

Summary
The murine epidermis with its hair follicles represents an invaluable model system for tissue regeneration and stem cell research. Here we used single-cell RNA-sequencing to reveal how cellular heterogeneity of murine telogen epidermis is tuned at the transcriptional level. Unbiased clustering of 1,422 single-cell transcriptomes revealed 25 distinct populations of interfollicular and follicular epidermal cells. Our data allowed the reconstruction of gene expression programs during epidermal differentiation and along the proximal-distal axis of the hair follicle at unprecedented resolution. Moreover, transcriptional heterogeneity of the epidermis can essentially be explained along these two axes, and we show that heterogeneity in stem cell compartments generally reflects this model: stem cell populations are segregated by spatial signatures but share a common basal-epidermal gene module. This study provides an unbiased and systematic view of transcriptional organization of adult epidermis and highlights how cellular heterogeneity can be orchestrated in vivo to assure tissue homeostasis.
Graphical Abstract
Highlights
-
•
Single-cell RNA-seq analysis identifies 25 populations of epidermal cells
-
•
Differentiation and spatial gene expression signatures can be defined
-
•
Interplay of differentiation and spatial signatures explains most heterogeneity
-
•
Stem cell populations are divided by spatial signatures and only share basal identity
Joost et al. use high-throughput single-cell RNA-seq to describe gene expression in mouse epidermis and hair follicles at unprecedented detail and explain epidermal heterogeneity as the interplay of differentiation-related and spatial gene expression signatures.
Introduction
The epidermis and its appendages form the outer layer of the mammalian skin and shield the body from external harm (Fuchs, 2007). Its regenerative capacity along with its accessibility and compartmentalized microanatomy has made the epidermis one of the most important model systems for stem cell biology (Hsu et al., 2014, Schepeler et al., 2014), and many paradigms of tissue maintenance and regeneration have been established or validated in the murine epidermis (Rompolas and Greco, 2014).
In mice, the epidermis consists of two main compartments with distinct physiological functions: the interfollicular epidermis (IFE), and the hair follicle (HF) including the sebaceous gland (SG) (Niemann and Watt, 2002). Cells of the IFE constitute the majority of epidermal cells and form a squamous, stratified, multilayered epithelium that plays the key role in securing the skin barrier function (Fuchs, 1990). In contrast, the main role of HFs lies in producing the hair shaft to maintain the murine fur. While the cells of IFE and SG are constantly replaced, the HF is subjected to cycles of rest (telogen), growth (anagen), and degeneration (catagen). The telogen HF exhibits a characteristic microanatomy including the bulge and hair germ fuelling hair growth, the isthmus and junctional zone encompassing the opening of the SG, and the infundibulum connecting the HF to the IFE (Figure 1B). The lower part of the HF closest to the hair-growth inductive dermal papilla is often referred to as the proximal part, and consequently the upper HF as distal (Müller-Röver et al., 2001).
Figure 1.

Defining the Main Epidermal Cell Populations
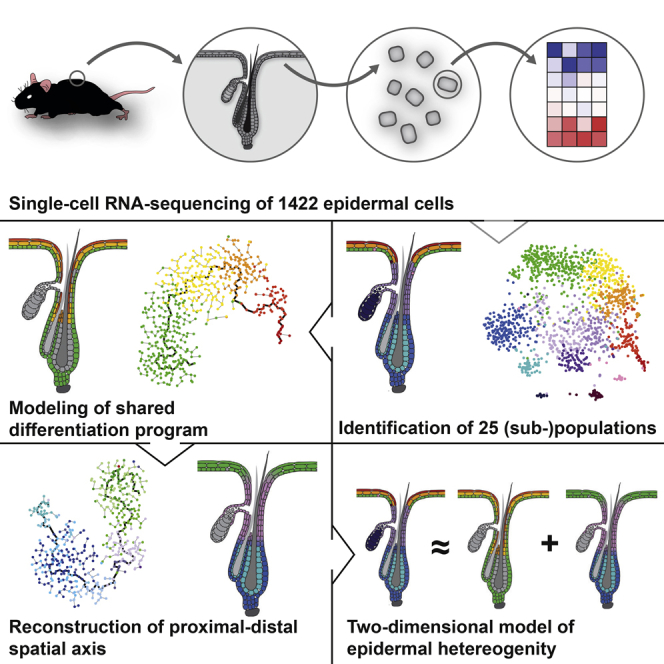
(A) Overview of the experimental workflow.
