

Abstract
Chromatin is composed of DNA and histones, which provide a unified platform for regulating DNA-related processes, mostly through their post-translational modification. During DNA replication, histone arrangement is perturbed, first to allow progression of DNA polymerase and then during repackaging of the replicated DNA. To study how DNA replication influences the pattern of histone modification, we followed the cell-cycle dynamics of 10 histone marks in budding yeast. We find that histones deposited on newly replicated DNA are modified at different rates: While some marks appear immediately upon replication (e.g., H4K16ac, H3K4me1), others increase with transcription-dependent delays (e.g., H3K4me3, H3K36me3). Notably, H3K9ac was deposited as a wave preceding the replication fork by ∼5–6 kb. This replication-guided H3K9ac was fully dependent on the acetyltransferase Rtt109, while expression-guided H3K9ac was deposited by Gcn5. Further, topoisomerase depletion intensified H3K9ac in front of the replication fork and in sites where RNA polymerase II was trapped, suggesting supercoiling stresses trigger H3K9 acetylation. Our results assign complementary roles for DNA replication and gene expression in defining the pattern of histone modification.
In eukaryotic cells, DNA is wrapped around histone octamers to form nucleosomes, the basic building blocks of the chromatin structure. This packing presents a unified platform for regulating processes that require DNA accessibility (Gossett and Lieb 2012), including gene transcription and DNA replication (Bannister and Kouzarides 2011). Central to this regulation is the covalent modification of histones by different chemical groups (e.g., acetyl or methyl) at defined sites. These modifications impact the binding affinity of histones to DNA and recruit specific factors that regulate DNA-dependent processes (Unnikrishnan et al. 2010; Rando and Winston 2012).
Histones are modified by regulatory enzymes that are recruited to particular positions either by binding to specific DNA sequences or by recruitment to other DNA-binding proteins (Bannister and Kouzarides 2011; Owen-Hughes and Gkikopoulos 2012). Transcription factors, for example, recruit histone modifiers to gene promoters, thereby regulating gene expression (Morse 2003; Rezai-Zadeh et al. 2003). In addition, modifiers are recruited by the general transcription machinery to modify histones along gene bodies as transcription progresses (Rodríguez-Navarro 2009).
Chromatin is also shaped by DNA replication. First, specific histone modifiers are recruited to the replication machinery to modify histones at replication origins (Li et al. 2008; Unnikrishnan et al. 2010). Furthermore, as replication progresses, histones are ejected and new histones are synthesized for wrapping DNA (Annunziato 2005; Groth et al. 2007; Radman-Livaja et al. 2010, 2011). Newly synthesized histones are acetylated on specific H3 and H4 residues but lack position-specific information (Sobel et al. 1995; Benson et al. 2006; Han et al. 2007a; Corpet and Almouzni 2009). Post-replication modification of these histones either occur immediately or occur with extended delays (Alabert et al. 2015).
The patterns of histone modifications therefore integrate the action of different DNA-related processes, in particular gene expression and DNA replication. For example, H3K4me3 and H3K9ac correlate with gene expression, H3K27me3 is found primarily in repressive regions (Pokholok et al. 2005; Boyer et al. 2006), and H3K56ac is deposited on newly replicated DNA (Li et al. 2008). Some histone marks may be associated with both transcription and replication and also with additional DNA-related processes such as DNA damage or repair (van Attikum and Gasser 2009). Histone modification profiles typically integrate all these effects, making it difficult to discern the contribution of individual processes.
Here, we describe the temporal dynamics of 10 histone marks along the budding yeast cell cycle. Simultaneously measuring changes in histone modifications, gene expression, and DNA replication allowed us to distinguish the individual contributions of transcription and replication to the modification pattern, as well as the interplay between them.
Results
Dynamics of histone modifications along the yeast cell cycle
To follow the temporal changes in histone modifications along the cell cycle, we synchronized cells to the beginning of S phase using hydroxyurea (HU; 3 h) and followed them for 90 min after release. Samples were taken every 10 min for profiling the genome-wide binding patterns of 10 histone modifications (Supplemental Table S1), genomic DNA sequencing, and gene expression (Fig. 1A). The synchronized progression along the cell cycle was verified by the coordinated expression of cell-cycle genes and by the increase in total DNA content (Fig. 1B; Supplemental Fig. S1A,B).
Figure 1.

Cell-cycle dynamics of chromatin marks. (A) Experimental scheme. Yeast cells were synchronized to the beginning of S phase using hydroxyurea (HU), released, and followed as they progressed through the cell cycle. Samples for DNA staining (using FACS), RNA-seq, and ChIP-seq were taken every 5–10 min. (B) Gene expression along the cell cycle. Average log2 changes in the expression of cell-cycle genes (G1, G2/M, and histones) and environmental stress response (ESR) genes at the indicated time-points. (C) Enrichment of histone marks at different positions along genes. Metagene representation of the indicated histone marks. Subsequent time-points are shown from top to bottom. Genes were aligned by their transcription start site (TSS) and transcription termination site (TTS) and are normalized (through binning) to the same length. (D) Modification pattern on all genes. Abundance of H3K4 methylations for all genes (transcript +250 bp from both sides), sorted by gene length and aligned to the middle of the transcript. Black line depicts TSS (left) and TTS (right). Data for each gene were averaged over the entire time-course. For other modifications, see Supplemental Figure S2C. (E) Correlations between changes in gene expression and changes in histone modifications. Changes were calculated between time-points separated by the indicated time lag (Δtime). Correlations are over all time-points with the indicated shift and are confined to genes of the ESR (Gasch et al. 2000), which showed a significant variation. For all individual correlations measured also for all genes, see Supplemental Figures S3 and S1D, and for normalization by different total modification dynamics, see Supplemental Figure S13. (F,G) Replication-associated dynamics of histone modifications. DNA content (top; genomic DNA) and the abundance of the indicated histone marks (bottom; as indicated) along chromosome IV are shown. Each plot depicts the signal along the chromosome (horizontal) at different times (vertical) relative to the average in the entire time-course. Assignment of regions to replication clusters is also shown (see also Supplemental Fig. S5A–C). Dashed line indicates the 40-min time-point when replication ends (see also Supplemental Fig. S5D). Histones marks are shown with or without normalization by DNA content, in G and F, respectively.
Histone marks concentrated at preferred positions along genes (Fig. 1C,D): H3K9ac and H3K4me3 peaked at gene start, while the other trimethylations were observed throughout the coding region, as previously described (Pokholok et al. 2005). These patterns were largely stable throughout the time-course, with the exception of H3K4me2 which shifted toward the transcription start site (TSS). Most patterns were independent of gene length, with few exceptions (e.g., lack of H3K4me1 on short genes) (Supplemental Fig. S2). Notably, while the abundance of H3K9ac, H3K4me3, and H3K36me3 on genes was correlated with gene expression (Pokholok et al. 2005), changes in these modifications showed little correlation with changes in gene expression (Fig. 1E; Supplemental Figs. S1C,D, S3).
