.edu/) and submitted for capture probe design by Roche NimbleGen. The probes were used to create a custom human exonic capture array that was assessed and validated by the Baylor College of Medicine Human Genome Sequencing Center using 24 Coriell HapMap samples. The resulting capture reagent was found to yield 91.2% of targeted bases covered at 20× or better. After validation, the capture array was utilized to enrich the targeted gene regions of the cohort DNA samples in preparation for high-throughput parallel next-generation sequencing.
Sample entry, library preparation, targeted capture, and sequencing
Prior to target capture, DNA samples were tested for quality and quantity by a combination of agarose gel electrophoresis and PicoGreen fluorescence on an Agilent Bioanalyzer 2100. Samples passing minimum standards were used to construct Illumina paired-end precapture libraries according to the manufacturer's protocol (Illumina Multiplexing_SamplePrep_Guide_1005361_D) with modifications as described in the BCM-HGSC protocol (https://www.hgsc.bcm.edu/content/protocols-sequencing-library-construction). BCM-HGSC exome capture methods, adapted from the manufacturer's protocol (NimbleGen SeqCap EZ Exome Library SR User's Guide Version 2.2), were further modified for targeted capture to allow for increased multiplexing in capture and sequencing. Briefly, 1 μg sample DNA was sheared into fragments of approximately 300–400 base pairs with the Covaris E210 system. End-repair, A-tailing, ligation of 9-bp barcode adaptors (24 barcodes in total), precapture ligation-mediated PCR (LM-PCR), as well as the SPRI bead purification (Agencourt AMPure XP beads) was automated on Biomek FXp robotic workstations (Adey et al. 2010). Uniquely barcoded precapture libraries were pooled in equimolar amounts (24 samples/pool, totaling 1 μg/pool) for cocapture. These library pools were then hybridized in solution to the custom NimbleGen capture design in the presence of human COT1 DNA to suppress repetitive genomic sequences. After post-capture LM-PCR amplification and a final SPRI bead purification, pooled samples were loaded on an Illumina MiSeq instrument for cluster formation. Library templates underwent bridge amplification to form clonal clusters, followed by hybridization with the sequencing primer. Sequencing runs were also performed on the Illumina MiSeq platform in paired-end mode, in which sequencing-by-synthesis reactions were extended for 101 cycles from each end, with an additional 10 cycles for the index read. Sequencing runs yielded an average of ∼79.4 Mb per sample and an average of 85.3% of the targeted bases covered to a depth of 20× or greater.
Primary data analysis
Initial sequence analysis was performed using the HGSC Mercury analysis pipeline (Reid et al. 2014). Briefly, the .bcl files produced on-instrument were first transferred into the HGSC analysis infrastructure by the HiSeq Real-time Analysis module and Mercury was then subjected to CASAVA, the primary analysis software, in order to demultiplex pooled samples and generate sequence reads and base-call confidence values (qualities). Reads were then mapped to the GRCh37 Human reference genome (http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/) using the Burrows–Wheeler aligner (Li and Durbin 2009). The resulting BAM (binary alignment/map) file underwent quality recalibration using GATK for BAM sorting, duplicate read marking, and realignment to improve indel discovery (Li et al. 2009; DePristo et al. 2011; Van der Auwera et al. 2013). Finally, BAM files were used by the ATLAS 2 suite to call SNVs and indel variants and to produce VCF files with variant annotation provided by the Cassandra pipeline (Challis et al. 2012).
Variant filtration
Demultiplexed and annotated variants were initially sorted by sequence quality. Variant sequences not meeting a computational “Pass” threshold for quality sequence coverage were excluded. Quality annotated variants were compiled in Excel spreadsheets and culled for possible mutations of significance based on variant type with initial inclusion criteria of nonsynonymous SNVs and indel variants in exon coding regions (Fig. 2). Candidate variants were further assessed by an initial comparison to literature and databases searches via Single Nucleotide Polymorphism Database (dbSNP) (Sherry et al. 2001), Online Mendelian Inheritance in Man database (OMIM) (http://www.ncbi.nlm.nih.gov/omim), and The Inherited Arrhythmias Database (http://www.fsm.it/cardmoc/). All benign, tolerated, common polymorphisms, VUS, and variants with no literature or identified in single individuals were removed from further analysis.
