

Abstract
We herein report the result of a large-scale, next generation sequencing (NGS)-based screen for embryonic lethal (EL) mutations in Belgian beef and New Zealand dairy cattle. We estimated by simulation that cattle might carry, on average, ∼0.5 recessive EL mutations. We mined exome sequence data from >600 animals, and identified 1377 stop-gain, 3139 frame-shift, 1341 splice-site, 22,939 disruptive missense, 62,399 benign missense, and 92,163 synonymous variants. We show that cattle have a comparable load of loss-of-function (LoF) variants (defined as stop-gain, frame-shift, or splice-site variants) as humans despite having a more variable exome. We genotyped >40,000 animals for up to 296 LoF and 3483 disruptive missense, breed-specific variants. We identified candidate EL mutations based on the observation of a significant depletion in homozygotes. We estimated the proportion of EL mutations at 15% of tested LoF and 6% of tested disruptive missense variants. We confirmed the EL nature of nine candidate variants by genotyping 200 carrier × carrier trios, and demonstrating the absence of homozygous offspring. The nine identified EL mutations segregate at frequencies ranging from 1.2% to 6.6% in the studied populations and collectively account for the mortality of ∼0.6% of conceptuses. We show that EL mutations preferentially affect gene products fulfilling basic cellular functions. The resulting information will be useful to avoid at-risk matings, thereby improving fertility.
Livestock productivity has dramatically increased over the last 50 years. Milk production in Holstein cows has doubled from ∼6000 in 1960 to ∼12,000 kgs in 2000, and ∼75% of this change was genetic (Dekkers and Hospital 2002). However, gains for producers were partially eroded by concomitant decreases in disease resistance and fertility. Pregnancy rate decreased by ∼6% in this population over the same period (Norman et al. 2009). It is assumed that the reduced fertility results from the negative energy balance of high-producing cows. A complementary explanation might be an increase in premature pregnancy termination due to homozygosity for embryonic lethal (EL) mutations.
This is supported by several observations. One is the recent positional cloning of a quantitative trait locus (QTL) for fertility in Nordic Red Cattle (Kadri et al. 2014). It was shown to be due to a 660-kb deletion on Chromosome 12 that causes early embryonic lethality in homozygotes. The deletion was shown to segregate at high frequencies in Nordic cattle (up to 16% in Finnish Ayrshire) as a result of its positive effect on milk yield in heterozygotes. Prior to its detection, it caused the death of up to ∼0.64% of conceptuses in these breeds. Also, the realization that all or a substantial proportion of embryos homozygous for the DUMPS (deficiency of uridine monophosphate synthase) (Robinson et al. 1983), CVM (complex vertebral malformation) (Thomsen et al. 2006), or BS (brachyspina syndrome) (Charlier et al. 2012) mutations die before birth and are therefore never reported suggests that other fully lethal (i.e., early mortality of all embryos) and hence unsuspected ELs might be segregating at fairly high frequencies. As an example, the BS mutation was shown to segregate at a frequency of 3.7% in Holstein Friesian and hence to cause the mortality of ∼0.14% of conceptuses. The 660-kb deletion, as well as the CVM and BS mutations, were identified using standard forward genetics approaches (Georges 2007). In the case of CVM and BS, this was possible because samples from affected individuals could be used for linkage and association analyses. The population frequency of the 660-kb deletion was high enough in Finnish Ayrshire to significantly affect the breeding values for fertility of carrier bulls, hence allowing QTL analysis. It is worth noting that in other Scandinavian breeds, in which the deletion was segregating at frequencies ≤6% (hence still causing mortality of 0.09% of conceptuses), QTL analysis was not possible, as the effect on the breeding values for fertility of this recessive EL was too modest. Thus, phenotype-driven forward genetic approaches are not suitable to identify ELs segregating at frequencies <∼10% which is likely to be the case for the majority.
An alternative, genotype-driven approach has recently been devised that takes advantage of the large cattle cohorts that have been genotyped with genome-wide SNP arrays for genomic selection. The signals that are sought are depletions in homozygotes (among live animals) for specific haplotypes assumed to be associated with EL mutations. This approach, combined with follow-up studies of the corresponding haplotypes, has led to the identification of six ELs in cattle (Fritz et al. 2013; Sonstegard et al. 2013; Daetwyler et al. 2014; Pausch et al. 2015). However, at least two conditions need to be met for this strategy to be effective: (1) Very large cohorts (tens to hundreds of thousands of animals) genotyped with medium- to high-density SNP arrays need to be available in the breeds of interest; and (2) linkage disequilibrium (LD) between the EL and the cognate haplotype needs to be very high, if not perfect (r2 ∼ 1). The former condition is only met for few very popular breeds, including Holstein-Friesian, in which three of six detected ELs were found. It is likely to remain a considerable bottleneck, as low-density (∼10K) SNP arrays (which are not suitable for haplotype-based analyses) are increasingly replacing medium-density (∼50K) ones. The latter condition is likely to be met for only part of the ELs, as most of the time LD between the EL and the haplotype will be complete (D′ ∼ 1) but not perfect (i.e., cognate haplotypes without the EL are also segregating in the population). Thus, it is almost certain that other as of yet unknown ELs still segregate in most livestock populations.