Next, we examined the spatial modification patterns by plotting their abundances along chromosomes (x-axis) at subsequent times (y-axis) (Fig. 1F). Triangle-shaped patterns were observed, largely resembling the progression of DNA replication (Fig. 1F, top). Indeed, clustering genes based on similarity in their histone modification dynamics essentially captured gene replication timings (Supplemental Fig. S4). This similarity between histone modification and DNA replication could reflect the deposition of histone modifications simultaneously with replication. Indeed, for some of the modifications, normalizing the histone modification pattern by DNA content (input) diminished the replication-like spatial pattern (e.g., H4K16ac or H3 itself). Other modifications, however, maintained the replication-like pattern even after normalization by DNA content (Fig. 1G). Examining the data more closely suggested that these modifications recovered from DNA replication with some delays. Below, we examine quantitatively these post-replication modification dynamics.
Histone modifications follow DNA replication with variable delays
We defined the time delay between DNA replication and histone modification using two measures. First, we grouped genomic regions into eight clusters based on their time of replication (Fig. 1F; Supplemental Fig. S5A–C). Comparing the cluster-averaged increase of DNA content and histone marks confirmed that some modifications are deposited immediately upon replication, while others recovered with a delay (Fig. 2A; Supplemental Fig. S6). Next, to obtain a measure of this delay, which is independent of cluster assignment, we performed a cross-correlation analysis, defining the time-shift that best aligns the genome-wide increase in DNA content and changes in each modification (Fig. 2B,C).
Figure 2.

Delayed recovery of histone modifications following DNA replication. (A) Replication-associated dynamics of histone marks. Abundances of genomic DNA (black) or histone marks were averaged over all regions assigned to replication cluster six (mid-late replicating) (for more clusters, see Supplemental Fig. S6). Shaded blue area denotes the time period in which this cluster was replicated. Initial decrease in signal resulted from data normalization, reflecting the increase in signal from earlier replicating loci (see Supplemental Note S1). All signals are log2-transformed, normalized by the signal in synchronized culture, and adjusted to show the same dynamic range. (B,C) Delayed post-replication recovery. Correlations were measured between changes in modification levels at time t and changes in DNA content at time t − τ. These cross-correlations are shown as a function of the delay τ (B). Only correlations higher than 0.2 are shown (see also Methods; Supplemental Fig. S7). The typical delay time between DNA replication and the indicated histone marks was defined as the time τ showing the maximal cross-correlation (C).
Histone H3 levels increased concomitantly with the genomic DNA, as expected (Shibahara and Stillman 1999; Li et al. 2008). A similar immediate post-replication increase was observed for H4K16ac and H3K4me1. Interestingly, neither H4K16ac nor H3K4me1 is enriched on newly synthesized histones (Sobel et al. 1995), suggesting their rapid deposition, perhaps through enzymes that are recruited to the replication fork. In contrast, all three trimethylations tested followed DNA replication with a significant delay of ∼20 min. In fact, these marks did not reach their prereplication value even after S phase was completed.
We noted that the heterogeneity (lower correlation) in the post-replication recovery times increased for marks that recovered with a delay. This heterogeneity may suggest a role for replication-independent processes, such as transcription. Indeed, highly expressed genes recovered faster than genes with low expression in four of the six histone marks showing a delayed recovery (Fig. 3). Further, fast recovery was associated with promoters that lack a TATA-box or that are depleted of a nucleosome near their TSS, features that correlate with slow histone turnover (Dion et al. 2007; Tirosh and Barkai 2008).
Figure 3.
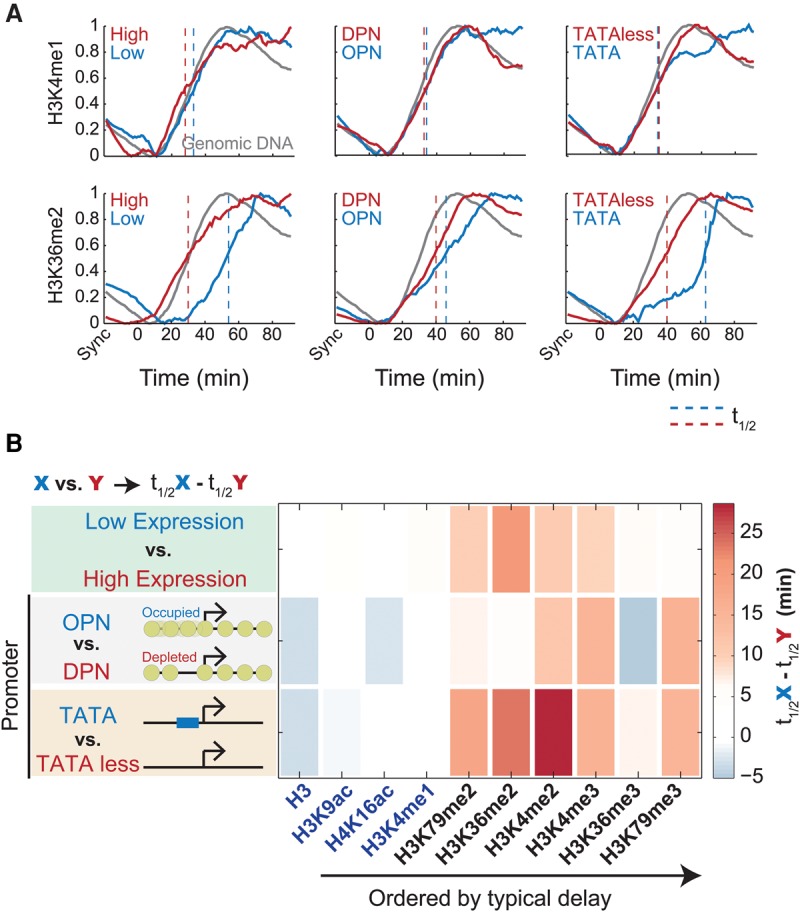
Post-replication delay in histone modifications depends on gene expression and promoter architecture. (A) Replication-associated dynamics of histone marks (as Fig. 2A) calculated separately for genes with the 25% highest or 25% lowest expression levels (left), genes containing occupied or depleted proximal nucleosome (OPN or DPN, respectively) promoter structure (center), or genes containing TATA/TATA-less promoters (right). Genomic DNA was plotted for comparison (gray). Typical half-life (t1/2) was defined as the time to reach half-maximal increase (dashed line), averaged over the three late-replicating clusters (5–7) for all modifications and features, and plotted in B.
A wave of H3K9ac precedes the replication fork
While the majority of modifications appeared simultaneously with DNA replication or after some delay, H3K9 acetylation preceded DNA replication by ∼9 min, being deposited in regions that are about to be replicated (Fig. 2B; Supplemental Fig. S6). To confirm that this prereplication dynamics was not specific to HU synchronization, we profiled cells released from G1 arrest induced by α-factor. Cells were followed for 39 min after release with samples taken every 3 min for profiling H3K9ac, gene expression, and DNA content (Fig. 4A). As a control, we profiled also H3K56ac, which is deposited on newly synthesized histones prior to DNA incorporation (Han et al. 2007b). The DNA replication program inferred from this experiment was essentially the same as that inferred from HU-synchronized cells (Supplemental Fig. S8A,B).
Figure 4.

