

Abstract
Background
One of the major complications of Human Immunodeficiency Virus (HIV) infection is the development of HIV-Associated Neurocognitive Disorders (HANDs) inpproximately 50–60% of HIV infected individuals. Despite undetectable viral loads in the periphery owing to anti-retroviral therapy, neuroinflammation and neurocognitive impairment are still prevalent in HIV infected individuals. Several studies indicate that the central nervous system (CNS) abnormalities observed in HIV infected individuals are not a direct effect of viral replication in the CNS, rather these neurological abnormalities are associated with amplification of HIV specific signals by unknown mechanisms. We propose that some of these mechanisms of damage amplification are mediated by gap junction channels, pannexin and connexin hemichannels, tunneling nanotubes and microvesicles/exosomes.
Objective
Our laboratory and others have demonstrated that HIV infection targets cell to cell communication by altering all these communication systems resulting in enhanced bystander apoptosis of uninfected cells, inflammation and viral infection. Here we discuss the role of these communication systems in HIV neuropathogenesis.
Conclusion
In the current manuscript, we have described the mechanisms by which HIV “hijacks” these host cellular communication systems, leading to exacerbation of HIV neuropathogenesis, and to simultaneously promote the survival of HIV infected cells, resulting in the establishment of viral reservoirs.
Keywords: HIV, gap junctions, connexin, tunneling nanotubes, exosomes
1. INTRODUCTION
The neurological manifestation of HIV/AIDS infection results in a debilitating clinical disorder that affects 50–60% infected population, and is collectively termed as HIV-Associated Neurocognitive Disorders (HANDs) [1–3]. Despite effective anti-retroviral therapy (ART), increasing levels of neurocognitive impairment has been observed in HIV/AIDS patients. HANDs can be categorized into Asymptomatic Neurocognitive Impairment (ANI), Minor Neurocognitive Disorder (MND), and HIV-Associated Dementia (HAD) [3]. While the incidence of HAD has reduced drastically in the post-ART era, the prevalence of milder forms of neurocognitive impairment has increased, and is a major public health concern [4, 5]. The HIV induced CNS damage is dependent on HIV infection, but not on replication. Although HIV infection in the CNS is limited, the mechanisms of exacerbation of HIV neuropathogenesis are poorly understated and require further exploration. We propose that gap junctions, chemical synapses and alternative mechanisms of cell-cell communication such as hemichannels, tunneling nanotubes and exosomes serve to amplify HIV associated CNS damage.
Furthermore, although ART has been successful in reducing HIV replication in the periphery, viral reservoirs are detected in several anatomical places including the brain [6]. The brain continues to serve as a viral ‘safe heaven’ which might aid in emergence of different evolutionary strains as well as drug-resistant viral strains. Our laboratory and others have shown that HIV infected astrocytes and macrophages are protected from apoptosis, which likely contributes to the persistence of HIV within the CNS [7–10]. Hence, there is an urgent need of elucidation of the mechanisms of viral persistence in the CNS and therapeutic interventions to target latent viral reservoirs in the CNS.
A critical area associated with the development of neurological deficits in HIV/AIDS patients is the high selectivity of the blood brain barrier (BBB) to several anti-retrovirals, which limits their CNS penetration. Since several anti-retrovirals achieve only marginal concentrations within the CNS, their effectiveness is greatly reduced [11, 12]. Moreover, certain drug transporters such as P-glycoprotein, serve as active efflux pumps and limit the bioavailability and brain entry for some anti-retrovirals [13, 14]. It is noteworthy to mention that probenecid, a uricosuric drug which blocks astroglial pannexin channels and possibly P-glycoproteins, has also been tested effectively to potentiate the anti-retroviral effects of several nucleoside reverse transcriptase inhibitors [15]. Probenecid has been extensively used in conjugation with anti-retrovirals such as Tenofovir, Zalcitabine and Zidovudine, due to its ability to enhance the clinical effects of HIV drugs by limiting their renal tubular secretion [16]. Hence, the effective concentration of anti-retrovirals is increased in the cells, leading to higher efficacy (Fig. 1). However, increased levels of anti-retrovirals may be intrinsically toxic in some populations. This is imperative since we have recently observed that African Americans and Hispanics have higher levels of pannexin channels (a main target for probenecid) as compared to Caucasians (unpublished data). Thus, pharmacokinetics of anti-retrovirals may be different according to ethnicity. Thus, use of probenecid requires critical re-evaluation or adjustment according to the ethnicity. With the recent discovery of mimetic peptides that specifically block pannexins channels sparing the drug efflux pumps (P-glycoproteins), the role of pannexin channels in altering the pharmacokinetics of anti-retrovirals can be accurately deciphered for therapeutic intervention.
Fig. 1. Probenecid: A key component regulating intercellular communication as well as anti-retroviral availability.
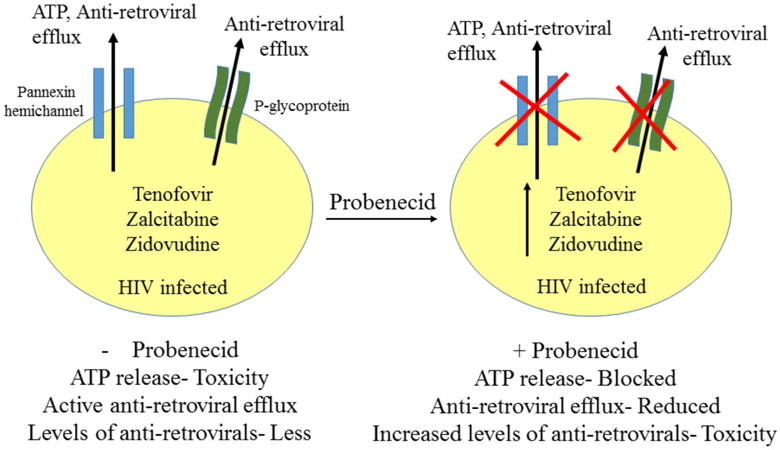
Probenecid, a pannexin channel blocker, has been extensively used along with anti-retrovirals during the treatment of HIV infection. Blocking pannexin channels by probenecid leads to inhibition of ATP release and further activation of purinergic receptors, thereby decreasing ATP-induced neurotoxicity. In addition to its effects on pannexin channels, probenecid leads to decreased renal clearance of anti-retrovirals, causing increased drug exposure. On the other hand, p-glycoproteins have been known to cause active efflux of anti-retrovirals from the cells, leading to lesser efficacy of these drugs. Probenecid, via possible interactions with p-glycoproteins, has been shown to enhance the clinical effects of Tenofovir, Zalcitabine and Zidovudine by limiting their renal tubular secretion. Although blocking pannexin channels by probenecid may lead to lesser ATP-induced neurotoxicity, increased levels of anti-retroviral may be toxic to cells. Hence, clinical usage of probenecid may be critically determined to achieve lesser anti-retroviral toxicity.
2. CLASSICAL MECHANISMS OF HIV INDUCED CNS TOXICITY
While the CNS enjoys the status of being ‘immune privileged’, monocytes and may be T-cells routinely transmigrate across the BBB as part of ‘immune surveillance’ to replenish the population of resident macrophages. HIV enters the CNS within 1–2 weeks of systemic infection [17] through a “Trojan Horse” mechanism [18], crossing the BBB inside of blood-borne monocytes that later differentiate into macrophages (Fig. 2). Circulating monocytes (CD14+CD16− cells) are non-susceptible to HIV infection; however, upon differentiation (CD14+CD16+ cells), these cells become highly susceptible to HIV infection [19]. Data from our collaborators indicates that HIV infected monocytes upregulate several adhesion and tight junction molecules as well as chemokine receptors that facilitate the transmigration of these HIV infected monocytes into the CNS [20].
Fig. 2. Model of HIV CNS infection.
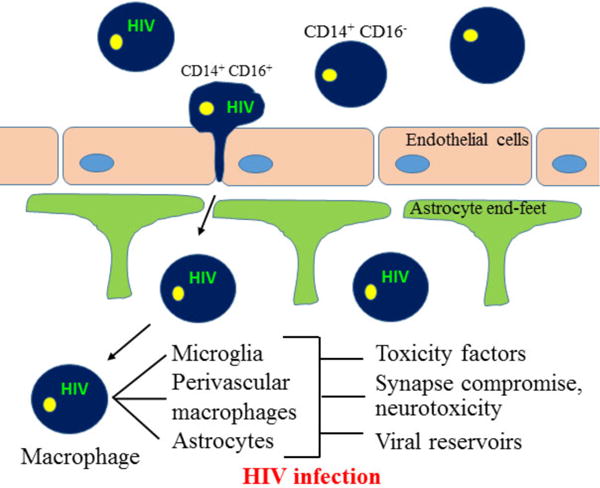
HIV enters the CNS early during the course of HIV infection. CD14+ CD16+ subset of monocytes are susceptible to HIV infection. This monocyte population (CD14+ CD16+) contributes to the development of HAND as they promote viral seeding into the brain and manifestation of neuroinflammation. Neuroinvasion by HIV infected CD14+ CD16+ monocytes is further promoted by expression of various junctional adhesion molecules on the transmigrating monocytes. The transmigration of HIV infected monocytes into the brain leads to alteration of brain homeostasis and results in further spread of the virus to macrophages, microglia as well as astrocytes. The release of viral as well as inflammatory mediators from infected cells results in further activation of non-infected cells, leading to neuroinflammation, neuronal compromise and establishment of viral reservoirs.
After transmigration, HIV infected macrophages establish their residence in the CNS secreting HIV proteins including HIV-Tat, HIV-gp120, and pro-inflammatory cytokines and chemokines such as Monocyte Chemoattractant Protein-1 (MCP-1 or CCL2), Tumor Necrosis Factor-α (TNF-α), Interleukin-1β (IL-1β) and Interferon-γ (IFN-γ) inducible Peptide-10 (IP-10 or CXCL10), to establish a chemotaxis gradient across the BBB, which recruits more monocytes from the periphery into the CNS [21], amplifying the cascade of neuroinflammation initiated from a few transmigrated monocytes.