To make further progress in the identification of ELs in cattle, we hereby apply a reverse genetics approach that takes advantage of the growing amount of whole-exome and whole-genome NGS data in livestock. The proposed approach consists in (1) mining available sequence data for predicted loss-of-function (LoF) and damaging missense (MS) variants, (2) genotyping large cohorts for the corresponding candidates and identifying putative ELs on the basis of a significant depletion in homozygotes, and (3) confirming the EL nature of the corresponding super-candidates on the basis of a significant depletion in homozygotes in carrier × carrier matings.
Results
Expectations for the number of EL mutations carried per individual
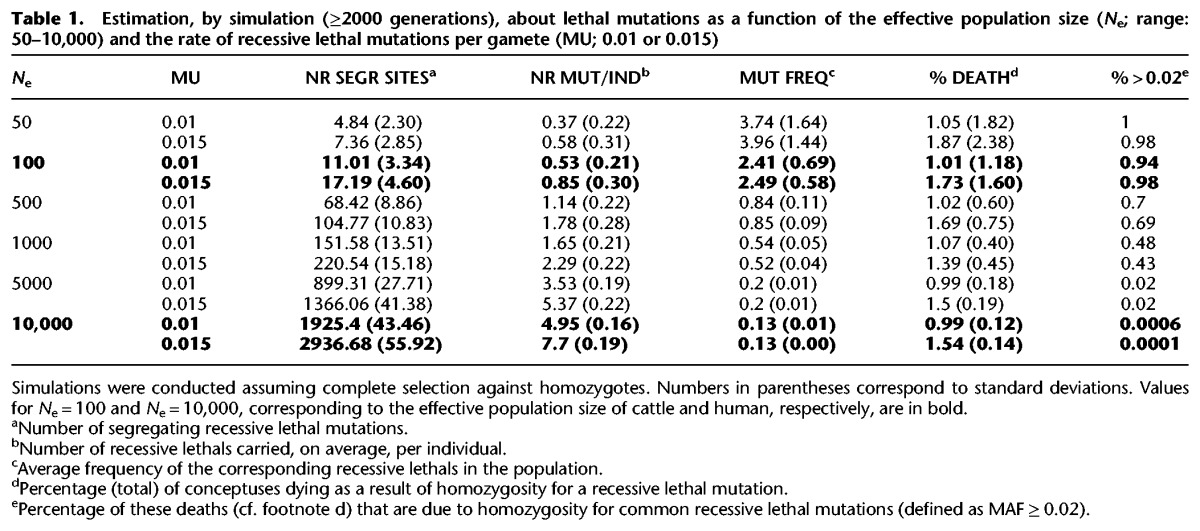
Diploidy has allowed the genome to increase in size while insuring at least one functional copy of each gene in the majority of individuals. Accordingly, most diploid individuals are assumed to carry a number of lethal mutations in the heterozygous state. In Drosophila melanogaster, this number has been estimated at ∼1.6 (e.g., Simmons and Crow 1977). Humans have been estimated to carry an average of the order of ∼0.29 recessive mutations that lead to complete postnatal sterility or death by reproductive age when homozygous (Gao et al. 2015), or ∼1.4 postnatal “lethal equivalents” (e.g., Sutter and Tabah 1953; Morton et al. 1956; Bittles and Neel 1994). It remains unknown, however, how many recessive mutations causing prenatal death when homozygous are carried, on average, by humans or any other mammal. The total number of recessive lethals (pre- and postnatal) carried by individuals is a function of the number of recessive lethals that segregate in the population as well as the frequency distribution of their occurrence in the population. The actual values of these parameters are unknown but can be estimated from the knowledge of (1) the genomic target size for recessive lethal mutations, (2) the rate of recessive lethal mutations in this target space, and (3) the present and past effective population size. Systematic knock-out programs conducted in the mouse indicate that ≤25% of mammalian genes are essential, i.e., defined as causing complete or partial preweaning lethality in homozygotes (International Mouse Phenotype Consortium [IMPC] at https://www.mousephenotype.org). This corresponds to a target space of ∼2,500,000 codons (or ∼7,500,000 nt), and ∼90,000 splice-sites (or ∼180,000 nt) (Ng et al. 2009). Assuming (1) a single nucleotide substitution rate of ∼10−8 per base pair and per gamete, (2) that 3% of single nucleotide substitutions in codon space cause illegitimate stop-gains (given the mammalian codon usage and a transition/transversion ratio of 2), (3) that all single nucleotide substitutions in splice-sites perturb splicing, and (4) a ∼25% proportion of stop-gains and splice-site variants among lethal mutations (deduced from the equivalent proportion among mutations causing known recessive genetic defects; see, for instance, The Human Gene Mutation Database [HGMD at http://www.hgmd.cf.ac.uk]), the rate of recessive lethal mutations can be estimated at ∼0.015 per gamete. We performed simulations under these assumptions and estimated that the number of recessive lethals (pre- and postnatal; hereafter collectively termed ELs) carried, on average, per individual increases with population size from ∼0.85 for an effective population size (Ne) of 100 to ∼7.7 for Ne = 10,000. Interestingly, the frequency of death as a result of homozygosity for EL remains nearly constant, diminishing only very slightly from ∼1.73% at Ne = 100 to ∼1.54 at Ne = 10,000. However, the proportion of these deaths due to “common” EL mutations (defined as having a minor allele frequency [MAF] ≥ 2%) ranges from ∼98% when Ne = 100 to ∼0% when Ne = 10,000 (Table 1). Despite an actual population of several tens of millions of animals, the effective population size of Holstein-Friesian dairy cattle has been estimated at ∼100, as a result of intense selection and widespread use of artificial insemination (de Roos et al. 2008). Despite an actual population size of billions, the effective population size of humans has been estimated at ∼10,000, as a result of past bottlenecks. Thus, our simulations indicate that the number of ELs segregating in dairy cattle populations may be of the order of tens, and that the population frequency of many of these may be of the order of 2% or more. Identifying these common ELs may be an effective first step to reduce the number of embryonic deaths from homozygosity for recessive lethals, thereby improving fertility.