H3K9 acetylation precedes replication. (A) Experimental scheme and gene expression. Cells were released from α-factor G1-arrest and sampled every 3 min for DNA staining, RNA-seq, and ChIP-seq of H3K9ac and H3K56ac. Gene expression changes are shown, as in Figure 1B. (B) Prereplication increase in H3K9ac. Same as Figure 2A for the indicated clusters (see more clusters in Supplemental Fig. S8C). (C,D) H3K9ac precedes DNA replication. Cross-correlation analysis as in Figure 2B, for H3K9ac and H3K56ac in wild-type cells (C), and in cells carrying the H3K9A or H3K56A histone variants (D). See replication progression in Supplemental Figure S8, F through H. (E) Prereplication increase in H3K9ac along 55 kb of chromosome V. Same as Figure 1F for wild-type cells released from α-factor synchronization. Data were normalized by the synchronized time-point and were plotted up to 21 min (for the full time-course, see Supplemental Fig. S8D). Autonomously replicating regions (ARSs) are indicated (top). Three 2-kb regions (I–III, marked by rectangles) were averaged for both H3K9ac and DNA content in each time-point and plotted together for comparison.
Cross-correlation analysis confirmed that also in this experiment, H3K9ac preceded replication and decreased following passing of the replication fork (Fig. 4B,C,E; Supplemental Fig. S8C,D). In these conditions, H3K9ac was observed ∼5 min before replication compared with the 9 min observed when releasing cells from HU arrest, likely reflecting faster replication. In contrast, H3K56ac, which is deposited on newly synthesized histones, increased practically together with the genomic DNA (Fillingham et al. 2008; Li et al. 2008).
To control for possible cross-reactivity of the H3K56ac and H3K9ac antibodies (Drogaris et al. 2012), we repeated our experiment in strains harboring mutated H3, incapable of being either K9- or K56-acetylated (depicted H3K9A, and H3K56A, respectively) (Dai et al. 2008). Indeed, in H3K9A mutant, H3K56ac increased concomitantly with the DNA content, while in the H3K56A mutant the prereplication induction of H3K9ac was maintained (Fig. 4D; Supplemental Fig. S8E–H).
Distinct roles of Rtt109 and Gcn5 in H3K9 acetylation in DNA replication and gene transcription
The prereplication pattern of H3K9ac described here complements the previously reported correlation of H3K9ac with gene expression (cf. Supplemental Fig. S1C; Pokholok et al. 2005). H3K9ac acetylation is catalyzed by two acetyltransferases, Gcn5 and Rtt109 (Fillingham et al. 2008). To distinguish which enzyme is required for the replication-associated H3K9ac, we repeated our experiment in cells individually deleted of either Rtt109 or Gcn5. Neither mutant detectably affected the replication program or S-phase duration (Supplemental Fig. S9A–C; Voichek et al. 2016).
Deletion of Rtt109 completely abolished the prereplication increase in H3K9ac, which now followed replication with ∼4-min delay (Fig. 5A; Supplemental Fig. S12). Deletion of Gcn5, on the other hand, strengthened the association of H3K9ac with prereplicated regions. Thus, Rtt109, but not Gcn5, is required for the replication-associated wave of H3K9ac.
Figure 5.
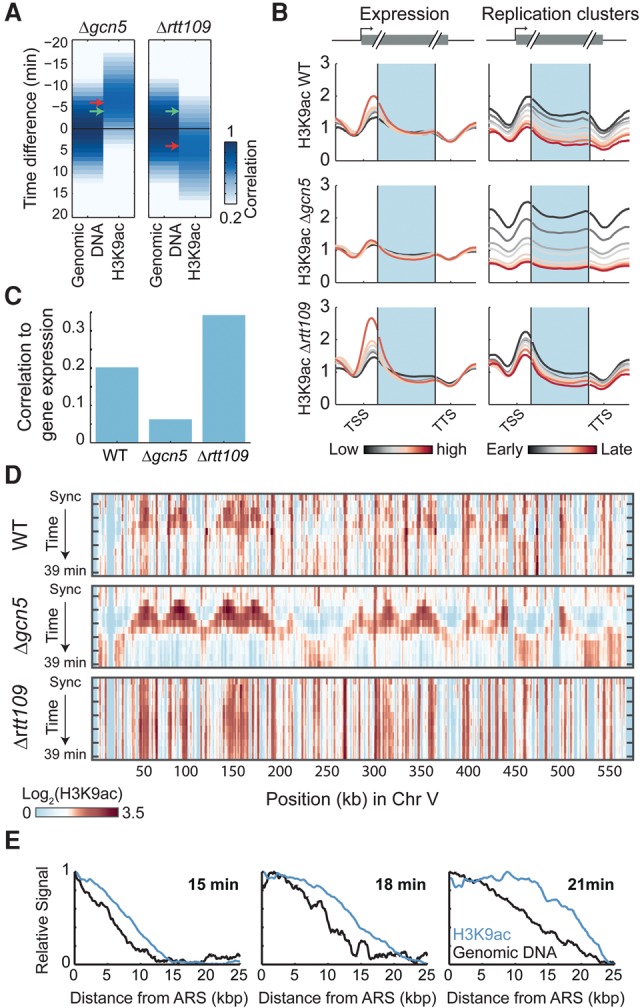
Replication-guided H3K9ac depends on Rtt109, while deletion of Gcn5 abolishes the expression-associated H3K9ac. (A) Prereplication H3K9ac depends on Rtt109. Same as Figure 2B for the indicated mutants. Arrows indicate the typical delay in the WT (green) or in the deletion strain (red). For H3K56ac in Δgcn5, see Supplemental Figure S9D. (B) Metagene pattern of H3K9ac in cells deleted of Gcn5 or Rtt109. Genes were separated into six groups based on their expression levels (left) or based on their replication clusters (right). The corresponding metagene patterns (cf. Fig. 1C) of H3K9ac in synchronized culture or in the middle of S phase (18 min) are shown, respectively. (C) Correlation between H3K9ac abundance and gene expression is lost in cells deleted of Gcn5. Correlations of gene expression to H3K9ac for the indicated strains at the synchronized time-point. Similar results were obtained at all other time-points. (D) The pattern of H3K9ac in wild type combines the replication- and transcription-associated patterns. H3K9ac (raw signal) in the indicated strains is plotted along chromosome V. (E) Spatial spreading of H3K9ac ahead of replication: All early ARS (<21 min in Yabuki et al. 2002) were aligned, and the average H3K9ac and genomic DNA signals were plotted over a 25-kb distance. Signal spreading is shown for three consecutive time-points in the gcn5-deleted strain and normalized by the synchronized time-point (Methods; see Supplemental Fig. S10 for H3K56ac).
Highly expressed genes show an elevated H3K9ac specifically at gene start sites, while H3K9ac in prereplicated regions increases throughout the gene bodies (Fig. 5B). Notably, deleting Gcn5 practically abolished the correlations between H3K9ac and gene expression (Fig. 5B,C) and diminished the characteristic H3K9ac peak at the gene 5′ end (Fig. 5B; Supplemental Fig. S9E). When the chromosome-wide H3K9ac signal is compared, the wild-type pattern appeared as a combination of the Rtt109-associated replication pattern and the Gcn5-associated expression pattern (Fig. 5D,E). Together, we conclude that Rtt109 and Gcn5 play distinct roles in catalyzing replication- and expression-associated H3K9 acetylation.
