

Abstract
The design, synthesis, and evaluation of methyl 1,2,8,8a-tetrahydrocyclopropa[c]imidazolo[4,5-e]indol-4-one-6-carboxylate (CImI) derivatives are detailed representing analogs of duocarmycin SA and yatakemycin containing an imidazole replacement for the fused pyrrole found in the DNA alkylation subunit.
Keywords: Duocarmycin, Yatakemycin, CC-1065, DNA alkylation
Graphical abstract
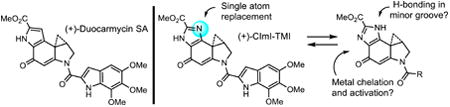
1. Introduction
Duocarmycin SA (1),1 yatakemycin (2),2 and CC-1065 (3)3 are the parent members of a class of compounds that derive their antitumor activity from their ability to alkylate DNA (Figure 1).4 The study of the natural products,5-8 their synthetic unnatural enantiomers,9 and key analogs10 has defined structural features that control their DNA alkylation selectivity,4,10 efficiency, rate, reversibility,11 and catalysis,12 providing a detailed understanding of the relationships between structure, reactivity, and biological activity.13 Central to these studies have been modifications in the DNA alkylation subunit of the natural products and the establishment of the impact that the deep-seated changes have on their functional reactivity and biological properties.13 Herein, we report the total synthesis and examination of the methyl 1,2,8,8a-tetrahydrocyclopropa[c]imidazolo[4,5-e]indol-4-one-6-carboxylate (CImI) alkylation subunit and its incorporation into key analogs of the natural products (Figure 1). The CImI alkylation subunit incorporates a single heavy atom change (aryl CH to N) to the alkylation subunit of duocarmycin SA and yatakemycin, replacing the fused pyrrole with a fused imidazole and complementing a now growing series of such heterocyclic analogs.14-22 In addition to the impact this change may have on the alkylation subunit intrinsic reactivity and reaction regioselectivity, the tautomeric imidazole presents the opportunity for H-bonding on the DNA bound face of the molecule (H-bond donor or acceptor) potentially impacting the DNA alkylation selectivity, and offers the potential for selective metal cation complexation and activation of the reactive cyclopropane analogous to the pyridine analogue CPyI.17 Finally, this change represents the composite structure of the duocarmycin SA and iso-duocarmycin SA alkylation subunits,20 the latter of which is a potent but isomeric variant of the natural alkylation subunit.
Figure 1.

Representative natural products in class and structure of CImI.
The alkylation subunits incorporate a vinylogous amide, which provides stability to what would otherwise be a reactive cyclopropane.10,12 Disruption of this vinylogous amide occurs upon a minor groove binding-induced conformational change, which brings the cyclopropane into conjugation with the cyclohexadienone and activates it for nucleophilic attack. Thus, the compounds are ordinarily unreactive, but are activated for adenine N3 alkylation upon target DNA binding.13,23 Moreover, this reactivity is still attenuated,9 allowing selective reaction by appropriately positioned adenines within the preferred AT-rich non-covalent binding sites such that it is the non-covalent binding selectivity of the compounds that controls the alkylation site selectivity.24 It was not apparent how the substitution of the fused imidazole of CImI for the pyrrole would impact this key structural feature of the natural products.
2. Results and discussion
2.1. Synthesis of CImI25
O-Benzylation of 2-hydroxy-4-nitroaniline (K2CO3, BnBr, acetone, reflux, 8 h, 88%) followed by iodination with ICl (THF, reflux, 3 h, 86%) provided 426 following a reported synthesis (Scheme 2). Acid-catalyzed amidine formation (4 N HCl, dioxane, 23 °C, 12 h, 88%) with methyl cyanoformate (4 equiv) cleanly and directly provided 5 in a reaction that is of special note for its effectiveness given the non-nucleophilic and hindered nature of the reacting amine. Intramolecular Cu(I)-catalyzed closure of 5 to the benzimidazole with C-N bond formation cleanly provided 6 in excellent conversion (0.05 equiv CuI, 0.1 equiv 1,10-phen, 1.25 equiv Cs2CO3, DME, 80 °C, 16 h).27 N-Benzyl protection (1.5 equiv Cs2CO3, 2 equiv BnBr, DMF, 23 °C, 12 h, 70%) of the imidazole 6, permitting protection during a later stage alkylation, provided a 1.6:1 mixture of isomeric N-benzyl products (7 + isomer) that in principle could both be carried through to the final products. The aryl nitro group of the isomeric mixture was reduced to the corresponding amine 8 (10 equiv Zn-nanopowder, 15 equiv NH4Cl, acetone/H2O, 23 °C, 10 min), the resulting anilines were chromatographically separated, and isomer 8 was protected to provide 9 (50% from 7) using Boc2O (2 equiv, THF, 70 °C, 12 h). Perhaps unnecessary, this did provide isomerically pure materials simplifying product characterizations and the work was carried forward using the major N3 isomer 9 depicted in Scheme 2. Regioselective C4-iodination using N-iodosuccinimide (1.2 equiv NIS, 0.1 equiv TsOH, THF/MeOH 1:1, 23 °C, 12 h, 86%) afforded 10. A single crystal X-ray structure determination conducted with 10 confirmed the assigned structure and unambiguously established the structures of the two N-benzyl regioisomers.28 Carbamate 10 was alkylated with 1,3-dichloropropene (2 equiv, 1.5 equiv NaH, DMF, 23 °C, 5 h, 56%) to afford 11. The final 5-membered ring of the alkylation subunit was constructed using a tris(trimethylsilyl)silane-mediated 5-exo-trig aryl radical–alkene cyclization (2 equiv (Me3Si)3SiH, 0.5 equiv AIBN, benzene, 80 °C, 4 h, 69%) of 11 to afford 12.29 An analogous Bu3SnH-mediated aryl radical– alkene cyclization (1.4 equiv Bu3SnH, 0.1 equiv AIBN, benzene, 70 °C, 7 h, 73%) also provided 12 in comparable conversions, but was more sensitive to the quality of commercial reagent available. Treatment of 12 with 10% Pd/C under an atmosphere of hydrogen gas (50 psi H2, THF, 25 °C, 2 h, 82%) effectively cleaved both the O- and N-benzyl protecting groups, affording 13. This could also be accomplished using transfer hydrogenolysis (20% w/w Pd(OH)2, 50 equiv 1,4 cyclohexadiene, EtOH, 80 °C, 2 h, 75%), albeit with a greater degree of competitive dechlorination. Resolution of 13 into its enantiomers was possible at this stage on a ChiralPak AD semi-preparative HPLC column (2 × 25 cm, 10% i-PrOH/hexane, 7 mL/min, α = 1.30). Spirocyclization was accomplished under exceptionally mild conditions upon treatment of 13 with aqueous 1 M NaHCO3 in THF (23 °C, 4 h, 59%) to afford 14 (Scheme 1, natural enantiomer shown).
Scheme 2.

Synthesis of CImI-TMI and CImI-indole2.
Scheme 1.

Synthesis of N-Boc-CImI.
Boc deprotection of 13 (4 N HCl/EtOAc, 23 °C, 2 h) followed by direct coupling of the resulting indoline hydrochloride salt with 5,6,7-trimethoxyindole-2-carboxylic acid30 (19, 1.3 equiv, 3.0 equiv EDCI, DMF, 23 °C, 48 h, 61%) afforded 15 (Scheme 2, natural enantiomer shown) that was spirocyclized under mild conditions (aqueous 1 M NaHCO3, THF, 23 °C, 4 h, 65%) to afford 16 (CImI-TMI). Similarly, sequential Boc deprotection of 13 (4 N HCl/EtOAc, 23 °C, 2 h), coupling with carboxylic acid 20 (1.3 equiv, 3.0 equiv EDCI, DMF, 23 °C, 48 h, 69%), and spirocyclization of 17 provided 18 (CImI-indole2, aqueous 1 M NaHC03, THF, 23 °C, 4 h, 60%) in good conversions.