Table 1.
Estimation, by simulation (≥2000 generations), about lethal mutations as a function of the effective population size (Ne; range: 50–10,000) and the rate of recessive lethal mutations per gamete (MU; 0.01 or 0.015)
Identification of ∼94,000 nonsynonymous variants in domestic cattle
We resequenced the whole genome of 496 animals from the New Zealand dairy cattle (NZDC) population and 50 Belgian Blue Cattle (BBC) at an average depth of 11 (range: 3–148). In addition, we resequenced the exome of 78 animals representing six cattle breeds (Bos taurus) at an average depth of 40 (range: 18–100). Sequencing was carried out using reversible terminator chemistry on HiSeq 2000 instruments (Illumina) and SureSelect Target Enrichment reagents (Agilent) for exome sequencing. Sequence reads were mapped to the Bostau6 bovine reference genome using BWA (Li and Durbin 2009). Exomic variants were identified using GATK and corresponding best practices (McKenna et al. 2010). Effects on gene function of the identified variants were predicted using Variant Effect Predictor (McLaren et al. 2010). We identified a total of 186,112 exonic variants, including 1377 stop-gain, 112 stop-loss, 3139 frame-shift, 1341 splice-site, 85,338 missense, and 92,163 synonymous variants (Supplemental Table S1). Of the missense variants, 22,939 were predicted by SIFT and/or PolyPhen to be disruptive/damaging (Kumar et al. 2009; Adzhubei et al. 2010). To ensure that the differences in nucleotide diversity observed between the human (BAM files downloaded from the 1000 Genomes Project) and bovine samples (sequenced at the University of Liège [ULg]) would not be merely technical artifacts, we compared the nucleotide diversity obtained with the 1000 Genomes BAM files with those obtained for 10 human samples sequenced at the ULg using the same experimental conditions (Supplemental Material S1).
Domestic cattle have a comparable LoF load as humans despite a more variable exome
It has been shown that humans carry, on average, ∼120 loss-of-function variants defined by MacArthur et al. (2012) as frame-shift, splice-site, stop-gains, and large deletions. To rigorously compare the mutational load of humans and domestic cattle, we selected 148,913 conserved coding exons from the human-bovine genome alignment (amounting to ∼58% of coding exon space) (see Methods) captured by Agilent's bovine SureSelect Target Enrichment assay. Within this sequence space, we called genetic variants in 59 exome-sequenced cattle and 60 humans using BAM files that were either generated in-house or downloaded from the 1000 Genomes Project (http://www.1000genomes.org/). From these data, we extrapolated (to the entire exome) that Yorubans are, on average, heterozygous at ∼9000 (9 K) synonymous (S) and ∼5.4 K nonsynonymous (NS) sites, while European and Asians are heterozygous at ∼6.3 K S and ∼4.0 K NS positions, in agreement with previous estimates (Fig. 1A; e.g., The 1000 Genomes Project Consortium 2010, 2012). In contrast, domestic cattle are, on average, heterozygous at 13.2 K S and 5.9 K NS positions (Fig. 1B). Thus, present-day domestic cattle are genetically more variable than humans, including Africans. The observed S/NS ratios are ∼4.6- and ∼6.3-fold larger than expected in humans and cattle, respectively, supporting enhanced purifying selection on NS variants as expected (more so in cattle; see hereafter). Humans were estimated to be heterozygous for 58 (range: 31–85) and homozygous for 9 (range: 0–21) LoF variants (excluding large deletions), which is also in agreement with previous studies (Fig. 1C; MacArthur et al. 2012). Domestic cattle were heterozygous for 51 (range: 25–82) and homozygous for 7 (range: 0–21) LoF variants (excluding large deletions), and this was significantly (P = 0.002) lower than humans (Fig. 1D). Thus, despite the higher overall genetic variation observed in domestic cattle, their load of LoF variants is equivalent, if not somewhat lower than that of humans.
Figure 1.

(A,B) Number of heterozygous synonymous (gray) and nonsynonymous (yellow) sites per individual (A: humans; B: bovine). (C,D) Number of heterozygous stop-gain (squares), splice-site (triangles), and frame-shift (circles) sites per individual. CHB: Chinese; JPT: Japanese; FIN: Finns; GBR: Britons; CEU: Northern Europeans; YRI: Yorubans; ANG: Angus; BBB: Belgian Blue; CHA: Charolais; HOL: Holstein-Friesian; JER: Jerseys; SIM: Simmentals.