(B) Illustrated microanatomy and compartmentalization of the murine epidermis including HF and SG, colored according to main populations (C).
(C) Identity and marker genes of cell populations defined during first-level clustering.
(D) Epidermal cell transcriptomes (n = 1,422) visualized with t-distributed stochastic neighbor embedding (t-SNE), colored according to unsupervised (first level) clustering (C).
(E) Expression of group-specific marker genes projected onto the t-SNE map.
(F) Immunostaining or single-molecule FISH for group-specific genes. Protein or mRNA (symbols italics) expression is pseudocolored corresponding to groups shown in (C). Cell nuclei are shown in white. Scale bars, 20 μm. See also Figure S2J.
(G) Hierarchical clustering (Ward’s linkage) of gene expression data averaged over each group.
The cellular composition of the epidermis has been extensively studied during the last decades. It has been shown that the keratinocytes of the IFE can be morphologically, molecularly, and functionally divided into basal cells, suprabasal spinous, and granular layer cells, which each play distinct roles in producing and maintaining the skin barrier (Fuchs, 1990). In a similar fashion, it has been established how SG cells differentiate to fulfill glandular functions or how HF keratinocytes maintain the hair shaft (Niemann and Horsley, 2012). More recently, reporter constructs and lineage tracing studies have characterized stem cell and progenitor populations in the IFE, the SG, and sub-compartments of the HF (Alcolea and Jones, 2014, Kretzschmar and Watt, 2014, Petersson and Niemann, 2012). The molecular relationship between the different stem and progenitor populations and “non-stem cell” populations is, however, still insufficiently addressed.
A large number of studies have investigated the transcriptomes of cell populations in the human and murine epidermis in vivo and in vitro. While a few pioneering studies were performed at single-cell resolution but were limited by low sensitivity or small numbers of analyzed genes (Jensen and Watt, 2006, Tan et al., 2013), most of the studies relied on bulk-sampling techniques and cell enrichment using pre-defined markers (Blanpain et al., 2004, Brownell et al., 2011, Füllgrabe et al., 2015, Greco et al., 2009, Jaks et al., 2008, Janich et al., 2011, Mascré et al., 2012, Page et al., 2013, Snippert et al., 2010, Tumbar et al., 2004). As nearly all of these studies were restricted to certain subpopulations or compartments of the epidermis, it has been difficult to directly compare results across studies and to analyze epidermal heterogeneity in a systematic fashion. In contrast, recent advances in single-cell RNA-sequencing (RNA-seq) technologies have made it possible to profile large numbers of cells in parallel (Hashimshony et al., 2012, Islam et al., 2014, Picelli et al., 2013) in order to comprehensively dissect the cellular composition of complex tissues (Sandberg, 2014). In addition to unveiling novel epidermal cell populations, high-throughput single-cell transcriptomics of the epidermis may also reveal heterogeneity within previously described populations in the murine skin (Jaks et al., 2010, Kretzschmar and Watt, 2014). However, such studies are lacking so far.
Here, we used quantitative single-cell RNA-seq to sequence 1,422 cells from the murine telogen epidermis to systematically dissect the cellular heterogeneity of epidermal cells during tissue homeostasis. We provide a high-resolution transcriptome map that is available online, present potential novel transcriptional regulators along the differentiation and spatial axes, and model the impact of each axis on transcriptional heterogeneity.
Results
Single-Cell Transcriptome Analysis of Mouse Epidermis
To study the transcriptional heterogeneity of the telogen epidermis, we isolated epidermal cells from dorsal skin of C57BL/6 wild-type mice during second telogen at around 8 weeks (Figures 1A, S1A, and S1B). The isolated cells of individual mice (n = 19 biological replicates) were, after one HF cell enrichment step, directly loaded into 96-well microfluidic C1 chips (Fluidigm) and randomly captured for sequencing. Because we expected higher cellular heterogeneity within HFs compared to IFE (Figure 1B), we used SCA-1 microbeads to enrich for HF cells and sampled HF (SCA-1−) and IFE/infundibulum (SCA-1+) cell numbers in a 2:1 ratio (Figures S1C–S1E). Although single-cell capturing in C1 chips showed a minor bias for larger cells, the whole size range of both cell fractions was represented in the dataset (Figure S1F). Through imaging of the C1 chips, chambers containing more than one cell were excluded. Next, we prepared and sequenced single-cell cDNA libraries using a quantitative single-cell RNA-seq protocol (Islam et al., 2014). Sequencing yield and quality was comparable to our previous studies (Figures S1G–S1N) (Zeisel et al., 2015). Single cells with <2,000 unique detected molecules failed to reach quality-control standards and were excluded, leaving 1,422 single-cell transcriptomes in the final dataset (Figure S1K).