2.2. Reactivity and reaction regioselectivity
Both the intrinsic reactivity of the alkylation subunits as assessed by the rate of acid-catalyzed solvolysis and the regioselectivity of addition to their activated cyclopropane have proven key to understanding the behavior of this class of compounds. The compared rates of solvolysis provide insights into the structural features that stabilize the reacting cyclopropane as well as into the source of catalysis for the DNA alkylation reaction, and their study defined a fundamental parabolic relationship between the alkylation subunit intrinsic reactivity and biological potency.31 The regioselectivity of the ring opening reaction involved in the DNA alkylation is controlled by the stereoelectronic alignment of the cyclopropane bonds with the cyclohexadienone π-system.32
The solvolysis reactivity of N-Boc-CImI (14) was followed spectrophotometrically by UV in pH 7 and pH 3 phosphate buffer (Figure 2). No measurable solvolysis was observed at pH 7 and it was found to exhibit a solvolysis rate constant of k = 5.52 × 10−6 s−1 and t1/2 = 34.7 h at pH 3. This reactivity is on par with that observed with N-Boc-MeCPI (k = 5.26 × 10−6 s−1, t1/2 = 37 h),31 the alkylation subunit of CC-1065. It is however, more reactive than N-Boc-DSA, which is also stable at pH 7, and exhibits an even lower solvolysis reactivity at pH 3 (k = 1.08 × 10−6 s−1, t1/2 = 177 h).10 Thus, the relative reactivity of CImI is essentially equal with that of CPI and it is 5-times more reactive than DSA toward acid-catalyzed solvolysis.
Figure 2.
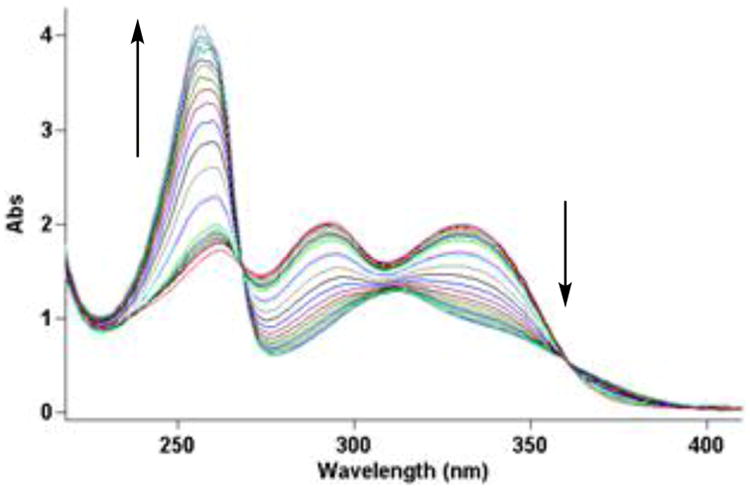
Solvolysis study (UV spectra) of N-Boc-CImI (14) in 50% MeOH–aqueous buffer (pH 3, 4:1:20 (v/v/v) 0.1 M citric acid, 0.2 M NaH2PO4, and H2O, respectively). The spectra were recorded at regular intervals from 0–187 h.
Treatment of 14 with 4 N HCl in EtOAc at −78 °C (1.5 h) provided a single chloride addition product 13 in near quantitative yield (Scheme 3). Exclusive chloride addition to the less substituted cyclopropane carbon was observed, and no ring expanded 6-membered ring addition product, resulting from attack on the more hindered carbon, was detected. Treatment of 14 with 0.35 equiv of CF3SO3H in MeOH at 0 °C slowly (24 h) led to cyclopropane ring opening, providing a 3:1 ratio of products 21:22, representing nucleophilic addition to the least substituted and more substituted cyclopropane carbons, respectively, and displaying a reaction regioselectivity under these conditions that is similar to that of N-Boc-DSA (4–6.5:1)10b but lower than that of N-Boc-CBI (>20:1).32
Scheme 3.

Acid-catalyzed Addition Reactions.
2.3. Metal-catalyzed nucleophilic addition
In a past study, we disclosed the metal-catalyzed nucleophilic addition reactions of CPyI, a similar alkylation subunit that possesses a fused pyridine in place of the imidazole of CImI or pyrrole of DSA. That alkylation subunit and analogs incorporating CPyI were unreactive toward conventional nucleophiles at pH 7, but were capable of metal cation-catalyzed reaction with nucleophiles. Moreover, this reactivity was tunable by the choice of metal and displayed relative rates that reflected the relative stability constants of the product 8-hydroxyquinoline metal complexes (Cu+2 > Ni+2 > Zn+2 > Mn+2 > Mg+2).17 Such behavior is not observed with the DSA alkylation subunit or CBI, indicating that it was intrinsic to the 8-ketoquinoline structure of CPyI. We anticipated that CImI might display this same behavior but exhibit an altered trend in reactivity, reflecting the relative stability constants of 7-hydroxybenzimidazole metal complexes (Cu+2 > Pb+2 > Cd+2 > Zn+2 > Ni+2).33 However, we found that N-Boc-CImI (14) failed to undergo solvolysis catalysis by any metal cation examined, rather that it behaved identical to N-Boc-DSA, being unaffected by treatment with Cu(acac)2, Ni(acac)2, Zn(acac)2, Pb(acac)2, or Cd(acac)2 (1 equiv) at 23 °C in MeOH.
2.4. Cell growth inhibition
Both enantiomers of compounds 14, 16, and 18 were assayed for cell growth inhibition (cytotoxic) activity against the L1210 tumor cell line (mouse leukemia cell line) that has been traditionally used initially to examine members in this class and the results are presented in Figure 3 alongside the comparison DSA-based compounds. The natural enantiomers of the compounds were found to display potent activity with IC50's of 70 nM, 50 pM and 40 pM, respectively. Although this represents a 5–10 fold drop in potency when compared to the corresponding DSA compounds themselves10 (IC50's = 6 nM, 10 pM, and 4 pM, respectively), the activity is on par with that observed with the CBI-based (e.g., CBI-TMI, IC50 = 30 pM)34 or CPI-based analogues and is consistent with the observation that CImI is five times more reactive than DSA. The unnatural enantiomers were ca. >20-fold less active, similarly following trends observed in prior alkylation subunit analogs.
Figure 3.

In vitro cytotoxic activity.
Moreover, this level of cytotoxic activity with the natural enantiomers is in line with expectations based on the relative stability of compounds and follows trends depicted in a parabolic relationship between reactivity and cytotoxic activity (Figure 4).31 This parabolic relationship, derived from data from more than 30 deep-seated modifications that span a 104–106 range of reactivity and activity, simply reflects the fact that the compound must be sufficiently stable to reach its biological target yet remain sufficiently reactive to alkylate DNA once it does.
Figure 4.