Estimating the proportion of EL among LoF and missense variants from population data
The observed number of ∼120 LoF variants per individual is ∼20-fold larger than the ∼1–5 recessive lethals estimated to be carried, on average, by individuals (see above). This discrepancy is thought to reflect the importance of molecular redundancy and the high proportion of developmentally nonessential genes. The identification of the minority of EL mutations among the many LoF variants remains a considerable challenge.
To gain insights into the proportion and nature of EL mutations among LoF variants in cattle, we mined the available lists of bovine variants for frame-shift, splice-site, and stop-gain variants. Moreover, we identified missense variants predicted by PolyPhen2 to be damaging and/or by SIFT to be deleterious (Kumar et al. 2009; Adzhubei et al. 2010). The corresponding list of candidate ELs was manually curated for possible sequencing or alignment artifacts using IGV (Robinson et al. 2011), including confirmation of the gene models using fetal RNA-seq data. We further selected variants for which none of the well-covered sequenced individuals were homozygous and which were breed-specific (see Methods). We selected 3779 candidate EL variants in the NZDC population (including 296 LoF and 3483 missense), and 1050 in the BBC population (108 LoF, 942 missense), and added them as custom variants to new designs of the Illumina bovine LD SNP arrays. Moreover, we added 200 breed-specific synonymous variants as “matched controls” to one of the BBC designs (Supplemental Table S2). We genotyped ∼35,000 NZDC and ≥6300 BBC healthy animals. For all variants on the array, we computed the statistical significance (log[1/p]) of the depletion in homozygosity for the minor allele (versus within-breed Hardy-Weinberg expectation) (Fig. 2; Supplemental Fig. S1; see Methods). We were struck by the occurrence, in both populations, of candidate EL variants without homozygote mutant animals despite population frequencies ≥1.3% (NZDC) and 1.8% (BBCB), while this was never observed for any one of the thousands of neutral (N) variants on the arrays. This suggested that the interrogated LoF and missense variants might indeed harbor EL mutations. Alternatively, the observed difference between candidate EL and N variants might reflect their distinct ascertainment scheme. As an example, interrogated LoF and missense variants were selected to be breed-specific and hence probably younger on average than the N variants shared by multiple breeds. To account for this possible discrepancy, we compared the behavior of candidate EL variants with a set of breed-specific synonymous variants, selected using the same criteria as the LoF and missense variants in BBC. Contrary to LoF and missense variants, there was not a single synonymous variant with population frequency ≥2.2% without homozygote individuals, again suggesting the occurrence of ELs among interrogated LoF and missense variants. The proportion of LoF variants without homozygotes was 0.348 (±0.050), while it was 0.228 (±0.038) for equally sized (50) sets of frequency-matched synonymous variants. The same numbers were 0.233 (±0.056) and 0.185 (±0.044) for frequency-matched sets of missense and synonymous variants. From this, we estimated the proportion of ELs at 15.5% of tested LoF variants and 5.9% of tested missense variants (see Methods).
Figure 2.

Statistical significance [−log(p): y-axis] of the depletion (positive values) or excess (negative values) in homozygotes for loss-of-function (red; defined as frame-shift, splice-site, and stop-gain variants), missense (yellow), matched synonymous (blue), and random neutral (small gray) variants ordered by minor allele frequency (MAF: x-axis), based on the genotyping of 6385 healthy BBC (A) and 35,219 healthy NZDC (B) animals. Variants that have been subsequently tested in carrier × carrier matings and proven to be embryonic lethals (EL) are labeled in italics and bold. WWP1, shown to affect muscularity, and GALNT2, shown to cause growth retardation, are labeled in italics. For NZDC (B), MAFs were computed across breeds (NZ Holstein-Friesian, NZ Jersey, and NZ cross-bred), explaining the differences with the within-breed MAF reported in Table 2, and the high proportion of variants with negative −log(p) values. Insets: loss-of-function-variants-alone graphs for the corresponding BBC (A) and NZDC (B) populations.
Confirming the embryonic lethality of nine common LoF variants in carrier-carrier matings
To provide direct evidence of their embryonic lethality, we retrospectively genotyped 25 trios (carrier sire, carrier dam, healthy offspring), on average (range: 8–50), for the (at the time) most significant four LoF and single missense variants in BBC, and for the (at the time) most significant three LoF and single missense variants in NZDC, all with MAF ≥1.2%, and without observed homozygotes. Using information from the matched S variants in BBC, we estimated the proportion of ELs among LoF and missense variants without homozygotes at 0.44 and 0.25, respectively (see Methods). Genotyping was done directly for 141/200 trios and by combining direct genotyping in the parents with linkage analysis for 59/200 trios (see Methods). No homozygous offspring were observed in the 200 offspring, supporting the embryonic lethality of the nine tested variants (four in NZDC and five in BBC). Ratios between homozygote wild-type and carrier animals did not depart significantly from the expected 1:2 in these crosses (P ≥ 0.13). Eight of these genes are broadly expressed and code for proteins fulfilling essential housekeeping processes, such as DNA replication, transcription, and RNA processing. Expected cis-eQTL effects were observed in mammary gland for the three LoF variants predicted to cause nonsense mediated RNA decay (OBFC1 frame-shift, TTF1 stop-gain, and RNF20 stop-gain) (Supplemental Material S2). Frequencies of the identified ELs averaged 3.2% and ranged from 1.2% to 6.6% (Table 2).