Unbiased Clustering Confirms Known Epidermal Cell Populations
First, we dissected the global structure of the dataset through unsupervised clustering with affinity propagation (Frey and Dueck, 2007) based on the expression of high variance genes (Figure S2A). Importantly, all clusters (representing distinct groups of cells) were derived without considering a priori knowledge from the literature. We robustly identified 13 highly distinct main groups of epidermal cells, which we visualized in two-dimensional space using t-distributed stochastic neighbor embedding (t-SNE) (Van der Maaten and Hinton, 2008) (Figures 1C, 1D, and S2B–S2F): SG cells marked by Scd1/Mgst1, inner and outer bulge keratinocytes characterized by expression of Krt6a/Krt75 and Cd34/Postn, respectively, predominantly IFE-derived basal cells with high expression levels of Krt14/Mt2, two stages of differentiated cells marked by Krt10/Ptgs1 and two stages of terminally differentiated keratinized layer cells expressing Lor/Flg2, three distinct groups of upper HF cells marked by different levels of Krt79/Krt17, and two immune cell populations Langerhans cells (Cd207+/Ctss+) and resident T cells (Cd3+/Thy1+). We subsequently used a negative binomial Bayesian regression model to identify group-specific gene expression signatures, and, as expected, each group of cells expressed a distinct set of genes (Figures 1C, 1E, and S2G–S2I; Table S1).
To confirm the existence of these cell populations with a sequencing-independent method, we selected known and newly derived marker genes and subsequently stained telogen skin tissue sections using immunohistochemistry (IHC) and/or single-molecule mRNA fluorescence in situ hybridization (FISH) (STAR Methods). This also allowed us to map the defined populations to their spatial location in the telogen epidermis (Figures 1F and S2J). Interestingly, comparing transcriptional similarity among the 13 epidermal groups revealed that the cell populations did not always cluster based on their physical location, raising the question whether similar cellular functions render cells more similar than location (Figure 1G). Overall, even though the first round (first level) of clustering did not reveal novel populations of cells, an outcome that is not unexpected given that the murine epidermis is one of the best studied mammalian organ systems (Fuchs, 2007, Niemann and Watt, 2002, Schepeler et al., 2014), it robustly recapitulated the expected main epidermal structures and cell populations.
Subclustering of Main Populations Reveals New Subpopulations
To further resolve cellular heterogeneity of HF and IFE cells, we selected all cells that were in the first-level clustering defined as having an outer bulge, inner bulge, upper HF, and basal IFE signature, respectively, and subjected them to a second round (second level) of unsupervised clustering (Figures S3A and S3B). We divided the upper HF into seven, the outer bulge into five, and the inner bulge as well as the basal IFE into three subpopulations, respectively (Figures 2A–2G and S3C–S3L; Table S2). To exclude that any population was merely the result of biological (e.g., variability between mice) or technical artifacts (e.g., variability in cell isolation, or cell doublets [Macosko et al., 2015]), we used three different validation strategies (STAR Methods): (1) verification that each cluster was formed by an adequate number of biological replicates, (2) resampling approach to test robustness of each cell cluster, (3) systematic staining of all populations by IHC and/or FISH. The results show that cells of at least eight different mice formed each cluster, the majority of clusters were highly robust (Figures S3G–S3J), and all populations could be identified by IHC and/or FISH staining.
Figure 2.

Subclustering of Epidermal Cell Populations
(A–D) Subclustering (second-level clustering) of epidermal cells from the IFE basal (A), upper HF (B), outer bulge (C), and inner bulge (D) compartments. Upper panel: projection of subpopulations onto the t-SNE map of the full dataset introduced in Figure 1D. Lower panel: barplots showing the expression of marker genes per subpopulation. Each bar represents a single cell, and the black line indicates the average expression over each subpopulation.