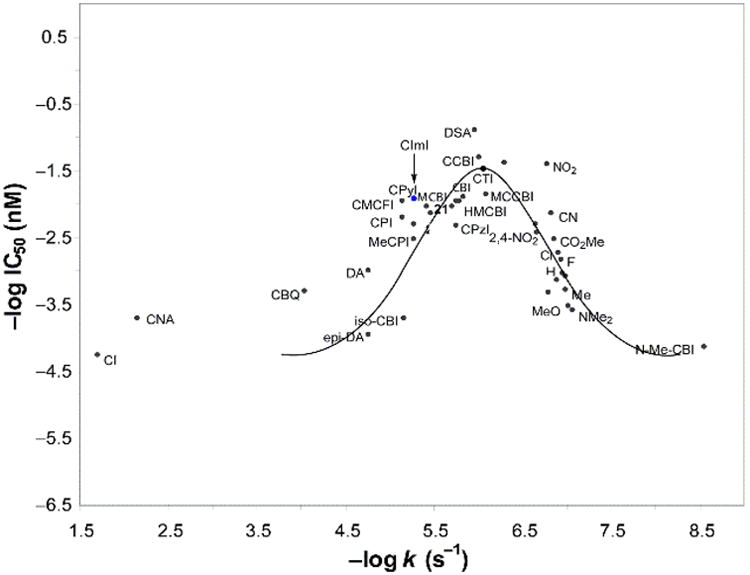
Plot of -log IC50 (nM) vs −log k (s−1) illustrating the parabolic relationship between reactivity and cytotoxic activity and the placement of CImI in this relationship.
2.5. DNA alkylation
The DNA alkylation selectivity and efficiency of the CImI analogues were examined within a 150 base-pair segment of DNA described previously (w794).35 The alkylation site identification and the assessment of the relative selectivity among the available sites were obtained by thermally-induced strand cleavage of the singly 5′-end-labeled duplex DNA after exposure to the compounds as detailed.35 The examination of (+)-CImI-TMI (16), conducted with the equivalent seco precursor 15, demonstrated that it alkylated the same single site as (+)-duocarmycin SA in w794, displaying the same characteristic DNA alkylation selectivity (Figure 5). In addition, the natural enantiomer (+)-16 alkylated DNA with an efficiency only 2-fold less than (+)-duocarmycin SA (data not shown), consistent with its 5-fold less potent cytotoxic activity. Like duocarmycin SA, the unnatural enantiomer of 16 proved less efficient at alkylating DNA than its natural enantiomer, and both unnatural enantiomers alkylated the same single major site. The results of these studies indicate that the incorporation of the additional nitrogen atom of the benzimidazole on the face of the compound that extends into the bottom of the minor groove does not alter the DNA alkylation selectivity despite its potential capabilities to serve as either a H-bond acceptor or H-bond donor, and that it has only a minor impact on the relative efficiency of DNA alkylation.
Figure 5.
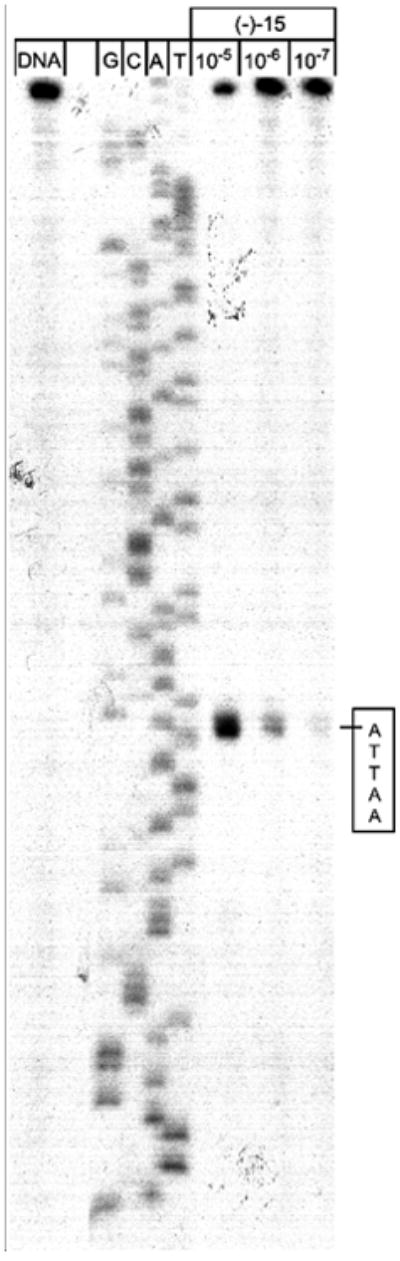
Thermally-induced strand cleavage of w794 DNA following DNA alkylation; DNA–agent incubation at 23 °C for 24 h, removal of unbound agent by EtOH precipitation, and 30 min of thermolysis (100 °C) followed by 8% denaturing PAGE and autoradiography. Lane 1, control DNA; lanes 2–5, Sanger G, C, A, and T sequencing reactions; lanes 6–8, (–)-15 (natural enantiomer at 1 × 10−5, 1 × 10−6, and 1 × 10−7 M−1).
3. Conclusions
A single heavy atom modification to the alkylation subunit of duocarmycin SA was examined with the synthesis and evaluation of analogs containing the CImI alkylation subunit, replacing the fused pyrrole with a fused imidazole and complementing a now growing series of such heterocyclic analogs. In addition to the impact this change could have on the alkylation subunit intrinsic reactivity and reaction regioselectivity, the tautomeric imidazole presented the opportunity for H-bonding on the DNA bound face of the molecule (H-bond donor or acceptor) potentially impacting the DNA alkylation selectivity, and offered the potential for selective metal cation complexation and activation of the reactive cyclopropane. It was found that this modification reduced the intrinsic stability 5-fold, correspondingly reduced the cell growth inhibition properties 5–10 fold, had no impact on the DNA alkylation selectivity but slightly reduced its efficiency, and provided a heterocyclic alkylation subunit that was not subject to a metal-cation catalyzed reactivity. Nonetheless, the CImI-based analogs remain remarkably potent (e.g., (+)-CImI-TMI IC50 = 50 pM), displaying activity on par with that observed with MeCPI-based analogs (e.g., CC-1065) and the widely used CBI-based analogs.
4. Experimental
4.1. Methyl 2-((2-(Benzyloxy)-6-iodo-4-nitrophenyl)amino)-2-iminoacetate (5)
A solution of 4 (5.71 g, 15.4 mmol) in 4 N HCl in dioxane (87 mL) was treated dropwise with methyl cyanoformate (5.25 g, 61.7 mmol). The reaction mixture was stirred at room temperature overnight and the solvent was removed under a stream of N2. The solid residue was dissolved and partitioned between EtOAc and saturated aqueous NaHCO3, and the organic phase was washed with saturated aqueous NaCl and dried (Na2SO4). The solvent was removed in vacuo and the residue was purified by column chromatography (SiO2, 30% EtOAc/hexanes) to afford 5 (6.20 g, 13.6 mmol, 88%) as an orange solid: 1H NMR (CD3OD, 400 MHz) δ 8.32 (d, J = 1.5 Hz, 1H), 7.89 (d, J = 1.6 Hz, 1H), 7.43 (d, J = 4.7 Hz, 2H), 7.35 (t, J = 4.7 Hz, 2H), 7.30 (t, J = 4.8 Hz, 1H), 5.18 (s, 2H), 3.91 (s, 3H); 13C NMR (CD3OD, 100 MHz) δ 163.0, 150.4, 149.0, 147.7, 146.0, 137.6, 129.6, 129.1, 128.4, 127.6, 109.2, 92.1, 72.0, 53.9; IR (film) νmax 3467, 3357, 3090, 2952, 1737, 1656, 1510, 1330, 1260 cm−1; ESI-TOF HRMS m/z 456.0054 (M+H+, C16H14IN3O5 requires 456.0051).