Table 2.
Main features of nine confirmed embryonic lethal (EL) mutations in cattle

Identifying nonlethal coding variants with phenotypic effects
Some variants were characterized by a pronounced depletion in homozygotes in the general population despite the occurrence of presumably healthy homozygous individuals. This suggests that selection acts against homozygotes, albeit without causing early death. Indeed, one of these variants proved to be a splice-site mutation in the GALNT2 gene, encoding polypeptide N-acetylgalactosaminyltransferase 2. It was recently identified by a standard forward genetic approach as the mutation causing “Small Calf Syndrome” in NZDC (M Littlejohn, pers. comm.). Another is a common (13% frequency in BBC) missense variant in the WWP1 gene, encoding the WW domain containing E3 ubiquitin protein ligase 1. The ≥6300 genotyped BBC animals included 581 bulls with an average of 331 (range: 1–4706) offspring records for more than eight traits pertaining to muscularity and stature, allowing computation of breeding values. A genome wide association study (GWAS) using these breeding values indicated that the R844Q WWP1 variant very significantly increased muscularity, while decreasing stature (Supplemental Fig. S2). This strongly suggests that its observed high frequency in BBC results from yet another example of balanced polymorphism operating in intensely selected livestock populations (Hedrick 2015).
Lack of evidence for synergistic epistasis
It has been suggested that deleterious variants are more effectively purged from populations as a result of synergistic epistasis, i.e., that multiple deleterious genetic variants have a larger cost on fitness than expected from their multiplicative effects. This hypothesis predicts that individuals carrying multiple deleterious variants will be fewer than expected assuming random assortment. Recent analyses of the GoNL sequence data suggest that synergistic epistasis might be operating in humans (Sohail et al. 2016). We tested the hypothesis using the large genotype database generated as part of this study. Analyses were conducted in the BBC population, separately for LoF and missense variants. We observed no evidence for an underrepresentation of animals carrying multiple LoF or missense variants in either of these populations (Supplemental Material S3).
Discussion
Making reasonable assumptions about the genomic target size for recessive lethal mutations (∼9 × 106 bp), the proportion of lethal mutations among all mutations in this space (∼15%), and a mutation rate per base pair of 10−8, we herein estimate by simulation that the number of ELs carried, on average, per individual increases with effective population size (Ne) from ∼0.5 for Ne = 100 to ∼5 for Ne = 10,000, corresponding to estimates of the effective population size for domestic cattle and humans, respectively. We show that the percentage of conceptuses that will die from homozygosity for EL mutations is independent of effective population size and on the order of ∼1% under our model. We show that the majority of these deaths involve on the order of tens of ELs segregating at frequencies >2% in domestic cattle, while likely involving a very large number of rare EL variants in human.
We then show that the exome of domestic cattle is more variable than that of humans, when considering both synonymous and nonsynonymous variants. These findings are in agreement with recent estimates of nucleotide diversity (based on whole-genome sequence data) shown to be higher in domestic cattle (1.44/kb) than in humans (Yoruba: 1.03/kb; European: 0.68/kb) (Daetwyler et al. 2014). The mutation rate in the cattle germ-line has recently been estimated at ∼1.1 × 10−8 per base pair per gamete (M Georges, unpubl.), hence near identical to human. This strongly suggests that the past effective population size of domestic cattle was larger than that of humans (e.g., MacEachern et al. 2009). Thus, against expectations, the bottlenecks undergone by cattle as the result of the domestication process appeared to have been less severe than the bottlenecks undergone by humans, including Africans. One explanation for this is that domestication of cattle has been a long-lasting process with a sustained flow of genes from the wild (with large effective population size) into the domestic populations. Another possible cause of the observed higher nucleotide diversity in domestic cattle when compared to humans is that domestication involved subspecies of wild bovids carrying highly divergent haplotypes. Thus, present-day domestic taurine cattle might in fact have a mosaic genome tracing back to distinct wild subspecies. This phenomenon is certainly well documented in European domestic pig breeds, in which alleles tracing back to Asian wild boars segregate in a genome with originates primarily from European wild boars (e.g., Van Laere et al. 2003; Groenen et al. 2012; Bosse et al. 2014).
When focusing on LoF variants, however, it appears that humans carry, on average, more such variants than cattle. We attribute this apparent conundrum to the fact that deleterious recessive alleles are being purged more effectively and more rapidly from the genome of present-day domestic cattle than from that of humans as a result of the rapid increase in inbreeding following breed creation and initiation of intense selection programs particularly in the nineteenth and twentieth centuries (including the widespread use of artificial selection) (Goddard et al. 2010). In agreement with this hypothesis, we observe that the S/NS ratios are larger in domestic cattle (∼6.3) than in humans (∼4.6), testifying to stronger purifying selection in cattle than in humans.