(E) Selection of immuno- and single-molecule FISH (symbols italics) stainings to visualize subpopulation localization within the tissue. Arrowheads highlight the position of the populations: IFE BI (filled arrowhead)/BII (empty arrowhead); uHF I (filled arrowhead)/II (empty arrowhead); OB III (filled arrowhead; dashed line marks lower end of KRT15 gap). HS, hair shaft. SG, sebaceous gland. CH, club hair. Scale bars, 10 μm. See also Figure S3L.
(F) Identity and marker genes of cell populations defined during second-level clustering.
(G) Summary of the approximate location of each defined subpopulation in the IFE, SG, and HF.
Upper HF
The cells of the upper HF could be separated into four known (uHF IV–VII), one indistinct (uHF III), and two new cell populations (uHF I and uHF II) (Figures 2B, 2E, 2G, S3D, and S3L). The new populations were located around the SG opening and could be distinguished by Rbp1 expression as well as high levels of Defb6 and Cst6. While uHF I cells showed additional expression of unique markers such as Klk10 and could be located to two suprabasal rings of cells around the SG opening, uHF II cells expressed a small subset of typical basal genes such as Krt14 (but not Krt5) and could be linked to the SG duct. The other subpopulations of uHF cells (uHF IV–VII) showed a typical uHF signature (high levels of Krt17, Krt79, Cd44, Cd200, and Lrig1 in the more basal cells) combined with expression of gene signatures linked to the basal (Krt5, Krt14), suprabasal (Krt10, Ptgs1), and keratinized layer (Flg2, Lor) of the IFE.
Outer Bulge
The outer bulge is the most well-investigated HF compartment and is characterized by high expression of Cd34, Krt15, and Lgr5 (Blanpain et al., 2004, Cotsarelis et al., 1990, Jaks et al., 2008, Morris et al., 2004). The degree of transcriptional heterogeneity within the outer bulge cells is, however, only partly explored (Blanpain et al., 2004, Janich et al., 2011, Tumbar et al., 2004). Subclustering cells with outer bulge signature revealed five subpopulations (Figures 2C and S3E). Most of the cells of the outer bulge belonged to either a Cd34hi, Postnhi, Lgr5hi, Krt24hi population (OB I) located in the proximal part of the outer bulge and the hair germ or a Cd34hi, Postnhi, Lgr5dim, Krt24dim population (OBII) that was mapped to the central part of the outer bulge (Figures 2G and S3L). The three additional OB-cell populations (OB III, IV, and V) were demarcated at the distal end of the bulge area and at the lower isthmus (Figures 2E, 2G, and S3L). OB III was characterized by a unique signature of genes including Aspn, Nrep, and Robo2 (Figures 2C and S3E), and, interestingly, this population also showed the strongest expression of Gli1 and Lgr6 in the HF indicating that this cluster includes cells from both the Gli1+ population defined by Brownell et al. and the Lgr6+ population described by Snippert et al. (Brownell et al., 2011, Snippert et al., 2010). In contrast to OB III, the cells of OB IV located distal to OB III did not express unique genes; instead, they were marked by an overlapping outer bulge (including Postn and Cd34) and upper HF signature (including Krt79, Krt17, Lrig1, and Cd44) (Figure 2E). OB V is a population of suprabasal cells, which expressed both an outer bulge signature and differentiation markers such as Krt10 and Ptgs1 (Figure 2E).
Inner Bulge
The majority of inner bulge cells belonged to a population (IB I) solely expressing the typical inner bulge signature (e.g., high levels of Krt6a, Krt75, Timp3, Fgf18). The second population (IB II) consisted of cells expressing both inner bulge and outer bulge markers and could be mapped to the outer bulge (Figure 2E). The third population (IB III) co-expressed an inner bulge and a differentiation signature (e.g., Krt10, Ptgs1) and was mapped to the distal end of the inner bulge compartment (Figure 2E).
Overall, we were able to resolve 16 distinct subpopulations of HF cells, of which many have not been previously described (Table S3). Intriguingly, only three of those subpopulations—the Gli1+ upper bulge population (OB III) and the upper HF populations located around the SG (uHF I and uHF II)—were defined by unique genetic signatures. In contrast, most heterogeneity in the HF seemed to result from the combination of recurring genetic signatures (Figures 2A–2D, S3C–S3F, and S3K; Table S2), suggesting that the vast complexity of cellular identities found in the HF might be the consequence of the coordinated interplay of just a few classes of genetic signatures. As a consequence, dividing lines (i.e., borders) between some populations (Figure S5E) became less distinct, exemplified by the overlap of genetic signatures in OB IV (upper HF and outer bulge signatures) and IB II (inner bulge and outer bulge signatures). Importantly, these observations were not limited to cells of the HF.