4.2. Methyl 7-(Benzyloxy)-5-nitrobenzimidazole-2-carboxylate (6)
A suspension of 5 (7.92 g, 17.4 mmol), Cs2CO3 (7.08 g, 21.7 mmol), CuI (165 mg, 0.87 mmol), 1,10-phenanthroline (326 mg, 1.8 mmol) in DME (170 mL) under Ar was stirred and warmed at 80 °C for 16 h. The reaction mixture was cooled to 0 °C and 93 mL of aqueous 0.4 N HCl was slowly added. The aqueous phase was extracted with EtOAc and the combined organic extracts were washed with saturated aqueous NaCl, dried (Na2SO4) and concentrated in vacuo. The crude product (quantitative yield) was typically advanced to next step without further purification. Material loss due to adhesion to SiO2 was observed if the reaction product was purified by column chromatography. For the reaction above, chromatography (SiO2, 20–40% EtOAc/hexane gradient) afforded 6 as a solid (3.66 g, 11.2 mmol, 64%): 1H NMR (DMSO-d6, 500 MHz, 70 °C) δ 8.13 (br s, 1H), 7.71 (s, 1H), 7.57 (d, J = 7.4 Hz, 2H), 7.43 (t, J = 7.0 Hz, 2H), 7.36 (t, J = 7.3 Hz, 1H), 5.47 (s, 2H), 3.99 (s, 3H); 13C NMR (DMSO-d6, 150 MHz) δ 158.9, 150.8, 145.2, 144.6, 137.6, 136.1, 134.4, 128.5, 128.1, 127.9, 102.5, 99.8, 70.4, 52.9; ESI-TOF HRMS m/z 328.0932 (M+H+, C16H13N3O5 requires 328.0928).
4.3. Methyl 1-Benzyl-7-(benzyloxy)-5-((tert-butyloxycarbonyl)amino)benzimidazole-2-carboxylate (9)
A solution 6 (1.03 g, 3.1 mmol) in DMF (20 mL) was treated with Cs2CO3 (1.55 g, 4.8 mmol) and the reaction mixture was stirred for 30 min at 23 °C. The resultant solution was treated with benzyl bromide (1.08 g, 6.3 mmol) and the reaction mixture was stirred at 23 °C overnight. The reaction mixture was diluted by addition of water and the aqueous portion was extracted with EtOAc. The combined organic extracts were washed with water, saturated aqueous LiCl and saturated aqueous NaCl, dried (Na2SO4) and concentrated in vacuo. Flash column chromatography afforded 7 (SiO2, 0.92 g, 2.2 mmol, 70%) as a 1:1.6 (minor: major (7) respectively) mixture of regioisomers.
The mixture of 7 and its isomer (1.0 g, 2.4 mmol) was dissolved in acetone (35 mL) and H2O (7 mL) containing NH4Cl (1.92 g, 35.9 mmol). This solution was treated with Zn-nano powder (1.56 g, 23.9 mmol) and the reaction mixture was vigorously stirred at 23 °C for 5–10 min. The reaction mixture was filtered and most of the acetone was removed in vacuo. The mixture was diluted with EtOAc and filtered again. The aqueous layer was separated and the organic phase was washed with saturated aqueous NaCl, dried (Na2SO4) and concentrated. The more polar isomer was isolated as a solid by column chromatography (SiO2, 1% MeOH/CH2Cl2). The amine product (472 mg, 1.2 mmol) was dissolved in THF (6.5 mL) and Boc2O (1.06 g, 4.9 mmol) was added. The reaction mixture was warmed at 70 °C overnight and cooled to room temperature. The solvent was removed in vacuo and the residual oil was purified by flash column chromatography (SiO2, 30% EtOAc/hexanes) to afford 9 (590 mg, 1.2 mmol, 50%) as a light yellow solid: 1H NMR (CDCl3, 400 MHz) δ 7.47 (br s, 1H), 7.26–7.32 (m, 3H), 7.18–7.20 (m, 3H), 7.13–7.15 (m, 3H), 6.87–6.89 (m, 2H), 6.66 (s, 1H), 6.10 (s, 2H), 5.09 (s, 2H), 3.96 (s, 3H), 1.53 (s, 9H); 13C NMR (CDCl3, 100 MHz) δ 160.1, 153.1, 146.8, 143.5, 140.9, 138.1, 135.8, 135.7, 128.6, 128.5, 128.4, 128.1, 127.2, 126.2, 123.1, 102.6, 101.1, 80.6, 70.9, 53.0, 50.2, 28.5 IR (film) νmax 2976, 1720, 1597, 1553, 1482, 1453 cm−1; ESI-TOF HRMS m/z 488.2178 (M+H+, C28H29N3O5 requires 488.2180).
4.4. Methyl 1-Benzyl-7-(benzyloxy)-5-((tert-butyloxycarbonyl)amino)-4-iodobenzimidazole-2-carboxylate (10)
A solution of 9 (684 mg, 1.4 mmol) and p-TsOH·H2O (27 mg, 0.14 mmol) in THF (10.5 mL) and MeOH (10.5 mL) was treated with NIS (378 mg, 1.7 mmol). The reaction vessel was covered in aluminum foil and the mixture was stirred overnight at 23 °C. A saturated solution of Na2S2O3 (6 mL) was added to the reaction mixture and stirred for 30 min. The reaction mixture was extracted with EtOAc and the combined organic phase was washed with saturated aqueous NaCl, dried (Na2SO4) and concentrated in vacuo. The residue was purified by flash column chromatography (SiO2, 2% MeOH/CH2Cl2) to afford 10 (742 mg, 1.2 mmol, 86%) as a pale yellow solid: 1H NMR (acetone-d6, 400 MHz) δ 7.74 (s, 1H), 7.50 (s, 1H), 7.30–7.33 (m, 5H), 7.22–7.26 (m, 3H), 6.94 (d, J = 4.8 Hz, 2H), 6.10 (s, 2H), 5.20 (s, 2H), 3.92 (s, 3H), 1.53 (s, 9H); 13C NMR (acetone-d6, 100 MHz) δ 160.8, 153.9, 147.8, 145.4, 141.9, 139.2, 137.7, 136.8, 129.3, 129.3, 129.1, 129.1, 128.0, 127.9, 127.0, 123.1, 104.6, 80.9, 71.6, 53.0, 51.2, 28.5; IR (film) νmax 1726, 1532, 1484, 1267, 1158 cm−1; ESI-TOF HRMS m/z 614.1144 (M+H+, C28H28IN3O5 requires 614.1146).
4.5. Methyl (Z)-1-Benzyl-7-(benzyloxy)-5-((tert-butyloxycarbonyl)(3-chloroallyl)amino)-4-iodo-benzimidazole-2-carboxylate (11)
A solution of 10 (640 mg, 1.04 mmol) in DMF (9.5 mL) was treated with NaH (60% dispersion in mineral oil, 60 mg, 1.5 mmol) at 0 °C. After stirring at this temperature for 30 min, cis-1,3-dichloropropene (231 mg, 2.1 mmol) was added and the mixture was stirred for 5 h at 23 °C. H2O (63 mL) was added to the reaction mixture and the mixture was extracted with EtOAc. The combined organic extracts were washed with H2O, saturated aqueous LiCl and saturated aqueous NaCl, dried (Na2SO4) and concentrated in vacuo. The residue was purified by flash column chromatography (SiO2, 20–35% EtOAc/hexanes, gradient) to afford 11 (398 mg, 0.58 mmol 56%) as a white solid: 1H NMR (CDCl3, 500 MHz, mixture of rotamers) δ 7.26–7.31 (m, 3H), 7.22 (s, 3H), 7.11 (d, J = 7.1 Hz, 2H), 6.90 (s, 1.4H) & 6.84 (s, 0.6 H), 6.75 (s, 0.3H) & 6.66 (s, 0.7H), 6.13 (s, 1.4H) & 6.10 (s, 0.6H), 5.98 (s, 2H), 5.10 (d, J = 11.4 Hz, 1H), 5.04 (d, J = 11.7 Hz, 1H), 4.50 (d, J = 13.5 Hz, 1H), 4.29 (d, J = 12.8 Hz, 0.7H) & 4.16 (d, J = 12.8 Hz, 0.3H), 3.97 (s, 3H), 1.56 (s, 3H) & 1.33 (s, 6H); 13C NMR (CDCl3, 125 MHz) δ 160.0, 154.1, 147.2 &146.7, 145.3 & 145.1, 141.5, 141.3 & 141.0, 137.7, 135.4, 128.8, 128.6, 128.5, 128.0 & 127.7, 127.5, 127.4, 126.2, 124.5 & 124.3, 120.7 &120.2, 109.4 & 109.2, 84.5, 81.3 & 80.8, 71.0, 53.6 & 53.1, 50.8, 47.5 & 46.3, 28.6 & 28.3; IR (film) νmax 2976, 1725, 1699, 1579, 1482, 1451, 1367 cm−1; ESI-TOF HRMS m/z 688.1070 (M+H+, C31H31IClN3O5 requires 688.1070).