The major cell types supporting HIV replication in the brain are perivascular macrophages and microglia [22, 23]. Though restricted, astrocyte infection by HIV has far reaching consequences in altering the brain microenvironment as they represent the most abundant brain cell type [24]. While most studies substantiate the notion of restricted or latent HIV infection in astrocytes that can be easily re-activated by TNF-α or IFN-γ [25–27], few have observed productive HIV infection also [28–30]. Several studies suggest that astrocytes serve as undetected HIV reservoirs in the brain [7, 10, 31–34]. Our group has extensively elucidated the role of astrocytes in mediating CNS dysfunction in HIV infection [7, 9, 10, 35]. We have shown that only a few infected astrocytes play a detrimental role in the breakdown of BBB by causing endothelial cell apoptosis and altered regulation in astrocytic end feet [10]. Moreover, we have shown that infected astrocytes amplify BBB toxicity by gap junction and purinergic receptor dependent mechanism, leading to further invasion by infected cells and aggravation of HIV neuropathogenesis. In addition, we showed that HIV infection of astrocytes leads to opening to connexin43 hemichannels, which results in secretion of ATP via purinergic receptors and aids in increasing neuroinflammation [36]. Interestingly, HIV-Tat can also lead to enhanced connexin43 expression in primary human astrocytes [37]. Hence, we postulated that gap junctional proteins, connexins as well as pannexins, play an important role in amplifying HIV associated toxicity in the CNS [7, 9, 10].
Neurons are not infected by HIV [21]. Neuronal loss has been observed in cortical and subcortical areas, basal ganglia and hippocampi in the brains of HIV infected individuals even with minor neuropathological changes [38–41], with Ketzler et al. reporting up to 18% decrease in neuronal density in AIDS brain [42]. The neurodegeneration and neurological symptoms in HAND can be explained by the concerted effects of the ‘direct’ and the ‘indirect’ model of HIV neuropathogenesis [7, 10, 21, 43–46].
The ‘direct’ model takes into account the direct effects of the virus and the secreted viral proteins (Tat, gp120) on neurons through various neuronal cell surface receptors: N-Methyl-D-Aspartate Receptor (NMDAR), low density Lipoprotein Receptor related Protein (LRP) and chemokine receptors (CCR5 and CXCR4). HIV-Tat leads to calcium cytotoxicity in neurons through NMDAR activation [46–48] and simultaneous mobilization of intracellular calcium stores in the endoplasmic reticulum via Phospholipase-C (PLC) driven activation of Inositol 1,4,5-triphosphate (IP3) [49].
The ‘indirect’ or the ‘bystander’ model proposes neuronal death as a consequence of the inflammatory response mounted by both infected and uninfected brain cells against HIV infection [7, 9, 21]. Activated macrophages, microglia and astrocytes release numerous soluble factors such as pro-inflammatory cytokines (TNF-α, IL-1β, IL-6, IFN-γ, IL-8), chemokines (CCL2, CCL5, CXCL10), Platelet Activating Factor (PAF), Nitric Oxide (NO), Matrix Metalloproteases (MMP), and arachidonic acid, which perturb neuronal homeostasis and lead to synapto-dendritic injury [43]. Astrocytes are a prime mediator of the ‘indirect’ damage to neurons as glutamate-glutamine homeostasis is perturbed in the HAND brain [50].
With the extensive use of anti-retroviral therapy and apparently low to undetectable levels of viral replication, it appears that HIV “hijacks” the host machinery and uses it to amplify virus associated inflammation and pathogenesis, leading to neurocognitive impairment. Hence, we propose that HIV utilizes the extensive host intercellular communication system (gap junctions, hemichannels, tunneling nanotubes, and exosomes) to spread toxicity, and also to establish viral reservoirs. In the present review, we will discuss these novel mechanisms mediating HIV induced neuropathology.
3. ROLE OF INTERCELLULAR COMMUNICATION SYSTEMS IN AMPLIFYING HIV INDUCED CNS DAMAGE IN HAND
3.1. Gap Junctions and Hemichannels
Under physiological conditions, gap junction (GJ) channels serve as a major communication system for astroglial and neuronal cross-talk. In addition to GJ, glial cells communicate with the extracellular space via single-membrane hemichannels that allow passage of small signalling molecules thereby exhibiting paracrine effects (Fig. 3A, B).
Fig. 3. Cell-cell communication systems and spread of toxic stimuli during HIV infection.
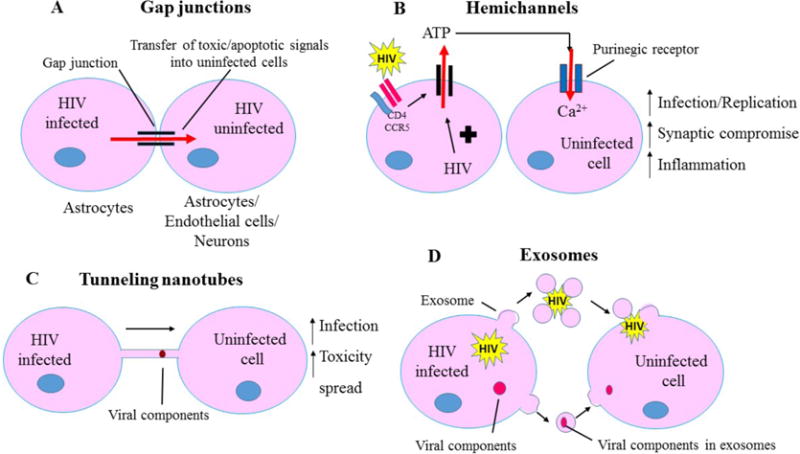
HIV “hijacks” the intercellular communication systems (Gap junctions, hemichannels, tunneling nanotubes) to transfer toxic stimuli from infected cells to adjacent uninfected cells. This allows the virus to evade the immune system as well as results in the spread of toxic metabolites (ATP, small molecules, glutamate) to the uninfected cells. A- Gap junction channels, composed of connexin proteins, allows HIV infected astrocytes to transfer toxic/apoptotic signals to physically adjacent cells (endothelial cells, astrocytes, neurons). B- Unopposed hemichannels, composed of connexins as well as pannexins, results in transfer of toxic signals, such as ATP, into the extracellular space which may further lead to activation of purinergic receptors on nearby uninfected cells and promote neuroinflammation as well as neuronal compromise. C- Tunneling nanotubes act as a long-range communication system between infected and uninfected cells and may assist in transfer of viral particles as well as viral proteins, successfully evading the host immune response. D- Exosomes contribute to HIV pathogenesis by aiding in transfer of HIV virions/viral components such as viral RNA and viral proteins from infected to uninfected cells.
Gap junctions are clusters of intercellular channels that connect the cytoplasm of two adjacent cells, and allow transfer of ions and intracellular messengers between them. Gap junctions are formed by head-to-head docking of two hemichannels (connexons), which are hexameric assemblies of homologous or heterologous subunit proteins termed as connexins (Cx) [51]. Gap junction channels can be either homotypic, composed of two identical hemichannel subunits, or heterotypic, composed of two different subunits of hemichannels. Moreover, unopposed hemichannels (uHC) formed by either homomeric or heteromeric connexons also serve as a means of metabolite exchange between the cytoplasm and the extracellular environment.
Apart from connexins, another family of proteins termed as pannexins (Panx) can also form unopposed hemichannels. Although connexins and pannexins share membrane topology and pharmacological characteristics, pannexins bear significant sequence homology to invertebrate gap junction proteins, innexins rather than vertebrate gap junction proteins, connexins [52]. Moreover, establishment of functional gap junction channels by pannexins is still under debate, and they are believed to form only single membrane channels [52]. Depending on the nature of connexin/pannexin present in a particular channel, GJs and uHCs differ in their biophysical properties and permeability. These channels have an internal pore diameter of approximately 12 A°, which facilitates the diffusion of ions and small molecules such as neurotransmitters, calcium, IP3, small RNA, cyclic nucleotides between the connected cells or from the cytoplasm to the extracellular space [53]. GJ channels serve as an important means of intercellular communication and coordinate various physiological functions such as neurotransmitter uptake and release, synaptic transmission, neuronal plasticity, glutamate homeostasis, and immune cross-talk [54, 55].
Connexins are widely expressed in all human tissues and exhibit cell-type specificity as well as tight developmental regulation [56]. Within the CNS, connexins are highly expressed in brain microvascular endothelial cells, astrocytes, neurons, oligodendrocytes, and under inflammatory conditions in macrophages and microglia [35]. Most reports indicate that exposure to LPS, ischemic injuries, cancer and inflammatory mediators such as IL-1β, ATP, Transforming Growth Factor-β (TGF-β), and NO decrease connexin expression as well as gap junctional communication [57–59]. Furthermore, several pathogens including swine-flu virus, borna virus, and Helicobactor pylori have also been reported to cause total shutdown of connexin mediated communication [60]. Thus, it is accepted that parenchymal connexins and gap junction are negatively regulated by inflammatory milieu except in macrophages [61–64].
Intercellular communication via astroglial gap junction channels is believed to protect neurons from neurotoxic substrates through spatial buffering of toxic metabolites such as glutamate, by allowing uptake of glutamate and intracellular diffusion between the connected cells [35, 65, 66]. Moreover, it has been shown that intercellular glial communication decreases neuronal vulnerability to oxidative stress by rapid calcium buffering [67]. As opposed to gap junctions, glial hemichannels are generally closed under physiological conditions as due to their high permeability, they can lead to initiation of cell death [65, 68]. However, they serve to regulate brain homeostasis during ischemia, several diseases and under conditions of cellular stress. Several reports have suggested that cytokines, oxidative stress, ischemia, hypoxia, and changes in calcium levels can result in opening of astrocytic Cx43 uHCs (the predominant connexin in astrocytes) that allows the release of glutamate, aspartate, ATP and prostaglandins into the extracellular space [69–74].