Topoisomerase depletion intensifies H3K9 acetylation
The prereplication wave of H3K9ac may be linked to the supercoiling stresses that accumulate in front of the replication fork. These stresses are relieved by DNA topoisomerases (Wang 2002). Notably, early studies have shown that topoisomerases relieve transcription inhibition caused by positive supercoiling, and implicated histone acetylation in this process (Krajewski and Luchnik 1991; Gartenberg and Wang 1992). We therefore profiled H3K9ac during replication also in strains depleted of topoisomerases (Wang 2002). In yeast, Top2 is the main enzyme functioning during replication, yet a top2-ts strain had no effect on DNA replication (Baxter and Diffley 2008) and maintained the normal H3K9ac pattern (Fig. 6E). We therefore analyzed a strain that harbors the temperature-sensitive top2-ts allele combined with top1 deletion. Cells were grown in the permissive temperature, arrested by α-factor for 2.5 h, and shifted to the restrictive temperature for half an hour before release from G1 arrest (Fig. 6A,B; Supplemental Fig. S11A).
Figure 6.

Topoisomerases depletion intensifies the prereplication H3K9ac. (A) Experimental scheme. Cells were grown in the permissive temperature (24°C), synchronized using α-factor, and 30 min before release transferred to the restrictive temperature (34°C). Cells were released from G1 arrest and sampled every 3 min for DNA staining, RNA-seq, and ChIP-seq of H3K9ac. Binding of RNA polymerase II was measured using ChIP-seq in the synchronized time-point. Gene expression changes of cell-cycle-regulated genes are shown in Supplemental Figure S11A. (B) S-phase progression. Flow cytometry analysis of DNA-stained cells collected at the indicated time-points. (C) Topoisomerase depletion arrests DNA replication. Same as Figure 1F (top) for the indicated strains on chromosome VII. Dashed lines denote confirmed replication origins (based on OriDB) that intersect with regions that were replicated in the double mutant (Nieduszynski et al. 2007). A high-resolution view of a 30-kb region is shown in the inset, together with the corresponding H3K9ac profile. Top panel represents genes. (Blue) Watson; (green) Crick. Dashed lines in inset indicate the replicated region and the replication origin. For all chromosomes, see Supplemental Figure S11B. (D) Topoisomerase depletion represses gene expression in replicated regions. Log2 changes in gene expression, relative to synchronized cells, averaged over all genes (∼400) positioned in regions that were replicated in the topoisomerase-depleted strain. (E) H3K9ac on chromosome VII. H3K9ac normalized by the synchronized time-point. The inset is a blow-up of the indicated region (also marked in C). (F) H3K9ac in replicated regions. All replicated regions in top2-tsΔtop1 were ordered according to their length. DNA abundance (left) and the H3K9ac levels (right) at these regions, 45 min after release, normalized by the synchronized time-point, are plotted. Dashed lines depict the edges of the replicated region. (G) H3K9ac precedes replication. Same as Figure 4B for one of the two clusters replicated in top2-tsΔtop1. For the second cluster, see Supplemental Figure S11C.
Replication of early origins initiated with some delay. Regions of ∼5–20 kb around early origins were replicated before replication arrested (Fig. 6C, bottom panel and inset; Supplemental Fig. S11B). Gene expression in these replicated regions was specifically reduced, consistent with the accumulation of supercoiling (Fig. 6D). Notably, also here, H3K9ac preceded replication and was observed at ∼5–6 kb in front of the fork (Fig. 6E–G; Supplemental Fig. S11C). The spread of H3K9ac stopped upon replication arrest, and it did not progress further into unreplicated regions. Therefore, the progression of H3K9ac in front of the replication fork depends on active replication or on other processes affected by the torsional stress. Notably, in contrast to the transient H3K9ac wave in wild-type cells, where replicated regions were deacetylated following the passing of the fork, H3K9ac remained stable in topoisomerase-depleted cells (Fig. 6E, bottom panel). This may indicate continuous acetylation, triggered by supercoiling, or an inability of histone deacetylases to access the supercoiled regions.
Topoisomerase depletion increases the association between H3K9ac and RNA polymerase II
Depletion of topoisomerase affected H3K9ac also outside S phase (Fig. 7A,C). As discussed above, replication-independent H3K9ac localizes to the TSS and correlates with gene expression (see Fig. 1). In contrast, upon topoisomerase depletion, H3K9ac was shifted downstream from the TSS and its correlation with gene expression was reduced (Fig. 7D,E).
Figure 7.

Replication-independent effects of topoisomerase depletion on H3K9ac and RNA polymerase II. (A,B) Colocalization of RNA polymerase II with replication-independent H3K9ac peaks. H3K9ac levels (A) and RNA pol II binding (B) in synchronized cells. Dashed lines highlight peaks of H3K9ac in the top2-tsΔtop1 strain. Gene positions are depicted in the center, as in Figure 6C (inset). (C) Topoisomerase depletion modifies the genome-wide H3K9ac pattern. The genome-wide correlations between H3K9ac in synchronized cells of the indicated strains. (D) Shift in H3K9ac upon depletion of topoisomerases. Metagene analysis of H3K9ac (as in Fig. 1C) for the indicated strains. (E) Shift in H3K9ac is associated with gene expression levels. Metagene analysis of H3K9ac for genes grouped according to their expression levels (as in Fig. 5B). Expression levels as measured in the relevant strain at the synchronized time-point. Correlation values (ρ) between gene average H3K9ac and gene expression levels are indicated. (F) H3K9ac colocalizes with RNA pol II. Peaks in H3K9ac were identified, the respective regions were aligned by the maximal H3K9ac signal, and the average RNA pol II measured over the aligned regions was plotted.
In addition to their role in replication, topoisomerases are required for relieving supercoiling stresses during transcription (Mondal 2003; Chong et al. 2014). We reasoned that in topoisomerase-depleted cells, RNA polymerase II may be trapped in supercoiled regions. We therefore profiled Pol II binding in wild-type and topoisomerase-depleted cells synchronized at G1. Indeed, Pol II binding was consistently modified by topoisomerase depletion, becoming highly concentrated at specific sites along genes. Further, Pol II binding became tightly correlated to H3K9ac (Fig. 7B,F). Together, our results suggest that H3K9ac and Pol II trapping are elevated in supercoiled regions, normally relieved by topoisomerases.
Discussion
DNA replication displaces nucleosomes and perturbs the pattern of histone modification. Our study reveals that gene transcription plays an important role in the post-replication recovery of histone marks. It further suggests a prereplication wave of histone modification, which may be guided by the supercoiling stresses developing in front of the replication fork.
Time resolved data enabled us to define the typical delays between DNA replication and associated histone modifications. Immediate retrieval of position-dependent modifications may suggest epigenetic mechanisms that rely on existing marks. Delayed recovery, on the other hand, is consistent with passive retrieval through replication-independent processes. Some histone marks appeared on newly replicated DNA practically immediately. This was expected for modifications such as H3K56ac, which is deposited on newly synthesized histones prior to DNA incorporation (Li et al. 2008), but was also observed for H3K4me1 and H4K16ac, which are not enriched on newly synthesized histones (Sobel et al. 1995). These modifications appear to be added uniformly concomitantly with, or immediately after, replication.
Newly deposited histones were di- or trimethylated rather slowly following replication, resembling the delayed kinetics reported for other methylations (Alabert et al. 2015). This delay was significantly shorter for highly expressed genes and for genes associated with slow histone turnover. These modifications are therefore added primarily during gene expression, with active epigenetic inheritance contributing little to their post-replication recovery.