We have mined exome sequence data from >500 animals and have identified >400 candidate LoF and >4400 deleterious missense variants which we have genotyped in large cohorts of 35,000 and 6300 animals in NZDC and BBC cattle, respectively. From the observed proportion of variants without homozygotes among healthy individuals, we have estimated that ∼15% of tested LoF variants and ∼6% of tested missense variants might be ELs. These percentages increase to 44% (LoF) and 25% (missense) when restricting the analysis to variants without homozygotes among healthy individuals. We have tested the ELs of nine of the most common of these candidate EL variants in carrier × carrier matings, indeed confirming their lethality. Not unexpectedly, the corresponding genes are broadly expressed and code for proteins fulfilling essential housekeeping functions, including DNA replication, transcription, and RNA processing. We estimated the proportion of affected conceptuses (i.e., homozygous for at least one of the nine reported ELs) to be ∼0.64% in the NZDC and ∼0.61% in the BBC populations, corresponding to ∼7600 and ∼3000 embryos, and an estimated cost of 13.8 million NZ$ and 2.7 million €, respectively. In offspring of sires that are carrying the most common ELs, these proportions reach ∼3.3% in the NZDC and ∼2.7% in the BBC populations, respectively. Knowing the genotypes of sires and dams for the corresponding EL variants will assist in avoiding at-risk matings, thereby improving fertility.
There remain two frame-shift and eight missense variants with population frequency >1% in the BBC population, of which the EL status has not yet been confirmed in carrier × carrier matings. At least four of these affect genes fulfilling essential functions (Supplemental Table S2). Our prediction is that these are likely ELs as well and work to test this is in progress.
Thus far, a number of ELs have been identified in livestock by taking advantage of large cohorts that were SNP-genotyped for genomic-selection purposes and identifying haplotypes never observed in homozygous form. The corresponding haplotypes are then sequenced to identify the putative EL mutations (e.g., Pausch et al. 2015). This approach is only effective if (1) the ELs are in complete linkage disequilibrium (r2 ∼ 1) with the corresponding haplotypes, and (2) large enough SNP-genotyped cohorts are available (which is the case for only very few breeds). Retrospective analyses indicate that only the single most common of the four EL mutations in NZDC (in OBFC1) would have been detected using this standard approach (Supplemental Material S4). For the remaining ones, the ELs are only in perfect LD (D′ ∼ 1; r2 < 1) with flanking haplotypes, indicating that equivalent wild-type haplotypes still segregate in the population, hence obscuring the signal. Thus, more ELs are likely to segregate in the studied populations than might be suspected from haplotype-based analyses alone. The absence of large SNP-genotyped cohorts in BBC (as in most other smaller breeds) precluded the use of the haplotype-based approach. Our results demonstrate the efficacy of an NGS-based reverse genetic screen even in smaller populations.
We observe a significant departure from Hardy-Weinberg equilibrium for some of the tested variants showing a depletion in (yet existence of) homozygotes. This could be due to the contamination of the supposedly healthy cohort with affected individuals, particularly for variants causing mild phenotypes. This was likely the case for the splice-site variant in the GALNT2 gene causing a form of dwarfism in the NZDC population. Alternatively, the expressivity may be variable up to the point of incomplete penetrance, such that a proportion of homozygotes may appear healthy and be included in the cohort. Selection should nevertheless act against such variants and drive them toward low frequencies. We observed a missense variant in the WWP1 gene showing a striking depletion in homozygotes yet having an allelic frequency as high as 13% in the BBC population. We provide evidence that this is likely due to the fact that it positively affects desirable phenotypes in the heterozygotes while being deleterious in the homozygote state. Thus, these variants, especially the most frequent ones, possibly encompass additional examples of balancing selection, which increasingly appear to be commonplace in domestic animals (e.g., Hedrick 2015).
In addition, this study yields a cohort of animals that appear normal at first glance despite being homozygous for obvious LoF variants in genes deemed essential. The list included homozygous mutants for NME7 (NDK7), SYNE2, SLC9A9, and FREM1 (Supplemental Table S2). Such animals will be deeply phenotyped to possibly uncover physiological perturbations that might be medically pertinent as illustrated by PCSK9, CCR5, ACTN3, CASP12, and SCN9A knockouts in humans (Kaiser 2014; Alkuraya 2015).
Methods
Simulations
To estimate the number of ELs carried on average per individual, we simulated the reproduction of panmictic populations with constant effective population size ranging from 50 to 10,000 for 10,000 generations. At every generation, gametes had a probability of 0.01 to be affected by a novel recessive lethal mutation, which was always considered to be distinct and affecting another gene compared to all other mutations already present in the population. All mutations were assumed to segregate independently of each other (no linkage). Individuals that were homozygous for any of the segregating mutations were removed from the population with compensatory reproduction.
Next generation sequencing
Genomic DNA was extracted from sperm or whole blood using standard procedures. For whole-genome resequencing, PCR-free libraries were generated and sequenced (100-bp paired ends) on HiSeq 2000 instruments by Illumina's FastTrack services for the NZDC samples, and at the CNAG (Barcelona) for the BBC samples. For exome sequencing, enrichment was conducted using the Sure Select Target Enrichment Reagents (Agilent), and sequencing conducted on HiSeq 2000 instruments at the GIGA Genomics platform at the University of Liège.