Since astrocytes serve as metabolic and trophic support to neurons, changes in astrocytic homeostasis and subsequent release of inflammatory mediators via uHCs may enhance neuronal susceptibility and potentiate indirect damage to neurons as observed in HIV neuropathogenesis. It has been shown that under inflammatory conditions, astroglial gap junctions and hemichannels have differential regulation that may be in part mediated by microglia. In fact, Retamal et al. have shown that IL-1β and TNF-α, released from microglia during inflammatory conditions, decrease astroglial gap junctional communication while simultaneously increasing extracellular communication via Cx43 hemichannels through a p38 mitogen-activated protein kinase-dependent mechanism [75]. In addition, the authors showed that exposure with pro-inflammatory cytokines reduced the total and cells surface levels of Cx43, which suggests that increased extracellular communication via Cx43 hemichannels was due to increased hemichannel activity.
Similar findings depicting increased Cx43 hemichannel activity and decreased intercellular communication via gap junctions has been reported in cortical astrocytes following hypoxia-induced ischemia [73]. The detrimental role of hemichannels for neuronal stability has been further strengthened by reports of neuronal pannexin1 hemichannels acting as site for convergence of neurotoxic signals (ATP and glutamate) released via astroglial Cx43 uHCs [72]. Moreover, it has been suggested that Cx43 hemichannel blockers may be neuroprotective against cytokine-induced NMDA mediated neurotoxicity [76]. Hence, it is likely that blocking of hemichannels especially under inflammatory conditions may serve to reduce neuronal loss under several pathological states such as viral infections, and also in cases of neurodegenerative disorders exhibiting chronic neuroinflammation.
As stated earlier, inflammatory milieu and infectious agents reduce connexin expression and diminish gap junction communication in several cell types [77]. However, in case of HIV, the expression of Cx43 and subsequently intercellular communication by gap junction channels is maintained in astrocytes. It has been shown that HIV infection in astrocytes upregulates Cx43 expression in infected cells and functional gap junction channels are maintained to preserve active communication with nearby non-infected cells [9, 37]. Moreover, functional gap junction channels are required to spread apoptotic signals to uninfected cells (neurons as well as astrocytes), as blocking gap junction communication led to reduced cell death in response to HIV infection. In addition to mediating neurotoxicity, gap junction communication also promoted inflammation by enhancing the release of CCL2 from infected astrocytes. Since, CCL2 is an important chemokine that promotes recruitment of monocytes into the brain and facilitates BBB disruption, gap junction communication directly contributes to HIV neuropathogenesis. Although astrocytes are minimally infected with HIV, gap junctions spread toxicity and inflammation to surrounding uninfected cells, including neurons and endothelial cells comprising BBB, and serve to amplify HIV associated neuropathology.
Further, during HIV infection, gap junctions also contribute to BBB disruption by perturbing the physiological interactions between astrocytic end feet and endothelial cells, mediating endothelial cell apoptosis, dysregulating lipoxygenase/cycloxygenase signaling, and ATP receptor activation in astrocytic end feet [10]. Interestingly, HIV infected astrocytes are resistant to apoptosis by virus-dependent mechanisms and contribute to establishment to viral reservoirs in the CNS. Even direct intracellular microinjection of cytochrome c into HIV infected astroglial cultures did not induce apoptosis in HIV infected astrocytes, suggesting viral mediated mechanism of cellular protection [7]. We have proposed that mitochondrial dysfunction post-HIV infection in astrocytes leads to release of cytochrome c in cytoplasm, further leading to dysregulation of IP3 signaling, impaired intracellular calcium metabolism, and subsequent diffusion of IP3 and Ca2+ into neighbouring astrocytes that results in apoptosis of uninfected astrocytes.
Recent findings from our laboratory suggest that HIV-Tat can also lead to enhanced expression of Cx43 in primary human astrocytes by directly binding to its promoter [37]. Whereas other viral proteins (Vif, Gag, Rev, Nef) did not alter Cx43 expression, gp120 negatively regulated Cx43 levels in astrocytes [37]. Furthermore, exposure of HIV-Tat led to increased gap junctional communication in astrocytes, suggesting that HIV-Tat can also contribute to the propagation of toxic stimuli generated within a few infected astrocytes to adjacent uninfected cells [37]. Since the production and secretion of HIV-Tat continues independent of effective viral replication even in the presence of anti-retrovirals, it is highly likely that HIV-Tat contributes to amplification of virus mediated neuropathology via gap junctions.
Although few studies have focussed on the role of gap junctions in HIV neuropathogenesis (mostly from our laboratory), the involvement of connexin/pannexin containing hemichannels has not been elucidated in detail. Recently, our laboratory has shown that HIV infection leads to opening of Cx43 hemichannels in human astrocytes [78]. This process does not involve chemokine receptor or mannose receptor interaction, but requires interaction of HIV with unknown receptors(s) on the surface of astrocytes. Although opening of Cx43 hemichannels did not affect viral replication or bystander apoptosis in astrocytes, microarray analysis revealed enhanced expression of Dickkopf-1 (DKK1) protein, a soluble Wnt pathway inhibitor. DKK1 participates in compromise of neuronal processes and substantiates astroglial Cx43 hemichannel mediated HIV induced neuronal damage. Interestingly, DKK1 expression has been found to be upregulated in astrocytes and in other cells in post-mortem human brain tissue samples from HIV-encephalitis cases [78]. Moreover, DKK1 has already been implicated as a key regulator of IFN-γ mediated enhancement of HIV replication and reactivation in astrocytes [79]. However, it remains to be seen whether Cx43 hemichannels also play a role in HIV reactivation in astrocytes.
DKK1 is involved in the embryonic development where it interacts with LP5/6 and Kremen1/2, triggering the internalization of LRP5/6, thereby preventing the formation of Wnt-LRP5/6-Frizzled complex, and subsequently inhibiting the cellular differentiation induced by Wnt signaling. Interestingly, connexin43 acts as a functional target for Wnt signaling as induction of Wnt1 has been shown to enhance electrical and chemical coupling, increase gap junctional communication as well as connexin43 expression [80]. Apart from DKK1, HIV also leads to alterations in the levels of various proteins associated with developmental pathways such as Wnt proteins, β-catenin, Notch, frizzled, LRPs [81, 82]. Our unpublished data using microarray analysis using U87 astrocytoma cells (transfected with CD4 and CCR5; leading to 80–90% HIV infection as opposed to 5–8% infection in primary astrocytes) infected with HIV for 2 days revealed altered regulation of several genes involved in developmental pathways such as Wnt, and β-catenin signaling (Table 1). The re-expression of developmental proteins during HIV infection, their regulation by connexin expression as well as the potential of Wnt/β-catenin signaling to regulate HIV replication [83], reveals a new facet of HIV infection which can deliver tremendous insights into various interconnected development/repair pathways.
Table 1.
mRNA expression of developmental proteins in HIV infection.
| Gene Description | Effect on HIV Infection |
|---|---|
| Dickkopf homolog 1 (DKK1) | Upregulated |
| Rho-associated, coiled-coil containing protein kinase 2 (ROCK2) | Upregulated |
| Low density lipoprotein receptor-related protein associated protein 1 (LRPAP1) | Upregulated |
| Low density lipoprotein receptor-related protein 5-like (LRP5L) | Downregulated |
| Low density lipoprotein-related protein 12 (LRP12) | Upregulated |
| Low density lipoprotein receptor-related protein 8, apolipoprotein e receptor (LRP8) | Downregulated |
| Catenin (cadherin-associated protein), alpha-like 1 (CTNNAL1) | Upregulated |
| Catenin, beta like 1 (CTNNBL1) | Downregulated |
| Catenin (cadherin-associated protein), delta 1 (CTNND1) | Downregulated |
| Catenin (cadherin-associated protein), alpha-like 1 (CTNNAL1) | Upregulated |
| Glycogen synthase kinase 3 beta (GSK3B) | Downregulated |
| Wingless-type MMTV integration site family, member 10A (WNT10A) | Downregulated |
Recently, our laboratory has demonstrated that HIV infection of CD4+ T lymphocytes and PBMCs leads to opening of pannexin1 hemichannels [36]. The opening of pannexin1 channels corresponds to the interaction of viral surface proteins with host CD4 and either CCR5 or CXCR4, followed by release of ATP and subsequent activation of purinergic receptors (Fig. 3B). Moreover, we have also shown that opening of pannexin1 hemichannels was required for viral entry as well as for viral replication in CD4+ T cells. We have proposed that opening of pannexin1 hemichannels leads to a surge in intracellular calcium levels and subsequent actin rearrangement, which is required for successful fusion of the viral particle with the host cell membrane [84]. Our findings are in concordance with observations from another group that in order to facilitate HIV infection, pannexin1 and purinergic receptors are physically recruited to the site of infection [85]. Interestingly, pharmacological or genetic inhibition of the biological activity of pannexin1 hemichannels was also shown to reduce HIV infection and further led to impaired viral replication. Thus, pannexin1 hemichannels, ATP and purinergic receptors play a detrimental role in HIV infection and replication, although the exact mechanism for their interplay with the life-cycle of virus are yet to be fully elucidated.