Similar to histone trimethylations, H3K9ac is deposited on highly expressed genes. However, in contrast to all the other modifications, H3K9ac preceded replication, being deposited ∼5–6 kb ahead of the replication fork. The two acetyltransferases, Gcn5 and Rtt109, played distinct roles in establishing the H3K9ac pattern: While the first catalyzed expression-dependent H3K9ac, the second was dedicated to the newly described replication-associated H3K9ac. Topoisomerase depletion modified H3K9ac by intensifying its abundance ahead of the replication fork and also by inducing replication-independent H3K9ac peaks in positions that trapped RNA polymerase. Together, our results suggest that supercoiling stresses such as the ones developing in front of the replication fork trigger H3K9ac, either by recruiting acetyltransferases or by promoting the incorporation of H3K9ac-containing histones.
Further studies are needed to define the function of H3K9ac during replication. Deletion of Rtt109 or mutating H3K9 did not perturb S-phase progression. This mutant was previously shown to increase genomic instability (Driscoll et al. 2007), although this effect was attributed to the loss of H3K56ac, which also controls expression homeostasis during S phase (Voichek et al. 2016). The prereplication H3K9ac wave may contribute to the smooth progression of the replication machinery, perhaps by recruiting or stabilizing nucleosome remodeling complexes.
Methods
Strains
Supplemental Table S2 summarizes all yeast strains described in this study. For ChIP-seq experiments in wild-type Saccharomyces cerevisiae, a BY4741 strain with C-terminal tagged Rpd3 and Whi5 was used. Rpd3 was tagged with nine repetitions of Myc peptide by transforming with a PCR fragment amplified from pFA6-9Myc-His3MX6 (Longtine et al. 1998) with primers 5′-TGCGAGGGACCTACATGTTGAGCATGACAATGAATTCTATGGTGGGGGCGGTGGTGGGTTAATTAACGGTGAACA and 5′-TCACATTATTTATATTCGTATATACTTCCAACTCTTTTTTAATCGACAGCAGTATAGCGA and selection on plates lacking histidine. Whi5 was tagged with three repetitions of HA peptide by transforming with a PCR fragment amplified from pYM24 (Janke et al. 2004) with primers 5′-ACGGACGAAACGGAGCCCGAGTCGGATACCGAAGTGGAGACGTCTGGCGGAGGTGGCGGAGGTGGAATCTTTTACCCATACGA and 5′-CTGCACTAACTCCGAGATTGCGGAGAAAAAACTCGTACTACCACATCCGCGGCCGCA and selection on plates with Hygromycin B and lacking histidine.
BY4741 strain was deleted of RTT109 or GCN5 by transforming with a PCR fragment amplified from pBS7 (Yeast Resource Center) with primers 5′-GTCACAAAACATCAGATTGAAGAGGATCACTTGGATAGATCTGTTTAGCTTGCCTCG and 5′-TTAATCAATAAGGTGAGAATATTCAGGTATTTCTTTTATCGATGAATTCGAGCTCG for GCN5 and 5′-TCACTGAATGACTTCCTAAGTTCCGTGCTACCTGTCAGATCTGTTTAGCTTGCCTCG and 5′-TCAAGTTTTAGGCAAGGCTTTAGCTTTTTTACGCGGTTTTAGCATCGATGAATTCGAGCTCG for RTT109 and selection on YPD+G418 plates.
Cell-cycle synchronization: HU synchronization
Yeast cells were synchronized to S phase using HU (NBS Biologicals, HB0528) as previously described (Slater 1973; Futcher 1999; Walker 1999). Briefly, cells were grown in YPD overnight at 30°C and inoculated in fresh medium to OD600 of 0.01. When reaching an OD600 of 0.1, HU was added to a final concentration of 0.2 M and grown for additional 3 h. Then, the culture was split to four vacuum-filtering bottles (0.45 µm), with an extra removable filter on top of them. Yeast stuck to the filter and then resuspended in fresh and warm media. Samples were washed again from the HU by centrifugation (4000 rpm, 2 min) and resuspended in warm, fresh YPD. Cells were collected at the following time-points: after 3 h of arrest (sync), 5 min after release, and every 10 min after release for 90 min. Due to the large volumes required for ChIP-seq, the HU experiment was split into eight experiments, done on different days and serving as biological replicates. In each experiment, two large quantities of culture (1 L) were fixated for ChIP. The pairs of ChIP-seq samples was as follows: time-points a, 10 and 80 min; b, 20 and 70 min; c, 40 and 90 min; d, 30 and 90 min; e, 5 and 60 min; f, 0 and 5 min; g, 0 min; and h, synchronized. An experiment of 50 min after release was excluded from the analysis.
In order to ensure different experiments are synchronized similarly, samples for DNA staining and RNA sequencing in all the time-points of each experiment (except for experiments g and h) were obtained.
Cell-cycle synchronization: α-factor synchronization
Synchronization using α-factor was done as previously described (Futcher 1999). Briefly, cells were grown in YPD overnight at 30°C and inoculated in fresh medium to OD600 of 0.05. When reaching an OD600 of 0.12, cells were washed from the media using 0.45-µm filters in order to remove secreted Bar1. Cells were then resuspended in an equal volume of fresh warm YPD with α-factor to a final concentration of 5 µg/mL. Next, the yeast culture was divided into 12 (wild-type experiment, H3K9A, and H3K56A) or 15 (HATs deletion experiment) separate 50-mL tubes with a ventilated cap (CELLSTAR CELLreactor filter top tube, Greiner Bio-One, 227245), each containing 35 mL of yeast culture. Each tube contained the material that will be used for ChIP-seq in a single time-point and DNA staining for flow cytometry. Cells were incubated for 3 h at 30°C with α-factor in an incubator and then transferred to a water bath orbital shaker 30 min before the end of synchronization (MRC, WBT-450). Every 3 min for 39 min, one tube was taken out of the bath orbital shaker and washed twice from α-factor by centrifugation (4000 rpm, 1 min) and resuspension in fresh, warm YPD. Following two washes, cells were resuspended in an equal volume of fresh, warm YPD and returned to the bath shaker to grow at 30°C. The first sample returned to the bath shaker is the last sample in the time-course (first released–last time-point in time-course). The second-to-last sample in the time-course was released and immediately fixated, termed the “0 minutes” sample. The last sample in the time-course was not released from α-factor and was termed “synchronized.” For the wild-type strain, samples were taken at time 0 and, after 9 min, every 3 min. Following release from synchronization, 5 mL of each sample was aliquoted to a different tube to be used for RNA sequencing.
For measuring replication progression of H3K9A and its corresponding wild type (W303 from Boeke library) (Supplemental Fig. S8), strains were synchronized using α-factor in a single Erlenmeyer and released by two washes with warm YPD, as described above. Each strain was released into a single Erlenmeyer shaking in a water bath orbital shaker (MRC, WBT-450). Samples for DNA staining were taken every 2 min (for 70 min) and fixated with 70% ethanol on ice.