Variant calling
Sequence reads were aligned to the bosTau6 reference genome using BWA (Li and Durbin 2009). PCR duplicates were identified using Picard (http://picard.sourceforge.net/). Local indel realignment and base quality score recalibration was conducted with GATK (McKenna et al. 2010). Variants were called using UnifiedGenotyper for the NZDC and exome samples, and using the GATK Haplotype caller (McKenna et al. 2010) for the BBC samples. Variant quality score recalibration was conducted using GATK VariantRecalibrator (McKenna et al. 2010) using the Illumina BovineHD Genotyping BeadChip variants and a subset of newly detected sequence variants showing correct Mendelian segregation within a large sequenced nuclear pedigree as reference sets.
Comparing the mutational load of human and bovine exomes
Bovine exome sequencing was generated as described above. Data from 60 unrelated human exomes were downloaded from ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/data/ and down-sampled to match the distribution of sequence depth of the bovine samples using GATK. Variants were called using GATK's UnifiedGenotyper (McKenna et al. 2010) as described above. The comparison of the mutational load was restricted to 148,913 coding exons that were nonredundant, 1:1 alignable, and of equal size in human and bovine, flanked by canonical splice-sites, and autosomal. Olfactory receptor genes were ignored. Variant sites were only considered if coverage ≥20 and mapping quality ≥30. Additional filters for qualifying SNPs were: QD < 2.0, MQ < 40.0, FS > 60.0, ReadPosRankSum <−8.0, MQRankSum <−12.5, and for qualifying indels: QD < 2.0, FS > 200.0, ReadPosRankSum <−20.0. Heterozygosity was calculated for each individual as the number of heterozygous sites divided by the total number of qualifying sites (coverage ≥ 20 and MQ ≥ 30). Variants were annotated as S, MS, SS, FS, and SG mutation based on the human RefSeq gene model.
Testing for depletion in homozygosity
The significance of the depletion in homozygosity was computed using a standard likelihood ratio test corresponding to LRT = 2ln(〈L|H1〉/〈L|H0〈 in which
and
In these, nxx corresponds to the number of animals with corresponding genotype (m: mutant, +: wild-type allele). LRT was assumed to have a χ2 distribution with one degree of freedom under the null.
Estimating the proportion of ELs among LoF and missense variants
The proportion of ELs among LoF (respectively, missense) variants, p, was estimated as p = b − a/1 − a, where b is the proportion of interrogated LoF (respectively, missense) variants without homozygotes and a is the average proportion of variants without homozygotes among size- and frequency-matched sets of control “S” variants (cf. main text). This is derived from the assumption that b = p + (1 − p)a.
The proportion of ELs among LoF (respectively, missense) variants without homozygous animals, q, was estimated as
where b and a are defined as above.
Testing for synergistic epistasis
To test for synergistic epistasis, we permuted (1000×) genotypes for LoF and/or missense variants among genotyped individuals (separately for each variant). We then examined the distribution of the number of individuals carrying 0, 1, 2, … n LoF/missense variants, looking for a depletion of individuals carrying multiple mutations when compared to the simulated data.
Data access
VCF files (GATK) and individual BAM files (BWA) from this study, corresponding to the annotated exonic sequences of the full bovine data set, have been submitted to the European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena/) under accession number PRJEB14827.
Supplementary Material
Acknowledgments
This work was supported by grants from the European Research Council (Damona), the Walloon Region (DGARNE Rilouke), and the EU Framework 7 program (GplusE). We used the supercomputing facilities of the Consortium des Equipements de Calcul Intensitf en Fédération Wallonie Bruxelles (CECI) funded by the F.R.S.-FNRS. We thank the Ministry for Primary Industries (Wellington, New Zealand) for financial support, who cofunded the work through the Primary Growth Partnership. We also thank the Asssociation Wallonne de l'Elevage and Herdbook Blanc-Bleu Belge for support. We thank Arnaud Sartelet, Xavier Hubin, and Kristof De Fauw for their assistance in sample collection.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.207076.116.