Basal IFE
While subclustering IFE basal cells, we found a subpopulation that expressed low levels of upper HF markers such as Krt79, the bulge marker Postn, and pan-HF markers like Sostdc1, Aqp3, and Fst in addition to the IFE basal signature (Figures 2A, 2E, and S3C). This unique combination of signatures turned out to mark basal cells of the infundibulum, the structure that connects the HF to the IFE, which was never transcriptionally resolved before. Moreover, we found two distinct basal IFE populations (IFE BI and II; Figure 2E) both expressing high levels of Krt14 and Krt5, and IFE BI additionally expressed high levels of Avpi1, Krt16, Thbs1, and the transcription factor Bhlhe40. Interestingly, Thrombospondin 1 (THBS1) was reported to inhibit angiogenesis and to modulate cell adhesion, motility, and growth (Guo et al., 1997), and BHLHE40 has been suggested to take part in the control of the circadian rhythm and counteract cell differentiation (Bi et al., 2015, Honma et al., 2002, Sato et al., 2004).
In summary, the observation that overlapping gene signatures frequently determine subpopulations justified the question whether the cellular heterogeneity in the epidermis was best represented as a set of distinct, clearly delineated clusters, or can be explained better by another model. Thus, we next sought to identify and characterize the biological processes that may give rise to HF and IFE keratinocyte heterogeneity.
Reconstruction of IFE Cell Differentiation by Pseudotemporal Ordering of Single-Cell Transcriptomes
Since the IFE is constantly renewed, it contains the whole range of basal to terminally differentiated keratinocytes (Fuchs, 1990, Toufighi et al., 2015). An advantage of sequencing single cells is that cells can be ordered along a path according to their transcriptional profile using a network-based approach (Trapnell et al., 2014). This allowed us to reconstruct the differentiation processes by ordering IFE cells along a pseudotemporal differentiation trajectory (Figures 3A and S4A). Increasing cell diameters with differentiation (data not shown), and expression levels of the well-known markers Krt14 (basal), Krt10 (mature), and Lor (terminally differentiated) along the defined pseudotime axis confirmed that our cell alignment was correct and in accordance with epidermal stratification (Fuchs, 1990). Mt4 marked a transitory stage, which we resolved in this study (Figure 3B).
Figure 3.

Reconstruction of the Epidermal Differentiation Process
(A) Pseudotemporal ordering of IFE cells (n = 536) in t-SNE space, using a minimum spanning tree. The longest path through the graph is highlighted and cells are colored according to first-level clustering.
(B) Validation of pseudotemporal ordering of IFE cells using the known basal (Krt14), mature (Krt10), and terminally differentiated (Lor) cell stage markers and Mt4, a transient marker defined in this study. Upper panel: gene expression in IFE cells plotted along pseudotime and fitted with a cubic smoothing spline (black line). Lower panel: gene expression projected onto the t-SNE map shown in (A).
(C) “Rolling wave” plot showing the spline-smoothed expression pattern of pseudotime-dependent genes (n = 1,627) clustered into eight groups (I–VIII) and ordered according to their peak expression.
(D) “Rolling wave” plot showing the spline-smoothed expression pattern of the 30 most significantly differentiation-related transcription factors (TFs). TFs were ordered according to group membership (left) and peak expression as shown in (C). P-values for pseudotime dependency are shown on the right. Red line marks Bonferroni-corrected significance threshold of 0.001. TFs marked in bold have not been previously described as relevant for epidermal stratification.
(E) Expression of differentiation-related genes in all epidermal subpopulations defined by either first- or second-level clustering. Bars show the percentage of genes expressed over baseline with 95% posterior probability (negative binomial regression model) in each of the populations for every differentiation group (I–VIII). Populations where the pseudotime model is not applicable are shaded gray.
(F) Position of epidermal cells from each subpopulation plotted on the differentiation axis (defined by highest Pearson correlation). Populations where the pseudotime model is not applicable are colored light gray.
(G) Summary illustrating the differentiation status of cells in the HF and IFE.