Variants were further assessed by the Medical Genetics Laboratories at Baylor College of Medicine (BCM-MGL), a Clinical Laboratory Improvement Amendments (CLIA)–accredited laboratory, by American College of Medical Genetic and Genomics (ACMG)–certified molecular geneticists (Richards et al. 2008). Briefly, minor allele frequencies for specific ethnic groups were assessed, and the phenotypic overlap between the particular putative pathogenic variant and the associated disease were compared to the phenotype of the decedent by comparison to the Single Nucleotide Polymorphism Database (dbSNP), the Exome Variant Server (http://evs.gs.washington.edu/EVS/), ClinVar, the 1000 Genomes Project Browser, Baylor College of Medicine Medical Genetics Laboratory database, and the Human Gene Mutation Database (McVean et al. 2012; Stenson et al. 2014). In silico tools including Polymorphism Phenotyping v2 (PolyPhen-2), Sorts Intolerant From Tolerant Substitutions tool (SIFT), and Protein Variation Effect Analyzer (PROVEAN) algorithmic tools to predict deleterious effects of mutation-induced structural changes on protein function were also considered during variant evaluation (Ng and Henikoff 2001; Adzhubei et al. 2010; Choi et al. 2012). Variants with phenotypes not consistent with autopsy findings were then eliminated in unequivocal cases. Samples were confirmed by Sanger sequencing, and the inheritance pattern was evaluated. For example, in cases of autosomal recessive disorders, two pathogenic changes needed to be detected in a particular gene. Extensive evaluations of published literature and comparisons to previously reported genotype/phenotype relationships for each sequence-confirmed putative pathogenic variant were carried out to determine the functional significance of the genetic mutation (Landrum et al. 2014). All literature available at the time of review was considered, and variants with minimal conflicting reports did not necessarily exclude the variant from further analysis. For all cases, including those with conflicting interpretations, the strength of the literature from those previous reports was considered.
Clinical genetic variant confirmation
A consortium of medical examiners, physicians, and researchers, including specialists in health policy and ethics, reviewed cases sent for CLIA laboratory evaluation in preparation for reporting genetic findings that contribute to cause of death to next-of-kin and supplement the respective autopsy reports. The genetic analysis, medical history, family history, and all gross anatomical and molecular equivocal and nonequivocal autopsy findings were reviewed. The multidisciplinary board made final diagnostic decisions of the clinical significance of each genetic variant as to cause of death. Variants were classified as (1) pathogenic (most likely related to cause and manner of death); (2) likely pathogenic (significant but not conclusively related to cause of death); or (3) incidental or VUS (unrelated or uncertain clinical significance). Classification was based on the variant evaluation by BCM-MGL, autopsy findings, available personal and family medical history, and terminal circumstances on a case-by-case basis. Pathogenic genetic defects were sequence variants previously reported and recognized as contributing to SD (Richards et al. 2008; Landrum et al. 2014). Incidental or VUS genetic variants were either benign or lacked sufficient evidence to assign pathogenicity at the time of review.
Only variants determined to be pathogenic or likely pathogenic were identified as reportable to families. Next-of-kin to decedents will receive a letter indicating either the genetic change was “likely related to cause of death” (pathogenic) or the genetic change “may or may not be related to cause death” (likely pathogenic). Families will be given a choice to receive further information. If families choose to receive the genetic results, the medical examiner will disseminate the results in the presence of a genetic counselor.
Data access
All nonsynonymous variants are listed in Supplemental Table S2. Confirmed pathogenic genetic variant data from this study have been submitted to the NCBI ClinVar database (http://www.clinvar.com/) under accession numbers SCV000263109–SCV000263122. Sequencing data from this study have been submitted to the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA320727.
Supplementary Material
Acknowledgments
We thank the contributions of the additional members of the Molecular Autopsy Consortium of Houston (MATCH), including Amy L. McGuire, Quianta Moore, Mary Majumder, Salma Nassef, Sandra Darilek, Katie Rutherford, Stacey Pereira, V. Reid Sutton, Art Beaudet, Eric Boerwinkle, L. Maximilian Buja, Thomas Caskey, Sherhonda Harper, Katelyn Weymouth, Albert Chu, Nicholas Lykos, and Thomas Wheeler. We also acknowledge the following BCM-HGSC contributors to the primary data generation and analysis: Jianhong Hu, Christian Buhay, and Jeffrey Reid. This project was supported by Award No. 2010-DN-BX-K230, awarded by the National Institute of Justice, Office of Justice Programs, US Department of Justice. The opinions, findings, and conclusions or recommendations expressed in this publication are those of the authors and do not necessarily reflect those of the Department of Justice.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.195800.115.