4.6. 3-(tert-Butyl) 7-Methyl 6-Benzyl-5-(benzyloxy)-1-(chloromethyl)-1,2-dihydro-3H-imidazo[4,5-e]indole-3,7-dicarboxylate (12)
Method A: Benzene (10 mL) degassed by the freeze-pump-thaw method three times was added to a sealed flask charged with 11 (200 mg, 0.29 mmol) and AIBN (20 mg, 0.12 mmol). (Me3Si)3SiH (145 mg, 0.58 mmol) was added and the flask was sealed and heated at 80 °C for 4 h. After cooling, the solvent was removed under a stream of N2 gas. The reaction mixture was purified by column chromatography (SiO2, 20% EtOAc/hexanes) to afford 12 (113 mg, 0.20 mmol, 69%) as a pale yellow solid: 1H NMR (CDCl3, 500 MHz) δ 7.85 (br s, 1H), 7.26–7.33 (m, 3H), 7.20 (t, J = 2.9 Hz, 3H), 7.14 (d, J = 7.1 Hz, 2H), 6.90 (d, J = 3.7 Hz, 2H), 6.11 (s, 2H), 5.10 (s, 2H), 4.28 (d, J = 11.0 Hz, 1H), 4.17–4.23 (m, 2H), 4.12 (br, 1H), 3.98 (s, 3H), 3.70 (br, 1H), 1.58 (s, 9H); 13C NMR (CDCl3, 125 MHz) δ 160.1, 147.3, 141.4, 139.8, 138.2, 135.8, 128.7, 128.6, 128.4, 128.3, 127.3, 126.2, 123.1, 97.5, 81.0, 71.2, 53.2, 50.2, 47.2, 41.1, 29.9, 28.6; IR (film) νmax 1726, 1697, 1455, 1419, 1339, 1264 cm−1; ESI-TOF HRMS m/z 562.2104 (M+H+, C31H32ClN3O5 requires 562.2103).
Method B: Benzene (42 mL) degassed by the freeze-pump-thaw method three times was added to a sealed flask charged with 11 (770 mg, 1.1 mmol) and AIBN (18 mg, 0.1 mmol). Bu3SnH (460 mg, 1.6 mmol) was added and the flask was sealed and heated at 80 °C for 7 h. After cooling, the solvent was removed under a stream of N2 gas. The reaction mixture was purified by column chromatography (SiO2, 30% EtOAc/hexanes) to afford 12 (457 mg, 0.81 mmol, 73%) as a yellow solid identical to the material described above.
4.7. 3-(tert-Butyl) 7-Methyl 1-(Chloromethyl)-5-hydroxy-1,2-dihydro-3H-imidazo[4,5-e]indole-2,6-dicarboxylate (13)
A solution of 12 (50 mg, 0.089 mmol) in THF (2 mL) under Ar was charged with 10% Pd/C (18 mg, 0.017 mmol) and placed under 50 psi pressure of H2. The reaction mixture was agitated in a Parr Shaker apparatus for 2 h at 23 °C. The reaction mixture was filtered, concentrated in vacuo and purified by flash chromatography (SiO2, 30% acetone/hexanes) to afford 13 (28 mg, 0.073 mmol, 82%) as a white solid: 1H NMR (acetone-d6, 400 MHz) δ 12.31 (br s, 1H), 8.96 (br s, 1H), 7.54 (br s, 1H), 4.25–4.31 (m, 1H), 4.10–4.18 (m, 1H), 4.06 (dd, J = 3.4, 10.7 Hz, 1H), 3.92–3.96 (m, 1H), 3.96 (s, 3H), 3.67 (t, J = 10.1 Hz, 1H), 1.56 (s, 9H); IR (film) νmax 1745, 1366 cm−1; ESI-TOF HRMS m/z 382.1175 (M+H+, C17H20ClN3O5 requires 382.1164).
Resolution: Racemic 13 was resolved into its enantiomers by HPLC on a ChiralPak AD semi-preparative column (2×25 cm, 10% i-PrOH/hexane, 7 mL/min, α = 1.3). Peak 1: [α]20D –52 (c 0.39, MeOH) for natural enantiomer; Peak 2: [α]20D +50 (c 0.42, MeOH) for unnatural enantiomer.
4.8. 2-(tert-Butyl) 6-Methyl 1,2,8,8a-Tetrahydrocyclopropa[c]imidazo[4,5-e]indol-4-one-2,6-dicarboxylate (N-Boc-CImI, 14)
A solution of 13 (8 mg, 0.021 mmol) dissolved in THF (0.8 mL) was treated with aqueous 1 M NaHCO3 (0.8 mL, 0.8 mmol). The reaction mixture was stirred at room temperature for 4 h before it was extracted with EtOAc, dried with anhydrous Na2SO4, filtered, and concentrated. The solid product was purified by PTLC (SiO2, 80% EtOAc/hexane) to yield 14 (4.3 mg, 0.012 mmol, 59%): 1H NMR (CDCl3, 400 MHz) δ 11.21 (br, 1H), 6.83 (s, 1H), 3.98–4.06 (m, 5H), 3.04 (q, J = 5.1, 7.5, 4.8 Hz, 1H), 2.10 (dd, J = 3.9, 7.8 Hz, 1H), 1.54 (s, 9H), 1.43 (t, J = 4.6 Hz, 1H); 13C NMR (CDCl3, 125 MHz) δ 178.0, 161.2, 158.9, 151.6, 148.6, 139.4, 129.3, 109.5, 83.7, 54.0, 53.4, 33.4, 28.3, 25.9, 23.6; IR (film) νmax 2923, 2852, 1721, 1603, 1393, 1370, 1345, 1277, 1206 cm−1; ESI-TOF HRMS m/z 346.1401 (M+H+, C17H19N3O5 requires 346.1397). [α]20D +115 (c 0.27, MeOH) for natural enantiomer; [α]20D –115 (c 0.27, MeOH) for unnatural enantiomer.