We identified 1,627 genes with statistically significant variation in expression levels along the differentiation trajectory (pseudotime-dependent genes, Figure S4B), and these genes clustered into eight groups according to their expression pattern during the differentiation process (Figures 3C and S4C), which also were linked to distinct functional terms (Figure S4D). Basal cells (group I) were defined by a low number of genes primarily involved in extracellular matrix deposition and interaction, cell proliferation, and tissue development. After a transitional stage (II), in which the basal signature was slowly reduced while ribosomal genes peak (III), we saw a first wave of genes linked to epidermal maturation, fatty acid metabolism and cholesterol synthesis, cell-cell junction formation, and protein transport (IV–VI). Toward the end of the cell’s life cycle, a second wave of genes involved in cornified envelope formation, ceramide synthesis, and proteolysis became active (VII and VIII) (Table S4). To gain insight into the molecular regulation of epidermal differentiation, we selected the 30 most pseudotime-dependent transcription factors (TFs) and analyzed their expression patterns during the differentiation process (Figures 3D and S4D). While only a few TFs (e.g., Bhlhe40, Zfp36l2) could be linked to the basal and intermediate signatures, we found a high number of new (e.g., Casz1, Klf3, Lrrfip2, Mllt4) and previously described (Gata3, Grhl1, Hes1, and Prdm1) (Kaufman et al., 2003, Kretzschmar et al., 2014, Mlacki et al., 2014, Wang et al., 2008) TFs that could play a role in the regulation of epidermal maturation and terminal differentiation (Figure 3D). In sum, our single-cell resolution data enabled the reconstruction of genetic programs during IFE differentiation in unprecedented detail.
A Majority of HF Subpopulations Express Large Sets of Pseudotime-Dependent Genes
Having defined the genetic program of differentiation in the IFE, we next asked to what degree this differentiation program was applicable to other epidermal cell populations. Interestingly, we observed that the vast majority of epidermal cell populations expressed large numbers of pseudotime-dependent genes in accordance with distinct stages in the differentiation process (Figures 3E, 3F, S4E, and S4F). For instance, most outer bulge subpopulations (OB I–OB V) robustly expressed a large subset of basal genes, while the cells of the upper HF seemed to traverse the complete differentiation program from basal (uHF IV) over intermediate (uHF V) to mature (uHF VI) and terminally differentiated (uHF VII). In order to further demonstrate that IFE and HF cells share core differentiation gene signatures, we identified and modeled the differentiation program independently in the upper HF and found large congruency with IFE differentiation (Figure S4G). The few cell populations (TC, LH, SG, uHF I–III, and IB I) that could not be robustly linked to a particular stage in the differentiation program (Figures 3E, 3F, S4E, and S4F), exhibited immune- and SG-related cellular functions, or underwent an entirely distinct differentiation path like the inner bulge cells (Hsu et al., 2011). Overall, the differentiation program that was identified from analyses of IFE cells seemed universal for most epidermal keratinocytes, summarized in Figure 3G, and accounted for one of the largest sources of cellular heterogeneity throughout the epidermis.
Identification of Spatial Gene Signatures along the Proximal-Distal HF Axis
To further dissect sources of cellular heterogeneity in the HF that are independent of the differentiation signature, we selected all basal IFE and basal HF cells and projected them into t-SNE space. Cells with IFE, uHF, OB, and IB signatures separated into four overlapping clusters positioned along a path, which was used to model a pseudospatial axis similar to the pseudotemporal ordering of the differentiation trajectory (Figures 4A and S5A). Intriguingly, this pseudospatial ordering robustly reproduced the spatial localization of basal subpopulations (Figure 2G) along the proximal-distal axis of the HF (Figures 4B and 4E).
Figure 4.

Defining Spatial Gene Expression Signatures
(A) Pseudospatial ordering of basal cells (n = 486) in t-SNE space, using a minimum spanning tree. The longest path through the graph is highlighted and cells are colored according to second-level clustering.
(B) Validation of pseudospatial ordering of basal cells using known and new IFE basal (Krt14), upper HF (Krt79), Gli1+ outer bulge (Aspn), general outer bulge (Postn), and inner bulge (Krt6a) markers. Upper panel: gene expression in basal cells plotted along the pseudospace trajectory and fitted with a cubic smoothing spline (black line). Lower panel: gene expression projected onto the t-SNE map shown in (A).