3.2. Tunneling Nanotubes
Another cell-cell communication system that facilities HIV pathogenesis and plays a role in neuroinflammation through direct long-range exchange of vesicles and organelles is tunneling nanotubes (TNT). Discovered in 2004 by Rustom and colleagues, TNTs are transient actin-rich projections that facilitate long distance intercellular communication [86, 87]. The formation of TNTs has been observed in cells of the immune system (B cells, T cells, NK cells, neutrophils and monocytes) as well as in neurons and astrocytes [88, 89]. Several studies have demonstrated the development of TNT-like structures forming bridges or channels between distant cells that allow transfer of endosomal cargo vesicles, calcium fluxes, and even pathogens such as bacteria, viruses and prions, through their cytoplasm or along the surface of nanotubes [90–92]. TNT like structures have also been identified in astrocytes upon treatment with H2O2, activated microglia in response to calcium, and in monocytes/macrophages exposed to Lipopolysaccharide (LPS) and IFNγ [62, 93, 94]. As a result of the direct cytoplasmic contact between two cells, TNTs coordinate several important biological processes such as development, homeostasis, metabolism and immune response.
Although gap junctions and TNTs both serve to synchronize intercellular communication, the foremost difference between them is the effective distance covered and size of the entities transferred. Whereas TNTs facilitate long-range communication through their extended processes, gap junctions allow only close cell-cell communication between physically adjacent cells. Based on the type of connexin present in the hemichannels, gap junctions enable trafficking of small molecules up to 1.2 kDa [95], whereas TNTs allow exchange of small signaling molecules as well as organelles, vesicles and several pathogens [88]. TNTs can sometimes extend up to 100 μm in length and can be classified into two categories. The ‘thin’ membrane nanotubes are less than 0.7 μm in diameter and are composed mostly of F-actin. These type of nanotubes generally carry portions of plasma membranes between the two connected cells [96]. The second type ‘thicker’ nanotubes are larger (diameter more than 0.7 μm), and are composed of F-actin as well as microtubules. In addition to portions of plasma membranes, the larger nanotubes carry some components of cytoplasm, such as vesicles, mitochondria and other organelles between the connected cells [87]. Although the complete mechanism that facilitates formation of TNTs has not been determined yet, M-Sec, a mammalian protein, in association with RalA (small GTPase) and the exocyst complex, has been shown to induce de-novo formation of membrane protrusions that extend from plasma membrane and tether onto adjacent cells forming TNT-like structures [97–99].
Prior to the discovery of TNTs, several groups reported that herpes virus and pseudo-rabies virus can be efficiently transmitted through elongated tube-like structures without any contact with the extracellular environment. Hence, it was suggested that viruses may have evolved to use TNTs as a mechanism to infect naïve cells, and to establish viral reservoirs bypassing the immune surveillance [100, 101]. Interestingly, prions have also been reported to hijack TNTs associated with dendritic cells to invade the CNS from periphery, and then to spread within neuronal populations at a faster rate [102, 103]. Similar transmission has also been reported for HIV, where ‘membrane nanotubes’ in T cells facilitated the intercellular transfer of HIV through virological synapses or through filopodia between the HIV infected T cell and the target uninfected T cell [104]. Transmission of HIV via T cell nanotubes has been estimated to be approximately 100–1000 fold more efficient than the classical viral transmission, and perhaps partly explains persistent viral spread even in the presence of neutralizing antibodies.
Our laboratory has also documented significant increase in the numbers of TNTs in primary human macrophages upon HIV infection [105]. Interestingly, formation of TNTs in macrophages correlated with the time-course of viral replication, reiterating virus mediated take-over of the host machinery for more efficient viral spread and establishment of reservoirs (Fig. 3C). In addition, we observed co-localization of viral capsid protein, HIV-p24 in TNTs suggesting that viral transmission in macrophages was occurring via TNTs. We also postulated that HIV may use two separate entry mechanisms during initial few days of infection [106]. Since the classical receptor binding mechanism requires several steps, TNTs may help to facilitate rapid infection of a larger population, while simultaneously evading host defence mechanisms or anti-retrovirals blocking early steps of viral entry.
Several other research groups have supported this notion of enhanced viral transfer from infected to uninfected T cells via intercellular structures termed as ‘virological synapses’. Although the relative contribution of virological synapses (VS) as compared to cell-free classical viral transfer has not been carefully evaluated, it was estimated that cell-associated VS mediated transfer of HIV between HIV-expressing Jurkat T cell and primary CD4+ T cell would be 18,000-fold more efficient as compared to cell-free receptor mediated virus uptake [107]. However, it is still not known if virological synapses are the dominant mode of viral transmission in vivo, or if they contribute to viral resistance to neutralizing antibodies, or to the generation of vast genetic diversity of HIV [108, 109].
HIV has also been shown to “hijack” TNTs for transferring viral negative factor, Nef, from infected macrophages to B cells, thereby interfering with IgG2 and IgA class-switching, and subsequently evading humoral immune response systemically and at mucosal sites of entry [110]. HIV Nef, which is known to be transferred to uninfected bystander cells through intercellular contacts and secreted microvesicles, further stimulates the formation of TNTs and virological synapses with nearby uninfected cells. Another study revealed the formation of an octameric complex consisting of Nef, components of the exocyst complex and Pak2 kinase that tethers vesicles at the plasma membrane and regulates their polarized exocytosis, while simultaneously recruiting membrane proteins required for nanotube formation [111].
3.3. Microvesicles/Exosomes
Another intercellular communication system that facilitates HIV dissemination as well as propagation of toxic viral factors are exosomes, a type of cell-secreted microvesicle. Although the effective contribution of exosomes to viral replication and pathogenesis remains largely unexplored, exosomes have been implicated to modulate host cell responses for certain RNA viruses such as HIV, Hepatitis C virus and dengue virus [112]. Exosomes are fluid filled vesicles enclosed by lipid bilayers and have an approximate diameter of 30–100 mm [113]. Exosomes are formed during the maturation of endosomes in the multivesicular bodies [114]. Exosomes are released by virtually all cell types, and their presence has been documented in several bodily fluids such as blood, saliva, CSF, and amniotic fluid [115–118]. Initially considered as a mechanism of discarding cellular waste, exosomes have been implicated as an important mediator for intercellular communication owing to their ability to transfer proteins, lipids, nucleic acids including regulatory RNAs, microRNAs, and small interfering RNAs [119].
Exosomes have been suggested to coordinate intercellular communication during several biological processes such as immune response, tissue homeostasis, neuronal development, and lactation, as well as during pathophysiological conditions such as liver diseases, cancer, neurodegenerative diseases, and viral infections [120–123]. Several recent studies have demonstrated that exosomes released from virally infected cells may carry viral nucleic acids (including miRNAs), proteins, and certain host genetic regulatory factors to uninfected bystander cells that may modulate host cellular response, helping the virus establish productive infection and cellular reservoirs [124–126].
In the context of exosome mediated facilitation of viral pathogenesis, a ‘Trojan exosome hypothesis’ has been proposed for HIV. According to this hypothesis, retroviruses such as HIV utilize the pre-existing host exosome biogenesis pathway for the formation of new virions, as well as for a receptor-independent and viral envelope-independent mode of infection [127]. It is interesting to note that both HIV and exosomes share biochemical properties, and both exploit the same cellular machinery for their assembly as well as for intercellular trafficking [128, 129]. The Trojan exosome hypothesis has been supported by a recent study by Kadiu et al, where they have reported that microvesicles and exosomes facilitated HIV infection in monocyte-derived macrophages [130]. In addition, the authors showed that microvesicles and exosomes were present at the site of viral assembly and assist in the transfer of virus as well as viral constituents from infected macrophages to adjacent uninfected cells. Moreover, HIV was shown to exploit the surface properties of exosomes for speeding up the infection, which could enable the virus to camouflage from the immune surveillance, and partly explain resistance to neutralizing antibodies. The authors also demonstrated that by entrapping or surrounding itself with exosomes, HIV accelerates its infection as well as dissemination to surrounding uninfected cells (Fig. 3D).
Recently, it has been shown that exosomes isolated from HIV-infected patient sera or from HIV-infected cells contain the trans-activation response (TAR) element RNA [131]. These exosomes played a crucial role in propagating HIV infection as prior exposure of naïve cells to exosomes from infected cells increased the susceptibility of HIV infection of the recipient cells as well as downregulated apoptosis by lowering Bim and cyclin-dependent kinase-9 (cdk9) levels. Other than the TAR RNA, exosomes isolated from HIV infected cells have also been shown to incorporate Gag [132] as well as Nef [133, 134] proteins. Interestingly, Nef stimulates its own cellular export via enhancing the release of exosomes from HIV infected cells and facilitates the activation-induced death of CD4+ T-cells, which is a hallmark of HIV pathogenesis [135].
Exosomes have also been shown to aid in HIV propagation via transfer of viral co-receptors, CCR5 as well as CXCR4, from infected cells to uninfected cells which are otherwise non-permissive to HIV owing to lack of these co-receptors [136, 137]. In addition, exosomes isolated from HIV infected patients have been shown to be enriched in cytokines as compared to uninfected donors, and exposure of peripheral blood mononuclear cells to these cytokine-enriched exosomes led to induction of CD38 on naïve and memory T cells, further contributing to inflammation and viral propagation [138]. Hence, exosomes are a crucial intercellular communication system, and play a central role in HIV pathogenesis via transport of virions/viral proteins between infected and uninfected cells, as well as in modulation of immune responses.
4. DISCUSSION
The extensive use of ART has translated AIDS from a death warrant to a chronic disease that is confounded by resistance to anti-retrovirals, associated neuronal toxicities, and chronic immune activation [139–141]. The increased life-span of HIV patients on ART results in chronic exposure of HIV virions and viral proteins in the brain causing neuroinflammation, as well as peripheral immune activation, leading to accumulation of neuropathological damage.