For W303, top2-ts, and top2-tsΔtop1, α-factor synchronization was done with slight modifications: Cells were grown in YPD overnight at 24°C (permissive temperature) and were inoculated to a fresh medium to OD600 of 0.02. When an OD600 of 0.2 was reached, cells were washed from the media using 0.45-µm filters in order to remove secreted Bar1 in the media. Cells were then resuspended in equal-volume fresh YPD prewarmed to 24°C, with α-factor in a final concentration of 5 µg/mL. Next, the yeast culture was divided into 15 separate 50-mL tubes with a ventilated cap as in the above-mentioned experiments. Cells were incubated for 2.5 h at 24°C and then switched to 34°C for an additional 30 min. Every 3 min for 45 min, one tube was taken out of the incubator and washed twice from the α-factor by centrifugation (4000 rpm for 1 min) and resuspension in fresh YPD prewarmed to 34°C. Following the two washes, the cells were resuspended in an equal volume of fresh YPD prewarmed to 34°C and returned to the incubator to grow at 34°C (restrictive temperature). Subsequent steps were done as in other strains, with an arrested sample (sync), immediately after release (time-point 0), and samples every 3 min starting at 6 min after release for 39 min (in total, 45 min after release). Samples for ChIP-seq of RNA pol II were taken at the synchronized time-point in the same experiment.
ChIP-seq
Chromatin immunoprecipitation was done as described previously (Liu et al. 2005), with modifications. In experiments that used HU, for each antibody, 50 mL of cells were used. In total, 1 L of yeast culture (equivalent to 20 antibodies) was cross-linked at the relevant time-point with 1% formaldehyde for 5 min in 30°C at 90 rpm shaking. Cross-linking was stopped by incubating cells with 125 mM glycine for 5 min at RT. Next, cells were washed twice with cold sterile water, frozen in liquid nitrogen, and moved to −80°C. Cells were resuspended in 1× Lysis buffer (Lysis buffer: 50 mM HEPES-KOH at pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate) with protease inhibitor cocktail (1:1000, Sigma-Aldrich, P8340). For every sample originating from 200 mL of yeast, 400 µL of zirconium oxide beads (0.5 mm) was added. Cells were then mechanically disrupted using a BBX24 bullet blender (Next Advance) for 1 min at intensity 8. Subsequently, cells were sonicated in a Branson digital sonifier S-250D (courtesy of Jacob Hanna's laboratory) for 10 min at 35% amplitude, in pulses of 0.7 sec on, 1.3 sec off. Cells were centrifuged twice to remove cell pellets. From each time-point, 50 µL of DNA was taken out to serve as input. To the remaining sample, 1 mg/mL BSA and NaCl to a final concentration of 275 mM were added. Sonicated DNA material was then precleared using magnetic beads (Dynabeads Protein G, Life Technologies) for 1 h in a 4°C tumbler. The remaining sonicated material was pooled together, and 300 µL of sonicated material was aliquoted to tubes containing Protein G magnetic beads bound to 10 µg of specific antibodies (for a list of antibodies, see Supplemental Table S1), and incubated in a tumbler at 4°C overnight.
After overnight incubation with antibodies, beads were washed five times with cold lysis buffer, twice with cold Buffer W1 (50 mM HEPES-KOH at pH 7.5, 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate), twice with cold Buffer W2 (10 mM Tris-HCl at pH 8.0, 250 mM LiCl, 0.5% NP-40, 0.5% sodium deoxycholate, 1 mM EDTA), and once with cold TE buffer (10 mM Tris-HCl at pH 8.0, 1 mM EDTA). Reversal of cross-linked was done by incubating beads in direct elution buffer (10 mM Tris-HCl at pH 8.0, 1 mM EDTA, 1% SDS, 150 mM NaCl, 5 mM DTT) overnight in 65°C. RNA was degraded using RNase A treatment for 60 min in 37°C. Proteins were degraded by incubating samples for 2 h in 37°C with Invitrogen Proteinase K in the presence of glycogen. ChIP libraries were indexed (Garber et al. 2012), pooled, and sequenced on Illumina HiSeq 2000.
ChIP-seq protocol for α-factor experiments was modified: For each time-point, 35 mL of yeast were used for ChIP-seq, with chromatin from 10 mL of culture used per antibody. Tubes were taken out of the bath shaker and fixated at the same time, as each sample has been released at a different time, in 3-min intervals. The cells were fixated and processed further with the same protocol as the HU experiments.
For details about addition of Schizosaccharomyces pombe cells to samples, for normalizing ChIP-seq data, see Supplemental Note S2.
RNA extraction and sequencing
Yeast culture was centrifuged at 4000 rpm for 1 min, supernatant was removed, and pellets were immediately frozen in liquid nitrogen. RNA was extracted using a modified protocol of the Nucleospin 96 RNA kit (Machrey-Nagel, 740709). Specifically, cell lysis was done in a 96 deep-well plate by adding 450 µL of lysis buffer containing 1 M sorbitol (Sigma-Aldrich), 100 mM EDTA, and 0.45 µL lyticase (10 IU/µL). The plate was incubated in 30°C for 30 min to break the cell wall and then centrifuged for 10 min at 3000 rpm, and supernatant was removed. From this stage, extraction proceeded as in the protocol of the Nucleospin 96 RNA kit, only substituting β-mercaptoethanol with DTT. Fragmented, poly(A)-selected, RNA extracts of size ∼200 bp were reverse-transcribed to cDNA using barcoded poly(T) primers. cDNA was amplified and sequenced with an Illumina HiSeq 2500 using a primer complementary to the opposite adaptor to the poly(A).
Flow cytometry
To assess cell-cycle synchronization efficiency and position along the cell cycle, we followed DNA staining of samples from every time-point using flow cytometry. Briefly, cells were washed twice with 50 mM Tris-HCl (pH 8), resuspended in RNase A for 40 min in 37°C, washed twice with 50 mM Tris-HCl (pH 8), and resuspended in Proteinase K for 1 h incubation at 37°C. Then, cells were washed twice again, resuspended in SYBR green (S9430, Sigma-Aldrich; 1:1000), and incubated in the dark at room temperature for 1 h. Then, cells were washed from the stain and resuspended in 50 mM Tris-HCl (pH 8) and sonicated in the Diagenode Bioruptor for three cycles of 10 sec on and 20 sec off in low intensity. Finally, cells were taken to FACS for analysis using the BD LSRII system (BD Biosciences).
Processing and analysis of RNA-seq data
For every RNA-seq sample we mapped the 50-bp/60-bp sequences to the S. cerevisiae genome (SGD, R64) using Bowtie (parameters: –best –a –m 2 –strata –5 10), with an average of 85% sequence alignment (Langmead et al. 2009). We then filtered the mapped reads for reads not mapped to rRNA and down-sampled the aligned filtered reads to 400,000 reads. Down-sampling the reads was done in order to have similar data from all samples. For every sequence, we normalized for PCR bias using the unique molecular identifier (UMI), scoring each position on the genome by the unique number of UMIs it had out of the 256 possible UMIs (Kivioja et al. 2012).
To get the expression of each gene, we summed all the reads aligned to 400 bp upstream of its 3′ end to 200 bp downstream. For genes having high sequence similarity in the region being summed and thus having sequences aligning as good to two genes, we separated the reads aligned to both genes according to the amount of uniquely mapped sequences: for example, if genes X and Y had 300 sequences aligning to both 3′ end regions, and 500, 1000 uniquely mapped to X, Y respectively. Then, the reads aligned twice will be divided in the same ratio (500/1500) × 300 versus (1000/1500) × 300. The final expression will be 600 for X and 1200 for Y.