4.9. Methyl 1-(Chloromethyl)-5-hydroxy-3-(5,6,7-trimethoxyindole-2-carbonyl)-1,2-dihydro-3H-imidazo[4,5-e]indole-2-carboxylate (15)
An oven dried vial containing 13 (5.0 mg, 0.013 mmol) was treated with 4 N HCl in EtOAc (1.6 mL). The reaction mixture was stirred at room temperature for 2 h. The solvent was evaporated under a stream of N2. Additional EtOAc was added and the solvent was evaporated again. The residue was placed under vacuum overnight. EDCI·HCl (7.4 mg, 0.039 mmol), 5,6,7-trimethoxyindole-2-carboxylic acid (19, 4.2 mg, 0.017 mmol) and DMF (140 μL) were added to the amine salt. The reaction mixture was stirred for 2 d at room temperature. The solvent was removed under a stream of N2. The crude product was purified by column chromatography (SiO2, 3% MeOH/CH2Cl2) to yield 15 (4.1 mg, 0.0079 mmol, 61%): 1H NMR (DMSO-d6, 500 MHz) δ 10.33 (s, 1H) 7.65 (br, 1H), 7.00 (s, 1H), 6.96 (s, 1H), 4.70 (t, J = 10.2 Hz, 1H), 4.40 (dd, J = 3.3, 11.2 Hz, 1H), 4.09–4.12 (m, 1H), 3.80–3.96 (m, 2H, embedded within –OMe singlets), 3.95 (s, 3H), 3.93 (s, 3H), 3.82 (s, 3H), 3.80 (s, 3H); ESI-TOF HRMS m/z 515.1327 (M+H+, C24H23ClN4O7 requires 515.1328). [α]26 D –14 (c 0.08, MeOH) for natural enantiomer; [α]26 D +15 (c 0.06, MeOH) for unnatural enantiomer.
4.10. Methyl 2-(5,6,7-Trimethoxy-1H-indole-2-carbonyl)-1,2,8,8a-tetrahydrocyclopropa[c]imidazo[4,5-e]indole-4-one-6-carboxylate (16)
A sample of 15 (2.0 mg, 3.88 μmol) was dissolved in THF (0.2 mL). Aqueous 1 M NaHCO3 (0.2 mL) was added and the reaction mixture was stirred at room temperature for 4 h. The reaction mixture was extracted with EtOAc and the combined organic phase was dried with anhydrous Na2SO4, filtered, and concentrated. The solid product was purified by PTLC (SiO2, 8% MeOH/CH2Cl2) to yield 16 (1.20 mg, 2.5 μmol, 65%): 1H NMR (CDCl3, 400 MHz) δ 11.02 (br, 1H), 9.26 (s, 1H), 7.12 (s, 1H), 6.95 (d, J = 2.3 Hz, 1H), 6.78 (s, 1H), 4.51 (dd, J = 4.9, 10.4 Hz, 1H), 4.45 (d, J = 10.4 Hz, 1H), 4.08 (s, 3H), 4.04 (s, 3H), 3.94 (s, 3H), 3.90 (s, 3H), 3.19–3.15 (m, 1H), 2.24 (dd, J = 4.1, 7.6 Hz, 1H), 1.58 (1H, embedded within H2O signal; appears as a triplet in CD3OD, 600 MHz at δ 1.71 (t, J = 5.4 Hz, 1H)); ESI-TOF HRMS m/z 479.1562 (M+H+, C24H22N4O7 requires 479.1561).
4.11. Methyl 6-(5-(Indole-2-carboxamido)indole-2-carbonyl)-1-(chloromethyl)-5-hydroxy-1,2-dihydro-3H-imidazo[4,5-e]indole-2-carboxylate (17)
An oven dried vial containing 13 (4.9 mg, 0.013 mmol) was treated with 4 N HCl in EtOAc (1.5 mL). The reaction mixture was stirred at room temperature for 2 h. The solvent was evaporated under a stream of N2. Additional EtOAc was added and the solvent was evaporated again. The residue was put under vacuum overnight. EDCI·HCl (7.4 mg, 0.038 mmol), 20 (5.3 mg, 0.017 mmol) and DMF (135 μL) were added to the amine salt. The reaction mixture was stirred for 2 d at room temperature. The solvent was removed under a stream of N2. The crude product was purified by column chromatography (SiO2, 4% MeOH/CH2Cl2) to yield 17 (5.14 mg, 0.009 mmol, 69%) and was carried forward without further characterization.
4.12. Methyl 2-(5-(Indole-2-carboxamido)indole-2-carbonyl)-1,2,8,8a-tetrahydrocyclopropa[c]imidazo-[4,5-e]indole-4-one-6-carboxylate (18)
A sample of 17 (1.58 mg, 2.71 μmol) was dissolved in THF (0.16 mL). Aqueous 1 M NaHCO3 (0.16 mL) was added and the reaction mixture was stirred at room temperature for 4 h. The reaction mixture was extracted with EtOAc and the combined organic phase was dried with anhydrous Na2SO4, filtered, and concentrated. The solid product was purified by PTLC (SiO2, 8% MeOH/CH2Cl2) to yield 18 (0.89 mg, 1.62 μmol, 60%): 1H NMR (CD3OD, 400 MHz) δ 8.16 (d, J = 1.8 Hz, 1H) 7.74 (d, J = 8.0 Hz, 1H), 7.63 (dd, J = 2.0, 8.8 Hz, 1H), 7.57 (d, J = 8.8 Hz, 1H), 7.40 (s, 1H), 7.33 (t, J = 7.6 Hz, 1 H), 7.26 (s, 1H), 7.18 (t, J = 7.6 Hz, 1H), 6.92 (s, 1H), 4.65 (dd, J = 4.6, 10.5 Hz, 1H), 4.58 (d, J = 10.6 Hz, 1H), 4.00 (s, 3H), 3.21 (dd, J = 4.8, 11.9 Hz, 1H), 2.15 (dd, J = 3.8, 7.6 Hz, 1H), 1.68 (t, J = 4.6 Hz, 1H); ESI-TOF HRMS m/z 547.1721 (M+H+, C30H22N6O5 requires 547.1724). [α]20D +74 (c 0.073, MeOH) for natural enantiomer; [α]20D –72 (c 0.068, MeOH) for unnatural enantiomer.
4.13. Aqueous Solvolysis of N-Boc-CImI (pH 3 Phosphate Buffer)
N-Boc CImI (14, 190 μg) was dissolved in MeOH (1.5 mL) and pH 3.0 buffer (1.5 mL, prepared by mixing 4:1:20 (v:v:v) 0.1 M citric acid, 0.2 M Na2HPO4, and water, respectively). After mixing of the reagents, the UV spectrum of the solution was measured against the reference solution containing MeOH (1.5 mL) and pH 3 phosphate buffer (1.5 mL) and this reading was used for the initial absorbance value (Ai). The UV spectrum was measured at regular intervals (every 1 h in first 8 h period and every 12 h for the remaining time) until no further change in absorbance was observed (Af). The decrease in the long-wavelength absorption at 331 nm and increase in the short-wavelength absorption at 262 nm were monitored. The solvolysis rate constant (k = 5.52 × 10-6 s-1) and half-life (t1/2 = 34.7 h) were calculated from the least-squares treatment of the slope of the plot of time versus ln [(Af-Ai)/Af-A)].
4.14. Addition of HCl to N-Boc-CImI
An oven-dried flask containing 14 (3.8 mg, 0.011 mmol) dissolved in THF (0.25 mL) at −78 °C was treated with 4 N HCl in EtOAc (5.5 μL, 22 μmol) dropwise. The reaction mixture was stirred at −78 °C for 1.5 h. Upon disappearance of 14 by TLC, the reaction mixture was concentrated by a stream of N2 keeping it at a cold temperature (−78 °C). The residue was purified by flash column chromatography (SiO2, 30% acetone/hexane) to yield 13 (3.8 mg, 0.01 mmol, 90%) identical with authentic material.
4.15. Acid-Catalyzed Addition of CH3OH to N-Boc-CImI
An oven-dried vial containing 14 (2.6 mg, 7.5 μmol) in MeOH (750 μL) was cooled to 0 °C and TfOH (0.08 μL, 0.903 μmol) was added. The reaction mixture was stirred at 0 °C for 3 h. Additional TfOH was added (0.08 μL, 0.903 μmol) and the reaction was stirred for another 3 h. Finally, additional TfOH was further added (0.08 μL, 0.903 μmol) and the mixture was stirred for another 18 h. At this point, no further change in TLC was observed. The reaction mixture was concentrated under a stream of N2 and the product was purified by PTLC (250 μm, SiO2, 2.5% MeOH/CH2Cl2, eluted 3 times). The purification provided 21 (1.7 mg, 4.50 μmol, 60%) and 22 (0.5 mg, 1.32 μmol 18%) and recovery of starting material (0.5 mg, 1.44 μmol, 19%).