We propose that intercellular communication systems such as gap junctions, hemichannels, tunneling nanotubes as well as exosomes, via the transfer of toxic signals from infected cells to the uninfected cells, aid in HIV induced cellular dysfunction and promote HIV pathogenesis (summarized in Fig. 3). Thorough understanding of the role of cell-cell communication modalities during HIV infection may lead to identification of novel therapeutic targets to subjugate HIV pathogenesis.
Acknowledgments
We would like to thank the PHRI Imaging Facility. This work was supported by the National Institutes of Mental Health grant, MH096625, and PHRI funding to E.A.E.
Biography

Shaily Malik
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
References
- 1.Cysique LA, Maruff P, Brew BJ. Prevalence and pattern of neuropsychological impairment in human immunodeficiency virus-infected/acquired immunodeficiency syndrome (HIV/AIDS) patients across pre- and post-highly active antiretroviral therapy eras: a combined study of two cohorts. Journal of neurovirology. 2004;10(6):350–7. doi: 10.1080/13550280490521078. [DOI] [PubMed] [Google Scholar]
- 2.Heaton RK, Clifford DB, Franklin DR, Jr, Woods SP, Ake C, Vaida F, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology. 2010;75(23):2087–96. doi: 10.1212/WNL.0b013e318200d727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007;69(18):1789–99. doi: 10.1212/01.WNL.0000287431.88658.8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McArthur JC, McDermott MP, McClernon D, St Hillaire C, Conant K, Marder K, et al. Attenuated central nervous system infection in advanced HIV/AIDS with combination antiretroviral therapy. Archives of neurology. 2004;61(11):1687–96. doi: 10.1001/archneur.61.11.1687. [DOI] [PubMed] [Google Scholar]
- 5.Valcour VG, Shikuma CM, Watters MR, Sacktor NC. Cognitive impairment in older HIV-1-seropositive individuals: prevalence and potential mechanisms. AIDS (London, England) 2004;18(Suppl 1):S79–86. doi: 10.1097/00002030-200401001-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Churchill M, Nath A. Where does HIV hide? A focus on the central nervous system. Current opinion in HIV and AIDS. 2013;8(3):165–9. doi: 10.1097/COH.0b013e32835fc601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eugenin EA, Berman JW. Cytochrome C dysregulation induced by HIV infection of astrocytes results in bystander apoptosis of uninfected astrocytes by an IP3 and calcium-dependent mechanism. Journal of neurochemistry. 2013;127(5):644–51. doi: 10.1111/jnc.12443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swingler S, Mann AM, Zhou J, Swingler C, Stevenson M. Apoptotic killing of HIV-1-infected macrophages is subverted by the viral envelope glycoprotein. PLoS pathogens. 2007;3(9):1281–90. doi: 10.1371/journal.ppat.0030134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eugenin EA, Berman JW. Gap junctions mediate human immunodeficiency virus-bystander killing in astrocytes. J Neurosci. 2007;27(47):12844–50. doi: 10.1523/JNEUROSCI.4154-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eugenin EA, Clements JE, Zink MC, Berman JW. Human immunodeficiency virus infection of human astrocytes disrupts blood-brain barrier integrity by a gap junction-dependent mechanism. J Neurosci. 2011;31(26):9456–65. doi: 10.1523/JNEUROSCI.1460-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Antinori A, Perno CF, Giancola ML, Forbici F, Ippolito G, Hoetelmans RM, et al. Efficacy of cerebrospinal fluid (CSF)-penetrating antiretroviral drugs against HIV in the neurological compartment: different patterns of phenotypic resistance in CSF and plasma. Clin Infect Dis. 2005;41(12):1787–93. doi: 10.1086/498310. [DOI] [PubMed] [Google Scholar]
- 12.Eisfeld C, Reichelt D, Evers S, Husstedt I. CSF penetration by antiretroviral drugs. CNS drugs. 2013;27(1):31–55. doi: 10.1007/s40263-012-0018-x. [DOI] [PubMed] [Google Scholar]
- 13.Kim RB, Fromm MF, Wandel C, Leake B, Wood AJ, Roden DM, et al. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. The Journal of clinical investigation. 1998;101(2):289–94. doi: 10.1172/JCI1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gimenez F, Fernandez C, Mabondzo A. Transport of HIV protease inhibitors through the blood-brain barrier and interactions with the efflux proteins, P-glycoprotein and multidrug resistance proteins. Journal of acquired immune deficiency syndromes (1999) 2004;36(2):649–58. doi: 10.1097/00126334-200406010-00001. [DOI] [PubMed] [Google Scholar]
- 15.Clemente MI, Alvarez S, Serramia MJ, Turriziani O, Genebat M, Leal M, et al. Non-steroidal anti-inflammatory drugs increase the antiretroviral activity of nucleoside reverse transcriptase inhibitors in HIV type-1-infected T-lymphocytes: role of multidrug resistance protein 4. Antiviral therapy. 2009;14(8):1101–11. doi: 10.3851/IMP1468. [DOI] [PubMed] [Google Scholar]
- 16.Massarella JW, Nazareno LA, Passe S, Min B. The effect of probenecid on the pharmacokinetics of zalcitabine in HIV-positive patients. Pharmaceutical research. 1996;13(3):449–52. doi: 10.1023/a:1016009029536. [DOI] [PubMed] [Google Scholar]
- 17.Gray F, Hurtrel M, Hurtrel B. Early central nervous system changes in human immunodeficiency virus (HIV)-infection. Neuropathology and applied neurobiology. 1993;19(1):3–9. doi: 10.1111/j.1365-2990.1993.tb00399.x. [DOI] [PubMed] [Google Scholar]
- 18.Meltzer MS, Skillman DR, Gomatos PJ, Kalter DC, Gendelman HE. Role of mononuclear phagocytes in the pathogenesis of human immunodeficiency virus infection. Annual review of immunology. 1990;8:169–94. doi: 10.1146/annurev.iy.08.040190.001125. [DOI] [PubMed] [Google Scholar]
- 19.Williams DW, Eugenin EA, Calderon TM, Berman JW. Monocyte maturation, HIV susceptibility, and transmigration across the blood brain barrier are critical in HIV neuropathogenesis. Journal of leukocyte biology. 2012;91(3):401–15. doi: 10.1189/jlb.0811394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams DW, Calderon TM, Lopez L, Carvallo-Torres L, Gaskill PJ, Eugenin EA, et al. Mechanisms of HIV entry into the CNS: increased sensitivity of HIV infected CD14+CD16+ monocytes to CCL2 and key roles of CCR2, JAM-A, and ALCAM in diapedesis. PloS one. 2013;8(7):e69270. doi: 10.1371/journal.pone.0069270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nature reviews. 2005;5(1):69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- 22.Cosenza MA, Zhao ML, Si Q, Lee SC. Human brain parenchymal microglia express CD14 and CD45 and are productively infected by HIV-1 in HIV-1 encephalitis. Brain pathology (Zurich, Switzerland) 2002;12(4):442–55. doi: 10.1111/j.1750-3639.2002.tb00461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiley CA, Achim CL, Christopherson C, Kidane Y, Kwok S, Masliah E, et al. HIV mediates a productive infection of the brain. AIDS (London, England) 1999;13(15):2055–9. doi: 10.1097/00002030-199910220-00007. [DOI] [PubMed] [Google Scholar]
- 24.Churchill MJ, Wesselingh SL, Cowley D, Pardo CA, McArthur JC, Brew BJ, et al. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Annals of neurology. 2009;66(2):253–8. doi: 10.1002/ana.21697. [DOI] [PubMed] [Google Scholar]
- 25.Carroll-Anzinger D, Kumar A, Adarichev V, Kashanchi F, Al-Harthi L. Human immunodeficiency virus-restricted replication in astrocytes and the ability of gamma interferon to modulate this restriction are regulated by a downstream effector of the Wnt signaling pathway. Journal of virology. 2007;81(11):5864–71. doi: 10.1128/JVI.02234-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tornatore C, Nath A, Amemiya K, Major EO. Persistent human immunodeficiency virus type 1 infection in human fetal glial cells reactivated by T-cell factor(s) or by the cytokines tumor necrosis factor alpha and interleukin-1 beta. Journal of virology. 1991;65(11):6094–100. doi: 10.1128/jvi.65.11.6094-6100.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawrence DM, Durham LC, Schwartz L, Seth P, Maric D, Major EO. Human immunodeficiency virus type 1 infection of human brain-derived progenitor cells. Journal of virology. 2004;78(14):7319–28. doi: 10.1128/JVI.78.14.7319-7328.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Canki M, Thai JN, Chao W, Ghorpade A, Potash MJ, Volsky DJ. Highly productive infection with pseudotyped human immunodeficiency virus type 1 (HIV-1) indicates no intracellular restrictions to HIV-1 replication in primary human astrocytes. Journal of virology. 2001;75(17):7925–33. doi: 10.1128/JVI.75.17.7925-7933.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ranki A, Nyberg M, Ovod V, Haltia M, Elovaara I, Raininko R, et al. Abundant expression of HIV Nef and Rev proteins in brain astrocytes in vivo is associated with dementia. AIDS (London, England) 1995;9(9):1001–8. doi: 10.1097/00002030-199509000-00004. [DOI] [PubMed] [Google Scholar]
- 30.Dewhurst S, Sakai K, Bresser J, Stevenson M, Evinger-Hodges MJ, Volsky DJ. Persistent productive infection of human glial cells by human immunodeficiency virus (HIV) and by infectious molecular clones of HIV. Journal of virology. 1987;61(12):3774–82. doi: 10.1128/jvi.61.12.3774-3782.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brack-Werner R. Astrocytes: HIV cellular reservoirs and important participants in neuropathogenesis. AIDS (London, England) 1999;13(1):1–22. doi: 10.1097/00002030-199901140-00003. [DOI] [PubMed] [Google Scholar]