References
- Ackerman MJ. 2005. Cardiac causes of sudden unexpected death in children and their relationship to seizures and syncope: genetic testing for cardiac electropathies. Semin Pediatr Neurol 12: 52–58. [DOI] [PubMed] [Google Scholar]
- Ackerman MJ, Splawski I, Makielski JC, Tester DJ, Will ML, Timothy KW, Keating MT, Jones G, Chadha M, Burrow CR, et al. 2004. Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals: implications for arrhythmogenic susceptibility and Brugada/long QT syndrome genetic testing. Heart Rhythm 1: 600–607. [DOI] [PubMed] [Google Scholar]
- Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R, et al. 2011. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace 13: 1077–1109.21810866 [Google Scholar]
- Adey A, Morrison HG, Asan, Xun X, Kitzman JO, Turner EH, Stackhouse B, MacKenzie AP, Caruccio NC, Zhang X, et al. 2010. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol 11: R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai R, Napolitano C, Bloise R, Monteforte N, Priori SG. 2009. Yield of genetic screening in inherited cardiac channelopathies: how to prioritize access to genetic testing. Circ Arrhythm Electrophysiol 2: 6–15. [DOI] [PubMed] [Google Scholar]
- Basso C, Burke M, Fornes P, Gallagher PJ, de Gouveia RH, Sheppard M, Thiene G, van der Wal A, Association for European Cardiovascular Pathology. 2010. Guidelines for autopsy investigation of sudden cardiac death. Pathologica 102: 391–404. [PubMed] [Google Scholar]
- Brion M, Blanco-Verea A, Sobrino B, Santori M, Gil R, Ramos-Luis E, Martinez M, Amigo J, Carracedo A. 2014. Next generation sequencing challenges in the analysis of cardiac sudden death due to arrhythmogenic disorders. Electrophoresis 35: 21–22. [DOI] [PubMed] [Google Scholar]
- Challis D, Yu J, Evani US, Jackson AR, Paithankar S, Coarfa C, Milosavljevic A, Gibbs RA, Yu F. 2012. An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinformatics 13: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. 2012. Predicting the functional effect of amino acid substitutions and indels. PLoS One 7: e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AJ. 2014. Managing the ethical challenges of next-generation sequencing in genomic medicine. Brit Med Bull 111: 17–30. [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorschner MO, Amendola LM, Turner EH, Robertson PD, Shirts BH, Gallego CJ, Bennett RL, Jones KL, Tokita MJ, Bennett JT, et al. 2013. Actionable, pathogenic incidental findings in 1,000 participants’ exomes. Am J Hum Genet 93: 631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudici V, Spanaki A, Hendry J, Mead-Regan S, Field E, Zuccotti GV, Abrams D, Lowe M, Kaski JP. 2014. Sudden arrhythmic death syndrome: diagnostic yield of comprehensive clinical evaluation of pediatric first-degree relatives. Pacing Clin Electrophysiol 37: 1681–1685. [DOI] [PubMed] [Google Scholar]
- Jind L, Elklit A, Christiansen D. 2010. Cognitive schemata and processing among parents bereaved by infant death. J Clin Psychol Med Settings 17: 366–377. [DOI] [PubMed] [Google Scholar]
- Klaver EC, Versluijs GM, Wilders R. 2011. Cardiac ion channel mutations in the sudden infant death syndrome. Int J Cardiol 152: 162–170. [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. 2014. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 42: D980–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberthson RR. 1996. Sudden death from cardiac causes in children and young adults. N Engl J Med 334: 1039–1044. [DOI] [PubMed] [Google Scholar]
- Loporcaro CG, Tester DJ, Maleszewski JJ, Kruisselbrink T, Ackerman MJ. 2014. Confirmation of cause and manner of death via a comprehensive cardiac autopsy including whole exome next-generation sequencing. Arch Pathol Lab Med 138: 1083–1089. [DOI] [PubMed] [Google Scholar]
- Matthews TJ, MacDorman MF. 2013. Infant mortality statistics from the 2010 period linked birth/infant death data set. In Vital health statistics, Vol. 62, pp. 1–26. National Center for Health Statistics, Hyattsville, MD. [PubMed] [Google Scholar]