For the HU experiments, we averaged the different biological replicates of the time-course to get one data set to work with.
Average changes in gene expression
Expression at each time-point was divided by the expression in the synchronized time-point, log2-transformed, and then averaged on the group of genes (for Figs. 1B, 4A, 6D; Supplemental Figs. S1A, S7A, S8E, S9B, S11A; and the clusters in Supplemental Fig. S4).
Processing and analysis of ChIP-seq and genomic DNA data
Genomic tracks representing the enrichment of every locus in the yeast genome were calculated for all ChIP-seq samples. The process was the same for the genomic DNA (input) and ChIP-seq samples, differing only for data of 50-bp paired-end sequencing (HU time-courses and WT α-factor time-courses) and data of 50-bp single-read sequencing (all other α-factor time-courses).
For paired-end data, reads were aligned using Bowtie (parameters: –m 1 –best –X 1000) to a genome containing both S. cerevisiae (SGD, R64) and S. pombe genomes (Supplemental Note S2). Genomic tracks for each sample were calculated by taking each location covered by a full read (including the region between the pairs) and adding +1 to these loci in total, so that every position increased by 1/(insert size).
For single-read data, reads were aligned using Bowtie (parameters: –best –m 1) to the combined S. cerevisiae and S. pombe genome (Supplemental Note S2). Genomic tracks were calculated by extending every aligned read to cover 200 bp and adding +1 to each covered location.
All tracks were normalized to have a total signal of 1,000,000. Enrichment level for each gene was calculated by averaging the signal over its coding region. In the HU time-course, we did not use samples having fewer than 500,000 reads.
Normalizing the log2-transformed bins of one time-point A by another time-point B was done using a linear fit: fitting B to A and taking the difference from the fitted line.
Normalization in Figure 1, F and G, was done by log2-transforming the 10-kb bin average and then subtracting the average signal of the bin along the time-course.
Time-points where there are two biological replicates were averaged in the level of the measurement used (e.g., bins, gene average), with the exception of analysis where extrapolation (using cubic spline) to a finer time-course was used. In those cases, the two biological replicates were entered as two measurements from the same time.
Metagene analysis
For a profile of a specific histone modification (time-course and a specific time-point) and a group of genes, metagene analysis was done as follows: taking the signal 400 bp upstream of the TSS to 400 bp downstream from the transcription termination site (TTS) for every gene in this profile (Pelechano et al. 2013). The signal found between the TSS+200 bp and the TTS−200 bp was binned into 20 equal-sized bins to be able to compare genes of different lengths (the binned area was then extended in size for visualization purposes). Signal for all genes was then averaged to get the average pattern.
Gene replication time for genes or 10-kb bins
Genome-wide replication timing data from Yabuki et al. (2002) was used to define gene replication time by assigning each gene the replication time closest to its 5′ end. For each 10-kb bin (Supplemental Fig. S4B), replication time was assigned as the average data points intersecting with the bin from Yabuki et al. (2002) data.
Plotting histone modifications for all genes
Plotting the modifications of all genes aligned to their middle (Supplemental Figs. S1D, S2C) or to the TSS (Supplemental Fig. S2D) was done by first sorting the genes according to their length and averaging the signal over all time-points. Then a small spatial filter (50 × 50 Gaussian with σ = 25) was used on this matrix.
Quantification of the time delays between DNA replication and histone modification
Correlations of changes in histone modifications to changes in genomic DNA are quantified as follows: First we extrapolated the temporal changes of a modification and genomic DNA to a 1-min resolution, using cubic spline. We then calculated the changes around each time t (±5 in HU, or ±2 in α-factor). For every time delay τ, we calculated the correlation of the modification changes in time t and changes in DNA content at time t − τ, for time-points for which t − τ is in S phase.
Normalization of genomic DNA profile to get absolute level
As each profile is normalized to the same total level, as explained in Supplemental Note S1, regions not replicated at a given time will show a decrease in their genomic DNA level due to replication of other regions. To normalize this effect in the genomic DNA data, we used the fact that DNA level can only increase during replication. Thus, a decrease in DNA can be corrected to no change (and thus a stronger increase in DNA content in replicated regions). We do so both for visualization purposes (Supplemental Fig. S5A) and also for more accurate calculation of the delays in the modification recovery (e.g., Fig. 2B).
For a log-binned genomic DNA profile along time, we clustered the bins according to their temporal changes in DNA content (as in Fig. 1F, “replication clusters” or Supplemental Fig. S5C). To avoid noise, we averaged the temporal changes in every cluster containing at least 50 bins. For each sequential time-point, we checked if at least one of the average clusters' value decreased; if so, we added a value to the later time-point so that the larger decrease will become the same value. The added value is an approximation of the absolute total DNA increase between the two time-points. The added values are added to all the bins in this time-point, creating a normalized genomic-DNA profile.
Groups of genes used in the enrichment analysis
For the enrichment analysis (Supplemental Fig. S4), we took the following coexpressed groups of genes from (1) the environmental stress response (ESR) from Gasch et al. (2000), (2) groups from module level 13 in Ihmels et al. (2004), and targets of different transcription factors from MacIsaac et al. (2006). For enrichment of genes replicating at different times, we separated the genes to six groups according to their time of replication (Yabuki et al. 2002).
Groups of genes used to plot the progression through the cell-cycle
“G2/M genes” and “G1 genes” are taken from Ihmels et al. (2004). ESR induce/reduce are taken from Gasch et al. (2000), and “Histones” contain the eight histone genes (e.g., Figs. 1B, 4A).
Identification of H3K9ac peaks
Peak calling was done with PeakFinder (https://www.mathworks.com/matlabcentral/fileexchange/25500) using the parameter sel = 1 (defining the threshold for calling a peak).
Data access
RNA-seq, ChIP-seq, and DNA-seq data from this study have been submitted to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/) under accession numbers PRJEB11501, PRJEB13262, and PRJEB11977.
Supplementary Material
Acknowledgments
We thank Ido Amit, Diego Jaitin, and Dalia Rosin for technical assistance and Anni Hangaard Andersen and Jim Karagiannis for yeast strains. This study was supported by the ERC and the Israel Science Foundation (ISF).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.201244.115.