- 32.Narasipura SD, Kim S, Al-Harthi L. Epigenetic regulation of HIV-1 latency in astrocytes. Journal of virology. 2011;88(5):3031–8. doi: 10.1128/JVI.03333-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tornatore C, Chandra R, Berger JR, Major EO. HIV-1 infection of subcortical astrocytes in the pediatric central nervous system. Neurology. 1994;44(3 Pt 1):481–7. doi: 10.1212/wnl.44.3_part_1.481. [DOI] [PubMed] [Google Scholar]
- 34.Gorry PR, Ong C, Thorpe J, Bannwarth S, Thompson KA, Gatignol A, et al. Astrocyte infection by HIV-1: mechanisms of restricted virus replication, and role in the pathogenesis of HIV-1-associated dementia. Current HIV research. 2003;1(4):463–73. doi: 10.2174/1570162033485122. [DOI] [PubMed] [Google Scholar]
- 35.Eugenin EA, Basilio D, Saez JC, Orellana JA, Raine CS, Bukauskas F, et al. The role of gap junction channels during physiologic and pathologic conditions of the human central nervous system. J Neuroimmune Pharmacol. 2012;7(3):499–518. doi: 10.1007/s11481-012-9352-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orellana JA, Velasquez S, Williams DW, Saez JC, Berman JW, Eugenin EA. Pannexin1 hemichannels are critical for HIV infection of human primary CD4+ T lymphocytes. Journal of leukocyte biology. 2013;94(3):399–407. doi: 10.1189/jlb.0512249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berman JW, Carvallo L, Buckner CM, Luers A, Prevedel L, Bennett MV, et al. HIV-tat alters Connexin43 expression and trafficking in human astrocytes: role in NeuroAIDS. Journal of neuroinflammation. 2016;13(1):54. doi: 10.1186/s12974-016-0510-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Everall IP, Luthert PJ, Lantos PL. Neuronal loss in the frontal cortex in HIV infection. Lancet. 1991;337(8750):1119–21. doi: 10.1016/0140-6736(91)92786-2. [DOI] [PubMed] [Google Scholar]
- 39.Meyerhoff DJ, MacKay S, Bachman L, Poole N, Dillon WP, Weiner MW, et al. Reduced brain N-acetylaspartate suggests neuronal loss in cognitively impaired human immunodeficiency virus-seropositive individuals: in vivo 1H magnetic resonance spectroscopic imaging. Neurology. 1993;43(3 Pt 1):509–15. doi: 10.1212/wnl.43.3_part_1.509. [DOI] [PubMed] [Google Scholar]
- 40.Weis S, Haug H, Budka H. Neuronal damage in the cerebral cortex of AIDS brains: a morphometric study. Acta neuropathologica. 1993;85(2):185–9. doi: 10.1007/BF00227766. [DOI] [PubMed] [Google Scholar]
- 41.Moore DJ, Masliah E, Rippeth JD, Gonzalez R, Carey CL, Cherner M, et al. Cortical and subcortical neurodegeneration is associated with HIV neurocognitive impairment. AIDS (London, England) 2006;20(6):879–87. doi: 10.1097/01.aids.0000218552.69834.00. [DOI] [PubMed] [Google Scholar]
- 42.Ketzler S, Weis S, Haug H, Budka H. Loss of neurons in the frontal cortex in AIDS brains. Acta neuropathologica. 1990;80(1):92–4. doi: 10.1007/BF00294228. [DOI] [PubMed] [Google Scholar]
- 43.Kaul M, Zheng J, Okamoto S, Gendelman HE, Lipton SA. HIV-1 infection and AIDS: consequences for the central nervous system. Cell death and differentiation. 2005;12(Suppl 1):878–92. doi: 10.1038/sj.cdd.4401623. [DOI] [PubMed] [Google Scholar]
- 44.Eugenin EA, King JE, Nath A, Calderon TM, Zukin RS, Bennett MV, et al. HIV-tat induces formation of an LRP-PSD-95- NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(9):3438–43. doi: 10.1073/pnas.0611699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Avdoshina V, Fields JA, Castellano P, Dedoni S, Palchik G, Trejo M, et al. The HIV Protein gp120 Alters Mitochondrial Dynamics in Neurons. Neurotoxicity research. 2016 doi: 10.1007/s12640-016-9608-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.King JE, Eugenin EA, Hazleton JE, Morgello S, Berman JW. Mechanisms of HIV-tat-induced phosphorylation of N-methyl-D-aspartate receptor subunit 2A in human primary neurons: implications for neuroAIDS pathogenesis. The American journal of pathology. 2010;176(6):2819–30. doi: 10.2353/ajpath.2010.090642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nath A, Psooy K, Martin C, Knudsen B, Magnuson DS, Haughey N, et al. Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. Journal of virology. 1996;70(3):1475–80. doi: 10.1128/jvi.70.3.1475-1480.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eugenin EA, D’Aversa TG, Lopez L, Calderon TM, Berman JW. MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. Journal of neurochemistry. 2003;85(5):1299–311. doi: 10.1046/j.1471-4159.2003.01775.x. [DOI] [PubMed] [Google Scholar]
- 49.Haughey NJ, Holden CP, Nath A, Geiger JD. Involvement of inositol 1,4,5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein tat. Journal of neurochemistry. 1999;73(4):1363–74. doi: 10.1046/j.1471-4159.1999.0731363.x. [DOI] [PubMed] [Google Scholar]
- 50.Wang Z, Pekarskaya O, Bencheikh M, Chao W, Gelbard HA, Ghorpade A, et al. Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology. 2003;312(1):60–73. doi: 10.1016/s0042-6822(03)00181-8. [DOI] [PubMed] [Google Scholar]
- 51.Saez JC, Contreras JE, Bukauskas FF, Retamal MA, Bennett MV. Gap junction hemichannels in astrocytes of the CNS. Acta Physiol Scand. 2003;179(1):9–22. doi: 10.1046/j.1365-201X.2003.01196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sosinsky GE, Boassa D, Dermietzel R, Duffy HS, Laird DW, MacVicar B, et al. Pannexin channels are not gap junction hemichannels. Channels (Austin) 2011;5(3):193–7. doi: 10.4161/chan.5.3.15765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bennett MV, Contreras JE, Bukauskas FF, Saez JC. New roles for astrocytes: gap junction hemichannels have something to communicate. Trends Neurosci. 2003;26(11):610–7. doi: 10.1016/j.tins.2003.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheung G, Chever O, Rouach N. Connexons and pannexons: newcomers in neurophysiology. Front Cell Neurosci. 2014;8:348. doi: 10.3389/fncel.2014.00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saez PJ, Shoji KF, Aguirre A, Saez JC. Regulation of hemichannels and gap junction channels by cytokines in antigen-presenting cells. Mediators Inflamm. 2014;2014:742734. doi: 10.1155/2014/742734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oyamada M, Oyamada Y, Takamatsu T. Regulation of connexin expression. Biochimica et biophysica acta. 2005;1719(1–2):6–23. doi: 10.1016/j.bbamem.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 57.Kielian T. Glial connexins and gap junctions in CNS inflammation and disease. Journal of neurochemistry. 2008;106(3):1000–16. doi: 10.1111/j.1471-4159.2008.05405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.De Maio A, Gingalewski C, Theodorakis NG, Clemens MG. Interruption of hepatic gap junctional communication in the rat during inflammation induced by bacterial lipopolysaccharide. Shock. 2000;14(1):53–9. doi: 10.1097/00024382-200014010-00010. [DOI] [PubMed] [Google Scholar]
- 59.Rouach N, Avignone E, Meme W, Koulakoff A, Venance L, Blomstrand F, et al. Gap junctions and connexin expression in the normal and pathological central nervous system. Biol Cell. 2002;94(7–8):457–75. doi: 10.1016/s0248-4900(02)00016-3. [DOI] [PubMed] [Google Scholar]
- 60.Ceelen L, Haesebrouck F, Vanhaecke T, Rogiers V, Vinken M. Modulation of connexin signaling by bacterial pathogens and their toxins. Cell Mol Life Sci. 2011;68(18):3047–64. doi: 10.1007/s00018-011-0737-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eugenin EA, Branes MC, Berman JW, Saez JC. TNF-alpha plus IFN-gamma induce connexin43 expression and formation of gap junctions between human monocytes/macrophages that enhance physiological responses. J Immunol. 2003;170(3):1320–8. doi: 10.4049/jimmunol.170.3.1320. [DOI] [PubMed] [Google Scholar]
- 62.Eugenin EA, Eckardt D, Theis M, Willecke K, Bennett MV, Saez JC. Microglia at brain stab wounds express connexin 43 and in vitro form functional gap junctions after treatment with interferon-gamma and tumor necrosis factor-alpha. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(7):4190–5. doi: 10.1073/pnas.051634298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eugenin EA, Gonzalez HE, Sanchez HA, Branes MC, Saez JC. Inflammatory conditions induce gap junctional communication between rat Kupffer cells both in vivo and in vitro. Cell Immunol. 2007;247(2):103–10. doi: 10.1016/j.cellimm.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Castellano P, Eugenin EA. Regulation of gap junction channels by infectious agents and inflammation in the CNS. Front Cell Neurosci. 2014;8:122. doi: 10.3389/fncel.2014.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bennett MV, Garre JM, Orellana JA, Bukauskas FF, Nedergaard M, Saez JC. Connexin and pannexin hemichannels in inflammatory responses of glia and neurons. Brain Res. 2012;1487:3–15. doi: 10.1016/j.brainres.2012.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ozog MA, Siushansian R, Naus CC. Blocked gap junctional coupling increases glutamate-induced neurotoxicity in neuron-astrocyte co-cultures. Journal of neuropathology and experimental neurology. 2002;61(2):132–41. doi: 10.1093/jnen/61.2.132. [DOI] [PubMed] [Google Scholar]
- 67.Blanc EM, Bruce-Keller AJ, Mattson MP. Astrocytic gap junctional communication decreases neuronal vulnerability to oxidative stress-induced disruption of Ca2+ homeostasis and cell death. Journal of neurochemistry. 1998;70(3):958–70. doi: 10.1046/j.1471-4159.1998.70030958.x. [DOI] [PubMed] [Google Scholar]