(C) “Rolling wave” plot showing the spline-smoothed expression pattern of pseudospace-dependent genes (n = 547) clustered into eight groups (I-VIII) and ordered according to their peak expression.
(D) “Rolling wave” plot showing the spline-smoothed expression pattern of the 30 most significant spatially expressed TFs. TFs were ordered according to group membership and peak expression as shown in (C). P-values for pseudospace dependency are shown on the right. Red line marks Bonferroni-corrected significance threshold of 0.001. TFs marked in bold have not been previously described as relevant for cellular heterogeneity along the proximal-distal axis.
(E) Peak positions of basal cell populations and IB I (defined in second-level clustering) on the spatial axis visualized by kernel density estimation. The organization of the cell populations confirms their spatial positioning in IFE and HF along the proximal-distal axis.
(F) Summary illustrating spatial signatures in epidermal cell populations.
We identified 547 significantly pseudospace-dependent genes and grouped these into eight spatial signatures (Figures 4C and S5B–S5D). A first group of pan-basal genes with peaked expression in the IFE (I), a group of genes most highly expressed in IFE basal (II), a group of genes shared by IFE and uHF basal cells (III), an exclusive uHF signature (IV), a group of genes linked to the Gli1+ population in the distal bulge region (V), an outer bulge signature (VI), a pan-bulge signature (VII), and an exclusive inner bulge signature (VIII) (Table S5). Screening for pseudospace-dependent TFs revealed that only a small number of TFs were linked to IFE and uHF basal signatures (e.g., Ahr, Ets2, Gata6, Tsc22d1) (Figures 4D and S5D). In contrast, TFs were overrepresented in bulge signature genes that can be roughly classified into three groups: TFs most strongly linked to upper bulge signatures (e.g., Gli1, Runx1), the outer bulge (e.g., Tbx1, Lhx2), and pan-bulge or pan-HF TFs (e.g., Foxp1, Sox9, Tfap2b). Overall, we identified well-known TFs in the HF and a variety of putatively new regulatory factors in the HF and IFE (Figures 4D and S5D). The fact that the proximal-distal axis spanning from the inner HF bulge to the IFE could be robustly recapitulated (Figures 4E and 4F) suggests that spatial cues generate gradient responses in keratinocyte populations along the proximal-distal axis (Figure S5E). Moreover, most spatial signatures in the HF were expressed independently of the differentiation state (Figures S5F–S5I). In sum, this analysis demonstrated that spatial gene signatures have a large influence on the overall cellular heterogeneity.
The Differentiation and Spatial Signatures Explain Most Epidermal Heterogeneity
To quantitatively assess to what extent differentiation and spatial gene signatures could explain the observed cellular heterogeneity in the epidermis, we modeled the gene expression profile of each cell as a combination of differentiation and spatial signatures, and five additional types of signatures (two SG signatures and three immune cell related signatures) (Figure 5). We first explored the positions of cells along the pseudotime- and pseudospace-axis (pseudospacetime model, Figures 5A and S6A), and most epidermal subpopulations were located in specific regions in pseudospacetime (Figure 5B). We divided the pseudospacetime model into 15 equally sized bins along each axis and used bin-membership of cells as predictors in a negative binomial regression model (STAR Methods). For each predictor, we were able to define distinct gene sets, which were expressed over the model baseline (i.e., the background expression found in all cells of the data) (Figure 5A, upper and left-hand side panel, and Figure 5C). To evaluate how well the model explained the observed single-cell data, we compared the in silico transcriptomes generated from the model for each cell with the experimentally observed number of molecules. We computed the numbers of molecules that were in agreement (explained molecules), and the numbers of molecules in excess (overexplained molecules) or lacking (underexplained molecules) in the modeled compared to the observed transcriptomes per cell (Figures S6B and S6C). In parallel, we used the same modeling strategy but binned cells based on the first- or second-level clustering. Intriguingly, the pseudospacetime model had an equally high “explanatory performance” as the first- and second-level clustering data (Figures 5D and S6D), suggesting that the differentiation and spatial signatures effectively covered all heterogeneity identified across the main populations (first-level clustering) and sub-populations (second-level clustering). The baseline signature explained around 50% of molecules in the dataset (Figure 5E), and we next investigated the additional “explanatory power” of the respective signatures. The differentiation signature could resolve additional 25%, and, together with the spatial signatures, more than 95% of transcriptome molecules could be explained. The remaining signatures had minor roles, as they were only important for certain cells such as immune cells (Figure 5E). When analyzed from a cell population perspective, the spatial signatures played larger roles in explaining gene expression in basal cells, and the differentiation signatures accounted for most of the non-baseline molecules in suprabasal cells (Figure S6E). We conclude that the gene expression programs associated with differentiation and the proximal-distal spatial axis explain most transcriptional heterogeneity within the epidermis.