- McGuire AL, Moore Q, Majumder M, Walkiewicz M, Eng CM, Belmont JW, Nassef S, Darilek S, Rutherford K, Pereira S, et al. 2016. The ethics of conducting molecular autopsies in cases of sudden death in the young. Genome Res (this issue) 26: 1165–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean GA, Donnelly P, Lunter G, Marchini J, Myers S, Gupta-Hinch A, Iqbal Z. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. 2001. Predicting deleterious amino acid substitutions. Genome Res 11: 863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadakis M, Raju H, Behr ER, De Noronha SV, Spath N, Kouloubinis A, Sheppard MN, Sharma S. 2013. Sudden cardiac death with autopsy findings of uncertain significance: potential for erroneous interpretation. Circ Arrhythm Electrophysiol 6: 588–596. [DOI] [PubMed] [Google Scholar]
- Reid JG, Carroll A, Veeraraghavan N, Dahdouli M, Sundquist A, English A, Bainbridge M, White S, Salerno W, Buhay C, et al. 2014. Launching genomics into the cloud: deployment of Mercury, a next generation sequence analysis pipeline. BMC Bioinformatics 15: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, Lyon E, Ward BE; Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee. 2008. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet Med 10: 294–300. [DOI] [PubMed] [Google Scholar]
- Ross LF, Saal HM, David KL, Anderson RR; American Academy of Pediatrics; American College of Medical Genetics and Genomics. 2013. Technical report: ethical and policy issues in genetic testing and screening of children. Genet Med 15: 234–245. [DOI] [PubMed] [Google Scholar]
- Shen WK, Edwards WD, Hammill SC, Bailey KR, Ballard DJ, Gersh BJ. 1995. Sudden unexpected nontraumatic death in 54 young adults: a 30-year population-based study. Am J Cardiol 76: 148–152. [DOI] [PubMed] [Google Scholar]
- Shephard R, Semsarian C. 2009. Advances in the prevention of sudden cardiac death in the young. Ther Adv Cardiovasc Dis 3: 145–155. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29: 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim SH, Ito M, Maher T, Milunsky A. 2005. Gene sequencing in neonates and infants with the long QT syndrome. Genetic Test 9: 281–284. [DOI] [PubMed] [Google Scholar]
- Skinner JR, Duflou JA, Semsarian C. 2008. Reducing sudden death in young people in Australia and New Zealand: the TRAGADY initiative. Med J Aust 189: 539–540. [DOI] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. 2014. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet 133: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Stahl-Herz J, Sampson BA. 2014. Molecular diagnostics of cardiovascular diseases in sudden unexplained death. Cardiovasc Pathol 23: 1–4. [DOI] [PubMed] [Google Scholar]
- Tester DJ, Ackerman MJ. 2006. The role of molecular autopsy in unexplained sudden cardiac death. Curr Opin Cardiol 21: 166–172. [DOI] [PubMed] [Google Scholar]
- Tester DJ, Ackerman MJ. 2009. Cardiomyopathic and channelopathic causes of sudden unexplained death in infants and children. Annu Rev Med 60: 69–84. [DOI] [PubMed] [Google Scholar]
- Tester DJ, Will ML, Haglund CM, Ackerman MJ. 2005. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm 2: 507–517. [DOI] [PubMed] [Google Scholar]
- Tester DJ, Medeiros-Domingo A, Will ML, Haglund CM, Ackerman MJ. 2012. Cardiac channel molecular autopsy: insights from 173 consecutive cases of autopsy-negative sudden unexplained death referred for postmortem genetic testing. Mayo Clin Proc 87: 524–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachtenberg FL, Haas EA, Kinney HC, Stanley C, Krous HF. 2012. Risk factor changes for sudden infant death syndrome after initiation of Back-to-Sleep campaign. Pediatrics 129: 630–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. 2013. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43: 11 10 11–11 10 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Driest SL, Wells QS, Stallings S, Bush WS, Gordon A, Nickerson DA, Kim JH, Crosslin DR, Jarvik GP, Carrell DS, et al. 2016. Association of arrhythmia-related genetic variants with phenotypes documented in electronic medical records. JAMA 315: 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Norstrand DW, Ackerman MJ. 2010. Genomic risk factors in sudden infant death syndrome. Genome Med 2: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Shah KR, Um SY, Eng LS, Zhou B, Lin Y, Mitchell AA, Nicaj L, Prinz M, McDonald TV, et al. 2014. Cardiac channelopathy testing in 274 ethnically diverse sudden unexplained deaths. Forensic Sci Int 237: 90–99. [DOI] [PubMed] [Google Scholar]
- Wong LC, Behr ER. 2014. Sudden unexplained death in infants and children: the role of undiagnosed inherited cardiac conditions. Europace 16: 1706–1713. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.