- 68.Orellana JA, Saez PJ, Shoji KF, Schalper KA, Palacios-Prado N, Velarde V, et al. Modulation of brain hemichannels and gap junction channels by pro-inflammatory agents and their possible role in neurodegeneration. Antioxid Redox Signal. 2009;11(2):369–99. doi: 10.1089/ars.2008.2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Contreras JE, Sanchez HA, Eugenin EA, Speidel D, Theis M, Willecke K, et al. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(1):495–500. doi: 10.1073/pnas.012589799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Retamal MA, Cortes CJ, Reuss L, Bennett MV, Saez JC. S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: induction by oxidant stress and reversal by reducing agents. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(12):4475–80. doi: 10.1073/pnas.0511118103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stout CE, Costantin JL, Naus CC, Charles AC. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. The Journal of biological chemistry. 2002;277(12):10482–8. doi: 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- 72.Orellana JA, Froger N, Ezan P, Jiang JX, Bennett MV, Naus CC, et al. ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. Journal of neurochemistry. 2011;118(5):826–40. doi: 10.1111/j.1471-4159.2011.07210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Orellana JA, Hernandez DE, Ezan P, Velarde V, Bennett MV, Giaume C, et al. Hypoxia in high glucose followed by reoxygenation in normal glucose reduces the viability of cortical astrocytes through increased permeability of connexin 43 hemichannels. Glia. 2010;58(3):329–43. doi: 10.1002/glia.20926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR. Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci. 2003;23(9):3588–96. doi: 10.1523/JNEUROSCI.23-09-03588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Retamal MA, Froger N, Palacios-Prado N, Ezan P, Saez PJ, Saez JC, et al. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J Neurosci. 2007;27(50):13781–92. doi: 10.1523/JNEUROSCI.2042-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Froger N, Orellana JA, Calvo CF, Amigou E, Kozoriz MG, Naus CC, et al. Inhibition of cytokine-induced connexin43 hemichannel activity in astrocytes is neuroprotective. Mol Cell Neurosci. 2010;45(1):37–46. doi: 10.1016/j.mcn.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 77.Hazleton JE, Berman JW, Eugenin EA. Novel mechanisms of central nervous system damage in HIV infection. HIV AIDS (Auckl) 2010;2:39–49. doi: 10.2147/hiv.s9186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Orellana JA, Saez JC, Bennett MV, Berman JW, Morgello S, Eugenin EA. HIV increases the release of dickkopf-1 protein from human astrocytes by a Cx43 hemichannel-dependent mechanism. Journal of neurochemistry. 2014;128(5):752–63. doi: 10.1111/jnc.12492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li W, Henderson LJ, Major EO, Al-Harthi L. IFN-gamma mediates enhancement of HIV replication in astrocytes by inducing an antagonist of the beta-catenin pathway (DKK1) in a STAT 3-dependent manner. J Immunol. 2011;186(12):6771–8. doi: 10.4049/jimmunol.1100099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.van der Heyden MA, Rook MB, Hermans MM, Rijksen G, Boonstra J, Defize LH, et al. Identification of connexin43 as a functional target for Wnt signalling. J Cell Sci. 1998;111(Pt 12):1741–9. doi: 10.1242/jcs.111.12.1741. [DOI] [PubMed] [Google Scholar]
- 81.Sharma M, Callen S, Zhang D, Singhal PC, Vanden Heuvel GB, Buch S. Activation of Notch signaling pathway in HIV-associated nephropathy. AIDS (London, England) 2010;24(14):2161–70. doi: 10.1097/QAD.0b013e32833dbc31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lodge R, Ouellet M, Barat C, Andreani G, Kumar P, Tremblay MJ. HIV-1 promotes intake of Leishmania parasites by enhancing phosphatidylserine-mediated, CD91/LRP-1-dependent phagocytosis in human macrophages. PloS one. 2012;7(3):e32761. doi: 10.1371/journal.pone.0032761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Narasipura SD, Henderson LJ, Fu SW, Chen L, Kashanchi F, Al-Harthi L. Role of beta-catenin and TCF/LEF family members in transcriptional activity of HIV in astrocytes. Journal of virology. 2012;86(4):1911–21. doi: 10.1128/JVI.06266-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Velasquez S, Eugenin EA. Role of Pannexin-1 hemichannels and purinergic receptors in the pathogenesis of human diseases. Front Physiol. 2014;5:96. doi: 10.3389/fphys.2014.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Seror C, Melki MT, Subra F, Raza SQ, Bras M, Saidi H, et al. Extracellular ATP acts on P2Y2 purinergic receptors to facilitate HIV-1 infection. The Journal of experimental medicine. 2011;208(9):1823–34. doi: 10.1084/jem.20101805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gurke S, Barroso JF, Gerdes HH. The art of cellular communication: tunneling nanotubes bridge the divide. Histochem Cell Biol. 2008;129(5):539–50. doi: 10.1007/s00418-008-0412-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH. Science. 5660. Vol. 303. New York, NY: 2004. Nanotubular highways for intercellular organelle transport; pp. 1007–10. [DOI] [PubMed] [Google Scholar]
- 88.Gerdes HH, Bukoreshtliev NV, Barroso JF. Tunneling nanotubes: a new route for the exchange of components between animal cells. FEBS letters. 2007;581(11):2194–201. doi: 10.1016/j.febslet.2007.03.071. [DOI] [PubMed] [Google Scholar]
- 89.Onfelt B, Nedvetzki S, Yanagi K, Davis DM. Cutting edge: Membrane nanotubes connect immune cells. J Immunol. 2004;173(3):1511–3. doi: 10.4049/jimmunol.173.3.1511. [DOI] [PubMed] [Google Scholar]
- 90.Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, et al. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001;2(4):361–7. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- 91.Sherer NM, Mothes W. Cytonemes and tunneling nanotubules in cell-cell communication and viral pathogenesis. Trends Cell Biol. 2008;18(9):414–20. doi: 10.1016/j.tcb.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Watkins SC, Salter RD. Functional connectivity between immune cells mediated by tunneling nanotubules. Immunity. 2005;23(3):309–18. doi: 10.1016/j.immuni.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 93.Martinez AD, Eugenin EA, Branes MC, Bennett MV, Saez JC. Identification of second messengers that induce expression of functional gap junctions in microglia cultured from newborn rats. Brain Res. 2002;943(2):191–201. doi: 10.1016/s0006-8993(02)02621-5. [DOI] [PubMed] [Google Scholar]
- 94.Zhu D, Tan KS, Zhang X, Sun AY, Sun GY, Lee JC. Hydrogen peroxide alters membrane and cytoskeleton properties and increases intercellular connections in astrocytes. J Cell Sci. 2005;118(Pt 16):3695–703. doi: 10.1242/jcs.02507. [DOI] [PubMed] [Google Scholar]
- 95.Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83(4):1359–400. doi: 10.1152/physrev.00007.2003. [DOI] [PubMed] [Google Scholar]
- 96.Onfelt B, Nedvetzki S, Benninger RK, Purbhoo MA, Sowinski S, Hume AN, et al. Structurally distinct membrane nanotubes between human macrophages support long-distance vesicular traffic or surfing of bacteria. J Immunol. 2006;177(12):8476–83. doi: 10.4049/jimmunol.177.12.8476. [DOI] [PubMed] [Google Scholar]
- 97.Hase K, Kimura S, Takatsu H, Ohmae M, Kawano S, Kitamura H, et al. M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nat Cell Biol. 2009;11(12):1427–32. doi: 10.1038/ncb1990. [DOI] [PubMed] [Google Scholar]
- 98.Kimura S, Hase K, Ohno H. Tunneling nanotubes: emerging view of their molecular components and formation mechanisms. Exp Cell Res. 2012;318(14):1699–706. doi: 10.1016/j.yexcr.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 99.Hashimoto M, Bhuyan F, Hiyoshi M, Noyori O, Nasser H, Miyazaki M, et al. Potential Role of the Formation of Tunneling Nanotubes in HIV-1 Spread in Macrophages. J Immunol. 2016;196(4):1832–41. doi: 10.4049/jimmunol.1500845. [DOI] [PubMed] [Google Scholar]
- 100.Favoreel HW, Van Minnebruggen G, Adriaensen D, Nauwynck HJ. Cytoskeletal rearrangements and cell extensions induced by the US3 kinase of an alphaherpesvirus are associated with enhanced spread. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(25):8990–5. doi: 10.1073/pnas.0409099102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sherer NM, Lehmann MJ, Jimenez-Soto LF, Horensavitz C, Pypaert M, Mothes W. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat Cell Biol. 2007;9(3):310–5. doi: 10.1038/ncb1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gousset K, Schiff E, Langevin C, Marijanovic Z, Caputo A, Browman DT, et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol. 2009;11(3):328–36. doi: 10.1038/ncb1841. [DOI] [PubMed] [Google Scholar]
- 103.Gousset K, Zurzolo C. Tunnelling nanotubes: a highway for prion spreading? Prion. 2009;3(2):94–8. doi: 10.4161/pri.3.2.8917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sowinski S, Jolly C, Berninghausen O, Purbhoo MA, Chauveau A, Kohler K, et al. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat Cell Biol. 2008;10(2):211–9. doi: 10.1038/ncb1682. [DOI] [PubMed] [Google Scholar]