References
- The 1000 Genomes Project Consortium. 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky V, Gerasimova A, Bork P, Kondrashov A, Sunyaev S. 2010. A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkuraya FS. 2015. Human knockout research: new horizons and opportunities. Trends Genet 31: 108–115. [DOI] [PubMed] [Google Scholar]
- Bittles AH, Neel JV. 1994. The costs of human inbreeding and their implications for variations at the DNA level. Nat Genet 8: 117–121. [DOI] [PubMed] [Google Scholar]
- Bosse M, Megens HJ, Frantz LA, Madsen O, Larson G, Paudel Y, Duijvesteijn N, Harlizius B, Hagemeijer Y, Crooijmans RP, et al. 2014. Genomic analysis reveals selection for Asian genes in European pigs following human-mediated introgression. Nat Commun 5: 4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlier C, Agerholm JS, Coppieters W, Karlskov-Mortensen P, Li W, de Jong G, Fasquelle C, Karim L, Cirera S, Cambisano N, et al. 2012. A deletion in the bovine FANCI gene compromises fertility by causing fetal death and brachyspina. PLoS One 7: e43085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daetwyler HD, Capitan A, Pausch H, Stothard P, van Binsbergen R, Brøndum RF, Liao X, Djari A, Rodriguez SC, Grohs C, et al. 2014. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat Genet 46: 858–865. [DOI] [PubMed] [Google Scholar]
- de Roos APW, Hayes BJ, Spelman RJ, Goddard ME. 2008. Linkage disequilibrium and persistence of phase in Holstein Friesian, Jersey and Angus cattle. Genetics 179: 1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers JCM, Hospital F. 2002. Multifactorial genetics: the use of molecular genetics in the improvement of agricultural populations. Nat Rev Genet 3: 22–32. [DOI] [PubMed] [Google Scholar]
- Fritz S, Capitan A, Djari A, Rodriguez SC, Barbat A, Baur A, Grohs C, Weiss B, Boussaha M, Esquerré D, et al. 2013. Detection of haplotypes associated with prenatal death in dairy cattle and identification of deleterious mutations in GART, SHBG and SLC37A2. PLoS One 8: e65550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Waggoner D, Stephens M, Ober C, Przeworski M. 2015. An estimate of the average number of recessive lethal mutations carried by humans. Genetics 199: 1243–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges M. 2007. Mapping, fine mapping, and molecular dissection of quantitative trait loci in domestic animals. Annu Rev Genomics Hum Genet 8: 131–162. [DOI] [PubMed] [Google Scholar]
- Goddard ME, Hayes BJ, Meuwissen TH. 2010. Genomic selection in livestock populations. Genet Res 92: 413–421. [DOI] [PubMed] [Google Scholar]
- Groenen MA, Archibald AL, Uenishi H, Tuggle CK, Takeuchi Y, Rothschild MF, Rogel-Gaillard C, Park C, Milan D, Megens HJ, et al. 2012. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 491: 393–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick PW. 2015. Heterozygote advantage: the effect of artificial selection in livestock and pets. J Hered 106: 141–154. [DOI] [PubMed] [Google Scholar]
- Kadri NK, Sahana G, Charlier C, Iso-Touru T, Guldbrandtsen B, Karim L, Nielsen US, Panitz F, Aamand GP, Schulman N, et al. 2014. A 660-kb deletion with antagonistic effects on fertility and milk production segregates at high frequency in Nordic Red cattle: additional evidence for the common occurrence of balancing selection in livestock. PLoS Genet 10: e1004049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser J. 2014. The hunt for missing genes. Science 344: 687. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4: 1073–1081. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, Jostins L, Habegger L, Pickrell JK, Montgomery SB, et al. 2012. A systematic survey of loss-of-function variants in human protein-coding genes. Science 335: 823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacEachern S, Hayes B, McEwan J, Goddard M. 2009. An examination of positive selection and changing effective population size in Angus and Holstein cattle populations (Bos taurus) using a high density SNP genotyping platform and the contribution of ancient polymorphism to genomic diversity in domestic cattle. BMC Genomics 10: 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. 2010. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26: 2069–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton NE, Crow JF, Muller HJ. 1956. An estimate of mutational damage in man from data on consanguineous marriages. Proc Natl Acad Sci 42: 855–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler EE, et al. 2009. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 461: 272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman HD, Wright JR, Hubbard SM, Miller RH, Hutchison JL. 2009. Reproductive status of Holstein and Jersey cows in the United States. J Dairy Sci 92: 3517–3528. [DOI] [PubMed] [Google Scholar]
- Pausch H, Schwarzenbacher H, Burgstaller J, Flisikowski K, Wurmser C, Jansen S, Jung S, Schnieke A, Wittek T, Fries R. 2015. Homozygous haplotype deficiency reveals deleterious mutations compromising reproductive and rearing success in cattle. BMC Genomics 16: 312–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JL, Drabik MR, Dombrowski DB, Clark JH. 1983. Consequences of UMP synthase deficiency in cattle. Proc Natl Acad Sci 80: 321–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons MJ, Crow JF. 1977. Mutations affecting fitness in Drosophila populations. Annu Rev Genet 11: 49–78. [DOI] [PubMed] [Google Scholar]
- Sohail M, Vakhrusheva OA, Sul JH, Pulit S, Francioli L, GoNL Consortium, Alzheimers Disease Neuroimaging Initiative, van den Berg LH, Veldink JH, de Bakker P, et al. 2016. Negative selection in humans and fruit flies involves synergistic epistasis. bioRxiv 066407 http://dx.doi.org/10.1101/066407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonstegard TS, Cole JB, Vanraden PM, Van Tassell CP, Null DJ, Schroeder SG, Bickhart D, McClure MC. 2013. Identification of a nonsense mutation in CWC15 associated with decreased reproductive efficiency in Jersey cattle. PLoS One 8: e54872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter J, Tabah L. 1953. Structure de la mortalité dans les familles consanguines. Population 8: 511–526. [Google Scholar]
- Thomsen B, Horn P, Panitz F, Bendixen E, Petersen AH, Holm LE, Nielsen VH, Agerholm JS, Arnbjerg J, Bendixen CA. 2006. Missense mutation in the bovine SLC35A3 gene, encoding a UDP-N-acetylglucosamine transporter, causes complex vertebral malformation. Genome Res 16: 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laere AS, Nguyen M, Braunschweig M, Nezer C, Collette C, Moreau L, Archibald AL, Haley CS, Buys N, Tally M, et al. 2003. A regulatory mutation in IGF2 causes a major QTL effect on muscle growth in the pig. Nature 425: 832–836. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.