For 21: 1H NMR (CD2Cl2, 600 MHz) δ 10.49 (s, 1H), 7.53 (s, 1H), 7.20 (s, 1H), 4.22 (t, J = 11.0 Hz, 1H), 3.99 (s, 3H), 3.80–3.84 (m, 1H), 3.70 (dd, J = 4.1, 8.5 Hz, 1H), 3.48–3.55 (m, 5H), 1.55 (s, 9H); 13C NMR (CD2Cl2, 150 MHz) δ 159.8, 152.5, 149.2, 143.9, 139.4, 132.2, 129.6, 106.8, 98.1, 76.9, 59.6, 53.5, 51.3, 39.2, 30.3, 28.7; ESI-TOF HRMS m/z 378.1667 (M+H+, C18H23N3O6 requires 378.1660).
For 22: 1H NMR (CD2Cl2, 600 MHz) δ 7.17 (s, 1H), 3.93–4.24 (m, 4H), 3.81 (s, 1H), 3.64 (d, J = 13.2 Hz, 1H), 3.44 (s, 3H), 3.14 (dd, J = 5.7, 16.7 Hz, 1H), 2.88 (d, J = 16.7 Hz, 1H), 1.53 (s, 9H); ESI-TOF HRMS m/z 378.1669 (M+H+, C18H23N3O6 requires 378.1660).
Supplementary Material
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (CA042056) and the Skaggs Institute for Chemical Biology. We especially wish to thank Yam Poudel and Dimitri Hirsch-Weil for intermediate efforts in the work, and Professor Arnold L. Rheingold and Dr. Curtis E. Moore (UCSD) for the single crystal X-ray structure determination of 10.
Footnotes
Supplementary Data. 1H NMR and 13C NMR are provided. The supplementary data associated with this article can be found in the online version at doi:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ichimura M, Ogawa T, Takahashi K, Kobayashi E, Kawamoto I, Yasuzawa T, Takahashi I, Nakano H. J Antibiot. 1990;43:1037. doi: 10.7164/antibiotics.43.1037. [DOI] [PubMed] [Google Scholar]
- 2.Igarashi Y, Futamata K, Fujita T, Sekine A, Senda H, Naoki H, Furumai T. J Antibiot. 2003;56:107. doi: 10.7164/antibiotics.56.107.. Structure revision: Tichenor MS, Kastrinsky DB, Boger DL. J Am Chem Soc. 2004;126:8396. doi: 10.1021/ja0472735.
- 3.Martin DG, Biles C, Gerpheide SA, Hanka LJ, Krueger WC, McGovren JP, Mizsak SA, Neil GL, Stewart JC, Visser J. J Antibiot. 1981;34:1119. doi: 10.7164/antibiotics.34.1119. [DOI] [PubMed] [Google Scholar]
- 4.Reviews: Boger DL, Johnson DS. Angew Chem Int Ed Engl. 1996;35:1438.Boger DL. Acc Chem Res. 1995;28:20.Boger DL, Johnson DS. Proc Natl Acad Sci U S A. 1995;92:3642. doi: 10.1073/pnas.92.9.3642.Boger DL, Garbaccio RM. Acc Chem Res. 1999;32:1043.Tichenor MS, Boger DL. Natural Prod Rep. 2008;25:220. doi: 10.1039/b705665f.Ghosh N, Sheldrake HM, Searcey M, Pors K. Curr Top Med Chem. 2009;9:1494. doi: 10.2174/156802609789909812.
- 5.Duocarmycin SA, Boger DL, Johnson DS, Yun W. J Am Chem Soc. 1994;116:1635. [Google Scholar]
- 6.Yatakemycin: Parrish JP, Kastrinsky DB, Wolkenberg SE, Igarashi Y, Boger DL. J Am Chem Soc. 2003;125:10971. doi: 10.1021/ja035984h.Trzupek JD, Gottesfeld JM, Boger DL. Nature Chem Biol. 2006;2:79. doi: 10.1038/nchembio761.
- 7.CC-1065: Hurley LH, Lee CS, McGovren JP, Warpehoski MA, Mitchell MA, Kelly RC, Aristoff PA. Biochemistry. 1988;27:3886. doi: 10.1021/bi00410a054.Hurley LH, Warpehoski MA, Lee CS, McGovren JP, Scahill TA, Kelly RC, Mitchell MA, Wicnienski NA, Gebhard I, Johnson PD, Bradford VS. J Am Chem Soc. 1990;112:4633.Boger DL, Johnson DS, Yun W, Tarby CM. Bioorg Med Chem. 1994;2:115. doi: 10.1016/s0968-0896(00)82007-6.Boger DL, Coleman RS, Invergo BJ, Sakya SM, Ishizaki T, Munk SA, Zarrinmayeh H, Kitos PA, Thompson SC. J Am Chem Soc. 1990;112:4623.
- 8.Duocarmycin A: Boger DL, Ishizaki T, Zarrinmayeh H, Munk SA, Kitos PA, Suntornwat O. J Am Chem Soc. 1990;112:8961.Boger DL, Ishizaki T, Zarrinmayeh H. J Am Chem Soc. 1991;113:6645.Boger DL, Yun W, Terashima S, Fukuda Y, Nakatani K, Kitos PA, Jin Q. Bioorg Med Chem Lett. 1992;2:759.Boger DL, Yun W. J Am Chem Soc. 1993;115:9872.Boger DL, Ishizaki T, Zarrinmayeh H. J Org Chem. 1990;55:4499.
- 9.(a) Boger DL, Coleman RS. J Am Chem Soc. 1988;110:1321. [Google Scholar]; (b) Boger DL, Coleman RS. J Am Chem Soc. 1988;110:4796. [Google Scholar]; (c) Boger DL, Coleman RS. J Am Chem Soc. 1987;109:2717. [Google Scholar]; (d) Boger DL, Machiya K. J Am Chem Soc. 1992;114:10056. [Google Scholar]; (e) Boger DL, Machiya K, Hertzog DL, Kitos PA, Holmes D. J Am Chem Soc. 1993;115:9025. [Google Scholar]; (f) Boger DL, McKie JA, Nishi T, Ogiku T. J Am Chem Soc. 1997;119:311. [Google Scholar]; (g) Boger DL, McKie JA, Nishi T, Ogiku T. J Am Chem Soc. 1996;118:2301. [Google Scholar]; (h) Tichenor MS, Trzupek JD, Kastrinsky DB, Shiga F, Hwang I, Boger DL. J Am Chem Soc. 2006;128:15683. doi: 10.1021/ja064228j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Tichenor MS, MacMillan KS, Trzupek JD, Rayl TJ, Hwang I, Boger DL. J Am Chem Soc. 2007;129:10858. doi: 10.1021/ja072777z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Boger DL, Bollinger B, Hertzog DL, Johnson DS, Cai H, Mesini P, Garbaccio RM, Jin Q, Kitos PA. J Am Chem Soc. 1997;119:4987. [Google Scholar]; (b) Boger DL, Hertzog DL, Bollinger B, Johnson DS, Cai H, Goldberg J, Turnbull P. J Am Chem Soc. 1997;119:4977. [Google Scholar]
- 11.Boger DL, Yun W. J Am Chem Soc. 1993;115:9872. [Google Scholar]
- 12.(a) Boger DL, Garbaccio RM. Bioorg Med Chem. 1997;5:263. doi: 10.1016/s0968-0896(96)00238-6. [DOI] [PubMed] [Google Scholar]; (b) Ambroise Y, Boger DL. Bioorg Med Chem Lett. 2002;12:303. doi: 10.1016/s0960-894x(01)00740-5. [DOI] [PubMed] [Google Scholar]
- 13.Review: MacMillan KS, Boger DL. J Med Chem. 2009;52:5771. doi: 10.1021/jm9006214.