- 105.Eugenin EA, Gaskill PJ, Berman JW. Tunneling nanotubes (TNT) are induced by HIV-infection of macrophages: a potential mechanism for intercellular HIV trafficking. Cell Immunol. 2009;254(2):142–8. doi: 10.1016/j.cellimm.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Eugenin EA, Gaskill PJ, Berman JW. Tunneling nanotubes (TNT): A potential mechanism for intercellular HIV trafficking. Commun Integr Biol. 2009;2(3):243–4. doi: 10.4161/cib.2.3.8165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen P, Hubner W, Spinelli MA, Chen BK. Predominant mode of human immunodeficiency virus transfer between T cells is mediated by sustained Env-dependent neutralization-resistant virological synapses. Journal of virology. 2007;81(22):12582–95. doi: 10.1128/JVI.00381-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen BK. T cell virological synapses and HIV-1 pathogenesis. Immunol Res. 2012;54(1–3):133–9. doi: 10.1007/s12026-012-8320-8. [DOI] [PubMed] [Google Scholar]
- 109.Dale BM, Alvarez RA, Chen BK. Mechanisms of enhanced HIV spread through T-cell virological synapses. Immunol Rev. 2013;251(1):113–24. doi: 10.1111/imr.12022. [DOI] [PubMed] [Google Scholar]
- 110.Xu W, Santini PA, Sullivan JS, He B, Shan M, Ball SC, et al. HIV-1 evades virus-specific IgG2 and IgA responses by targeting systemic and intestinal B cells via long-range intercellular conduits. Nat Immunol. 2009;10(9):1008–17. doi: 10.1038/ni.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mukerji J, Olivieri KC, Misra V, Agopian KA, Gabuzda D. Proteomic analysis of HIV-1 Nef cellular binding partners reveals a role for exocyst complex proteins in mediating enhancement of intercellular nanotube formation. Retrovirology. 2012;9:33. doi: 10.1186/1742-4690-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chahar HS, Bao X, Casola A. Exosomes and Their Role in the Life Cycle and Pathogenesis of RNA Viruses. Viruses. 2015;7(6):3204–25. doi: 10.3390/v7062770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes) The Journal of biological chemistry. 1987;262(19):9412–20. [PubMed] [Google Scholar]
- 114.Vlassov AV, Magdaleno S, Setterquist R, Conrad R. Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochimica et biophysica acta. 2012;1820(7):940–8. doi: 10.1016/j.bbagen.2012.03.017. [DOI] [PubMed] [Google Scholar]
- 115.Thery C, Regnault A, Garin J, Wolfers J, Zitvogel L, Ricciardi-Castagnoli P, et al. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. The Journal of cell biology. 1999;147(3):599–610. doi: 10.1083/jcb.147.3.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Almqvist N, Lonnqvist A, Hultkrantz S, Rask C, Telemo E. Serum-derived exosomes from antigen-fed mice prevent allergic sensitization in a model of allergic asthma. Immunology. 2008;125(1):21–7. doi: 10.1111/j.1365-2567.2008.02812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Keryer-Bibens C, Pioche-Durieu C, Villemant C, Souquere S, Nishi N, Hirashima M, et al. Exosomes released by EBV-infected nasopharyngeal carcinoma cells convey the viral latent membrane protein 1 and the immunomodulatory protein galectin 9. BMC Cancer. 2006;6:283. doi: 10.1186/1471-2407-6-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Street JM, Barran PE, Mackay CL, Weidt S, Balmforth C, Walsh TS, et al. Identification and proteomic profiling of exosomes in human cerebrospinal fluid. J Transl Med. 2012;10:5. doi: 10.1186/1479-5876-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Keller S, Sanderson MP, Stoeck A, Altevogt P. Exosomes: from biogenesis and secretion to biological function. Immunol Lett. 2006;107(2):102–8. doi: 10.1016/j.imlet.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 120.Masyuk AI, Masyuk TV, Larusso NF. Exosomes in the pathogenesis, diagnostics and therapeutics of liver diseases. J Hepatol. 2013;59(3):621–5. doi: 10.1016/j.jhep.2013.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bard MP, Hegmans JP, Hemmes A, Luider TM, Willemsen R, Severijnen LA, et al. Proteomic analysis of exosomes isolated from human malignant pleural effusions. Am J Respir Cell Mol Biol. 2004;31(1):114–21. doi: 10.1165/rcmb.2003-0238OC. [DOI] [PubMed] [Google Scholar]
- 122.Romagnoli GG, Zelante BB, Toniolo PA, Migliori IK, Barbuto JA. Dendritic Cell-Derived Exosomes may be a Tool for Cancer Immunotherapy by Converting Tumor Cells into Immunogenic Targets. Front Immunol. 2014;5:692. doi: 10.3389/fimmu.2014.00692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schorey JS, Bhatnagar S. Exosome function: from tumor immunology to pathogen biology. Traffic. 2008;9(6):871–81. doi: 10.1111/j.1600-0854.2008.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Meckes DG, Jr, Gunawardena HP, Dekroon RM, Heaton PR, Edwards RH, Ozgur S, et al. Modulation of B-cell exosome proteins by gamma herpesvirus infection. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(31):E2925–33. doi: 10.1073/pnas.1303906110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Meckes DG, Jr, Shair KH, Marquitz AR, Kung CP, Edwards RH, Raab-Traub N. Human tumor virus utilizes exosomes for intercellular communication. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(47):20370–5. doi: 10.1073/pnas.1014194107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vojtech L, Woo S, Hughes S, Levy C, Ballweber L, Sauteraud RP, et al. Exosomes in human semen carry a distinctive repertoire of small non-coding RNAs with potential regulatory functions. Nucleic Acids Res. 2014;42(11):7290–304. doi: 10.1093/nar/gku347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gould SJ, Booth AM, Hildreth JE. The Trojan exosome hypothesis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(19):10592–7. doi: 10.1073/pnas.1831413100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pelchen-Matthews A, Raposo G, Marsh M. Endosomes, exosomes and Trojan viruses. Trends Microbiol. 2004;12(7):310–6. doi: 10.1016/j.tim.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 129.Booth AM, Fang Y, Fallon JK, Yang JM, Hildreth JE, Gould SJ. Exosomes and HIV Gag bud from endosome-like domains of the T cell plasma membrane. The Journal of cell biology. 2006;172(6):923–35. doi: 10.1083/jcb.200508014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kadiu I, Narayanasamy P, Dash PK, Zhang W, Gendelman HE. Biochemical and biologic characterization of exosomes and microvesicles as facilitators of HIV-1 infection in macrophages. J Immunol. 2012;189(2):744–54. doi: 10.4049/jimmunol.1102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Narayanan A, Iordanskiy S, Das R, Van Duyne R, Santos S, Jaworski E, et al. Exosomes derived from HIV-1-infected cells contain trans-activation response element RNA. The Journal of biological chemistry. 2013;288(27):20014–33. doi: 10.1074/jbc.M112.438895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fang Y, Wu N, Gan X, Yan W, Morrell JC, Gould SJ. Higher-order oligomerization targets plasma membrane proteins and HIV gag to exosomes. PLoS biology. 2007;5(6):e158. doi: 10.1371/journal.pbio.0050158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Campbell TD, Khan M, Huang MB, Bond VC, Powell MD. HIV-1 Nef protein is secreted into vesicles that can fuse with target cells and virions. Ethnicity & disease. 2008;18(2 Suppl 2):S2–14-9. [PMC free article] [PubMed] [Google Scholar]
- 134.Raymond AD, Campbell-Sims TC, Khan M, Lang M, Huang MB, Bond VC, et al. HIV Type 1 Nef is released from infected cells in CD45(+) microvesicles and is present in the plasma of HIV-infected individuals. AIDS research and human retroviruses. 2011;27(2):167–78. doi: 10.1089/aid.2009.0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lenassi M, Cagney G, Liao M, Vaupotic T, Bartholomeeusen K, Cheng Y, et al. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic. 2010;11(1):110–22. doi: 10.1111/j.1600-0854.2009.01006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mack M, Kleinschmidt A, Bruhl H, Klier C, Nelson PJ, Cihak J, et al. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: a mechanism for cellular human immunodeficiency virus 1 infection. Nature medicine. 2000;6(7):769–75. doi: 10.1038/77498. [DOI] [PubMed] [Google Scholar]
- 137.Rozmyslowicz T, Majka M, Kijowski J, Murphy SL, Conover DO, Poncz M, et al. Platelet- and megakaryocyte-derived microparticles transfer CXCR4 receptor to CXCR4-null cells and make them susceptible to infection by X4-HIV. AIDS (London, England) 2003;17(1):33–42. doi: 10.1097/00002030-200301030-00006. [DOI] [PubMed] [Google Scholar]
- 138.Konadu KA, Chu J, Huang MB, Amancha PK, Armstrong W, Powell MD, et al. Association of Cytokines With Exosomes in the Plasma of HIV-1-Seropositive Individuals. The Journal of infectious diseases. 2015;211(11):1712–6. doi: 10.1093/infdis/jiu676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nath A, Sacktor N. Influence of highly active antiretroviral therapy on persistence of HIV in the central nervous system. Current opinion in neurology. 2006;19(4):358–61. doi: 10.1097/01.wco.0000236614.51592.ca. [DOI] [PubMed] [Google Scholar]
- 140.Lindl KA, Marks DR, Kolson DL, Jordan-Sciutto KL. HIV-associated neurocognitive disorder: pathogenesis and therapeutic opportunities. J Neuroimmune Pharmacol. 2010;5(3):294–309. doi: 10.1007/s11481-010-9205-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.McArthur JC, Steiner J, Sacktor N, Nath A. Human immunodeficiency virus-associated neurocognitive disorders: Mind the gap. Annals of neurology. 2010;67(6):699–714. doi: 10.1002/ana.22053. [DOI] [PubMed] [Google Scholar]