References
- Alabert C, Barth TK, Reverón-Gómez N, Sidoli S, Schmidt A, Jensen ON, Imhof A, Groth A. 2015. Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev 29: 585–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziato AT. 2005. Split decision: What happens to nucleosomes during DNA replication? J Biol Chem 280: 12065–12068. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. 2011. Regulation of chromatin by histone modifications. Cell Res 21: 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter J, Diffley JFX. 2008. Topoisomerase II inactivation prevents the completion of DNA replication in budding yeast. Mol Cell 30: 790–802. [DOI] [PubMed] [Google Scholar]
- Benson LJ, Gu Y, Yakovleva T, Tong K, Barrows C, Strack CL, Cook RG, Mizzen CA, Annunziato AT. 2006. Modifications of H3 and H4 during chromatin replication, nucleosome assembly, and histone exchange. J Biol Chem 281: 9287–9296. [DOI] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. 2006. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441: 349–353. [DOI] [PubMed] [Google Scholar]
- Chong S, Chen C, Ge H, Xie XS. 2014. Mechanism of transcriptional bursting in bacteria. Cell 158: 314–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpet A, Almouzni G. 2009. Making copies of chromatin: the challenge of nucleosomal organization and epigenetic information. Trends Cell Biol 19: 29–41. [DOI] [PubMed] [Google Scholar]
- Dai J, Hyland EM, Yuan DS, Huang H, Bader JS, Boeke JD. 2008. Probing nucleosome function: a highly versatile library of synthetic histone H3 and H4 mutants. Cell 134: 1066–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion MF, Kaplan T, Kim M, Buratowski S, Friedman N, Rando OJ. 2007. Dynamics of replication-independent histone turnover in budding yeast. Science 315: 1405–1408. [DOI] [PubMed] [Google Scholar]
- Driscoll R, Hudson A, Jackson SP. 2007. Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science 315: 649–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drogaris P, Villeneuve V, Pomiès C, Lee E-H, Bourdeau V, Bonneil E, Ferbeyre G, Verreault A, Thibault P. 2012. Histone deacetylase inhibitors globally enhance h3/h4 tail acetylation without affecting h3 lysine 56 acetylation. Sci Rep 2: 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillingham J, Recht J, Silva AC, Suter B, Emili A, Stagljar I, Krogan NJ, Allis CD, Keogh M-C, Greenblatt JF. 2008. Chaperone control of the activity and specificity of the histone H3 acetyltransferase Rtt109. Mol Cell Biol 28: 4342–4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futcher B. 1999. Cell cycle synchronization. Methods Cell Sci 21: 79–86. [DOI] [PubMed] [Google Scholar]
- Garber M, Yosef N, Goren A, Raychowdhury R, Thielke A, Guttman M, Robinson J, Minie B, Chevrier N, Itzhaki Z, et al. 2012. A high-throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals. Mol Cell 47: 810–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartenberg MR, Wang JC. 1992. Positive supercoiling of DNA greatly diminishes mRNA synthesis in yeast. Proc Natl Acad Sci 89: 11461–11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11: 4241–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossett AJ, Lieb JD. 2012. In vivo effects of histone H3 depletion on nucleosome occupancy and position in Saccharomyces cerevisiae. PLoS Genet 8: e1002771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth A, Rocha W, Verreault A, Almouzni G. 2007. Chromatin challenges during DNA replication and repair. Cell 128: 721–733. [DOI] [PubMed] [Google Scholar]
- Han J, Zhou H, Horazdovsky B, Zhang K, Xu R-M, Zhang Z. 2007a. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 315: 653–655. [DOI] [PubMed] [Google Scholar]
- Han J, Zhou H, Li Z, Xu R-M, Zhang Z. 2007b. The Rtt109-Vps75 histone acetyltransferase complex acetylates non-nucleosomal histone H3. J Biol Chem 282: 14158–14164. [DOI] [PubMed] [Google Scholar]
- Ihmels J, Bergmann S, Barkai N. 2004. Defining transcription modules using large-scale gene expression data. Bioinformatics 20: 1993–2003. [DOI] [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, et al. 2004. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21: 947–962. [DOI] [PubMed] [Google Scholar]
- Kivioja T, Vähärautio A, Karlsson K, Bonke M, Enge M, Linnarsson S, Taipale J. 2012. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods 9: 72–74. [DOI] [PubMed] [Google Scholar]
- Krajewski WA, Luchnik AN. 1991. Relationship of histone acetylation to DNA topology and transcription. MGG Mol Gen Genet 230: 442–448. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Zhou H, Wurtele H, Davies B, Horazdovsky B, Verreault A, Zhang Z. 2008. Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell 134: 244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CL, Kaplan T, Kim M, Buratowski S, Schreiber SL, Friedman N, Rando OJ. 2005. Single-nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol 3: e328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961. [DOI] [PubMed] [Google Scholar]
- MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, Fraenkel E. 2006. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics 7: 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal N. 2003. Elongation by RNA polymerase II on chromatin templates requires topoisomerase activity. Nucleic Acids Res 31: 5016–5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse RH. 2003. Getting into chromatin: how do transcription factors get past the histones? Biochem Cell Biol 81: 101–112. [DOI] [PubMed] [Google Scholar]
- Nieduszynski CA, Hiraga SI, Ak P, Benham CJ, Donaldson AD. 2007. OriDB: a DNA replication origin database. Nucleic Acids Res 35: D40–D46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen-Hughes T, Gkikopoulos T. 2012. Making sense of transcribing chromatin. Curr Opin Cell Biol 24: 296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Wei W, Steinmetz LM. 2013. Extensive transcriptional heterogeneity revealed by isoform profiling. Nature 497: 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, et al. 2005. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell 122: 517–527. [DOI] [PubMed] [Google Scholar]
- Radman-Livaja M, Liu CL, Friedman N, Schreiber SL, Rando OJ. 2010. Replication and active demethylation represent partially overlapping mechanisms for erasure of H3K4me3 in budding yeast. PLoS Genet 6: e1000837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radman-Livaja M, Verzijlbergen KF, Weiner A, van Welsem T, Friedman N, Rando OJ, van Leeuwen F. 2011. Patterns and mechanisms of ancestral histone protein inheritance in budding yeast. PLoS Biol 9: e1001075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando OJ, Winston F. 2012. Chromatin and transcription in yeast. Genetics 190: 351–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezai-Zadeh N, Zhang X, Namour F, Fejer G, Wen YD, Yao YL, Gyory I, Wright K, Seto E. 2003. Targeted recruitment of a histone H4-specific methyltransferase by the transcription factor YY1. Genes Dev 17: 1019–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Navarro S. 2009. Insights into SAGA function during gene expression. EMBO Rep 10: 843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibahara K, Stillman B. 1999. Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 96: 575–585. [DOI] [PubMed] [Google Scholar]
- Slater ML. 1973. Effect of reversible inhibition of deoxyribonucleic acid synthesis on the yeast cell cycle. J Bacteriol 113: 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel RE, Cook RG, Perry CA, Annunziato AT, Allis CD. 1995. Conservation of deposition-related acetylation sites in newly synthesized histones H3 and H4. Proc Natl Acad Sci 92: 1237–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Barkai N. 2008. Two strategies for gene regulation by promoter nucleosomes. Genome Res 18: 1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unnikrishnan A, Gafken PR, Tsukiyama T. 2010. Dynamic changes in histone acetylation regulate origins of DNA replication. Nat Struct Mol Biol 17: 430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Attikum H, Gasser SM. 2009. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol 19: 207–217. [DOI] [PubMed] [Google Scholar]
- Voichek Y, Bar-Ziv R, Barkai N. 2016. Expression homeostasis during DNA replication. Science 351: 1087–1090. [DOI] [PubMed] [Google Scholar]
- Walker GM. 1999. Synchronization of yeast cell populations. Methods Cell Sci 21: 87–93. [DOI] [PubMed] [Google Scholar]
- Wang JC. 2002. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol 3: 430–440. [DOI] [PubMed] [Google Scholar]
- Yabuki N, Terashima H, Kitada K. 2002. Mapping of early firing origins on a replication profile of budding yeast. Genes Cells 7: 781–789. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.