- 14.MeCFI: Mohamadi F, Spees MM, Staten GS, Marder P, Kipka JK, Johnson DA, Boger DL, Zarrinmayeh H. J Med Chem. 1994;37:232. doi: 10.1021/jm00028a005.
- 15.CFI and CTI: Muratake H, Okabe K, Takahashi M, Tonegawa M, Natsume M. Chem Pharm Bull. 1997;45:799.Muratake H, Hayakawa A, Natsume M. Tetrahedron Lett. 1997;38:7577.Muratake H, Hayakawa A, Natsume M. Chem Pharm Bull. 2000;48:1558. doi: 10.1248/cpb.48.1558.
- 16.CPzI: Baraldi PG, Cacciari B, Romagnoli R, Spalluto G, Boyce CW, Boger DL. Bioorg Med Chem Lett. 1999;9:3087. doi: 10.1016/s0960-894x(99)00533-8.
- 17.CPyI: Boger DL, Boyce CW. J Org Chem. 2000;65:4088. doi: 10.1021/jo000177b.Boger DL, Wolkenberg SE, Boyce CW. J Am Chem Soc. 2000;122:6325.Ellis DA, Wolkenberg SE, Boger DL. J Am Chem Soc. 2001;123:9299. doi: 10.1021/ja010769r.
- 18.MeCTI: Tichenor MS, MacMillan KS, Stover JS, Wolkenberg SE, Pavani MG, Zanella L, Zaid AN, Spalluto G, Rayl TJ, Hwang I, Baraldi PG, Boger DL. J Am Chem Soc. 2007;129:14092. doi: 10.1021/ja073989z.
- 19.CTI: MacMillan KS, Lajiness JP, Cara CL, Romagnoli R, Robertson WM, Hwang I, Baraldi PG, Boger DL. Bioorg Med Chem Lett. 2009;19:6962. doi: 10.1016/j.bmcl.2009.10.063.
- 20.Iso-DSA: MacMillan KS, Nguyen T, Hwang I, Boger DL. J Am Chem Soc. 2009;131:1187. doi: 10.1021/ja808108q.
- 21.COI: Boyle KE, MacMillan KS, Ellis DA, Lajiness JP, Robertson WM, Boger DL. Bioorg Med Chem Lett. 2010;20:1854. doi: 10.1016/j.bmcl.2010.01.145.
- 22.Iso-CFI: Patil PC, Lee M. Tetrahedron Lett. 2014;55:3283.Patil P, Cousins K, Smith M, Wieskamp S, Ferrara M, Bruce CD, Lee M. Tetrahedron Lett. 2013;54:4756.Purnell B, Lingerfelt B, Scott A, Townes H, Summerville K, Hudson S, Kiakos K, Hartley JA, Lee M. Med Chem. 2006;2:139. doi: 10.2174/157340606776056188.
- 23.Reviews: Searcey M. Curr Pharm Des. 2002;8:1375. doi: 10.2174/1381612023394539.Wolkenberg SE, Boger DL. Chem Rev. 2002;102:2477. doi: 10.1021/cr010046q.Tse WC, Boger DL. Chem Biol. 2004;11:1607. doi: 10.1016/j.chembiol.2003.08.012.Tse WC, Boger DL. Acc Chem Res. 2004;37:61. doi: 10.1021/ar030113y.
- 24.(a) Boger DL, Coleman RS, Invergo BJ, Zarrinmayeh H, Kitos PA, Thompson SC, Leong T, McLaughlin LW. Chem-Biol Interact. 1990;73:29. doi: 10.1016/0009-2797(90)90107-x. [DOI] [PubMed] [Google Scholar]; (b) Boger DL, Zarrinmayeh H, Munk SA, Kitos PA, Suntornwat O. Proc Natl Acad Sci U S A. 1991;88:1431. doi: 10.1073/pnas.88.4.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Boger DL, Munk SA, Zarrinmayeh H. J Am Chem Soc. 1991;113:3980. [Google Scholar]; (d) Boger DL, Johnson DS. J Am Chem Soc. 1995;117:1443. [Google Scholar]
- 25.Patil PC, Satam V, Lee M. Anti-Cancer Agents Med Chem. 2015;15:616. doi: 10.2174/1871520615666141216144116. [DOI] [PubMed] [Google Scholar]; Boger DL, Boyce CW, Garbaccio RM, Goldberg JA. Chem Rev. 1997;97:787. doi: 10.1021/cr960095g. [DOI] [PubMed] [Google Scholar]
- 26.Hiroya K, Matsumoto S, Sakamoto T. Org Lett. 2004;6:2953. doi: 10.1021/ol0489548. [DOI] [PubMed] [Google Scholar]
- 27.(a) Goodbrand HB, Hu NX. J Org Chem. 1999;64:670. [Google Scholar]; (b) Evindar G, Batey RA. Org Lett. 2003;5:133. doi: 10.1021/ol027061h. [DOI] [PubMed] [Google Scholar]
- 28.The X-ray structure of 10 conducted on white needles obtained from EtOAc/Et2O has been deposited with the Cambridge Crystallographic Data Centre with accession code CCDC 1457717.
- 29.Boger DL, Boyce CW, Garbaccio RM, Searcey M. Tetrahedron Lett. 1998;39:2227. [Google Scholar]
- 30.(a) Boger DL, Ishizaki T, Kitos PA, Suntornwat O. J Org Chem. 1990;55:5823. [Google Scholar]; (b) Boger DL, McKie JA, Han N, Tarby CM, Riggs HW, Kitos PA. Bioorg Med Chem Lett. 1996;6:659. [Google Scholar]
- 31.(a) Parrish JP, Hughes TV, Hwang I, Boger DL. J Am Chem Soc. 2004;126:80. doi: 10.1021/ja038162t. [DOI] [PubMed] [Google Scholar]; (b) Parrish JP, Trzupek JD, Hughes TV, Hwang I, Boger DL. Bioorg Med Chem. 2004;12:5845. doi: 10.1016/j.bmc.2004.08.032. [DOI] [PubMed] [Google Scholar]; (c) Boger DL, Ishizaki T. Tetrahedron Lett. 1990;31:793. [Google Scholar]; (d) Boger DL, Munk SA, Ishizaki T. J Am Chem Soc. 1991;113:2779. [Google Scholar]; (e) Boger DL, Yun W. J Am Chem Soc. 1994;116:5523. [Google Scholar]
- 32.(a) Boger DL, Turnbull P. J Org Chem. 1997;62:5849. [Google Scholar]; (b) Boger DL, Mesini P. J Am Chem Soc. 1995;117:11647. [Google Scholar]
- 33.Lane TJ, Sam A, Kandathil AJ. J Am Chem Soc. 1960;82:4462. [Google Scholar]
- 34.Boger DL, Yun W. J Am Chem Soc. 1994;116:7996. [Google Scholar]
- 35.(a) Boger DL, Munk SA, Zarrinmayeh H, Ishizaki T, Haught J, Bina M. Tetrahedron. 1991;47:2661. [PubMed] [Google Scholar]; (b) Boger DL, Munk SA. J Am Chem Soc. 1992;114:5487. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.