Figure 5.

Modeling Transcriptional Heterogeneity Using Space and Time Signatures
(A) Pseudospacetime: matrix showing each cell’s (dots) identity along the differentiation- and spatial-axis, in which both axes were divided into 15 equally sized bins. The numbers of genes expressed over baseline (95% posterior probability, negative binomial regression model) for each bin are shown in barplots (upper and left panels). Cells with expression patterns that could not be placed along the differentiation- and spatial-axes are presented in a separated bar to the right.
(B) The pseudospacetime positions of cells from each cell population defined by either first- or second-level clustering, visualized as percentage of cells per bin.
(C) The number of genes expressed over baseline (95% posterior probability) for the additional signatures used for modeling the transcriptomes of all cells (including SG-related and immune populations).
(D) Model accuracy for the model (including all signature model predictors) in comparison with model accuracy based on either grouping cells according to the first- or second-level clustering or after shuffling the model-predictor matrix (negative control). The model accuracy was computed as the ratio of explained molecules (present in both the simulated and observed) to the sum of explained and unexplained molecules. For each model, the mean and SD of the model accuracy over each group are shown. See Figure S6D for results of each individual cell population.
(E) Percentage of molecules (averaged over all cells) explained by models of increasing complexity. The explained molecules are indicated in green, underexplained in red, and overexplained in blue.
Stem Cells Share a Basal Transcriptional Signature
In the last two decades, numerous studies have described and transcriptionally profiled distinct murine epidermal cell populations in the HF and the IFE with long-term self-renewal capabilities (Blanpain et al., 2004, Brownell et al., 2011, Füllgrabe et al., 2015, Greco et al., 2009, Jaks et al., 2008, Mascré et al., 2012, Page et al., 2013, Snippert et al., 2010). These studies have identified important gene signatures, but they were inherently limited to measuring averages across cell populations due to predefined marker-based sorting strategies. Therefore, it is still unknown what distinguishes cells that express stem cell and progenitor markers (SCMs) from cells that do not. To this end, we selected cells expressing the established SCMs Cd34, Lgr5, Lgr6, Gli1, Lrig1, or high levels of Krt14 (Krt14hi). As expected, we found that most of the SCM+ cells exhibited a basal phenotype (Figure 6A). We next selected all basal cells (STAR Methods), projected them into t-SNE space (Figures 6B and S7B), and marked Cd34, Lgr5, Lgr6, Gli1, Lrig1, or Krt14hi cells on this t-SNE map to display their location (Figures 6B and 6C). As a control, pre-sorted Lgr5-EGFP+ keratinocytes (Jaks et al., 2008) were processed in the same way as the 1,422 cells in this study and found to occupy the same locations in the t-SNE plot as Lgr5-expressing cells did in Figure 6C (data not shown). Interestingly, we observed that, although showing clear peaks in distinct compartments, the expression of most SCMs was scattered over several basal compartments (Figures 6B, 6C, S7A, and S7B), and SCM expression alone was not sufficient to clearly delineate basal cell populations in our dataset. It needs to be determined whether or not these observations could have implications when using SCM-promoter-based lineage tracing (Kretzschmar and Watt, 2014). However, when analyzing each heterogeneous SCM+ population for shared gene expression, we identified robust SCM-linked signatures that were independent of differentiation stages (Figures S7C–S7F; Table S6), underlining the strong impact of niches on gene expression.
Figure 6.

Single-Cell Analyses of Epidermal Stem Cell Populations
(A) Percentage of basal (pseudotime ≤300) and non-basal cells, in each population of cells expressing Lgr5, Cd34, Gli1, Lgr6, Lrig1, or Krt14, respectively. For basal cells, the percentage and the number of cells per total cells are given.
(B) Selection of all basal cells. Right panel: projection of all basal cells (pseudotime ; with and without SCM expression) onto t-SNE space, colored according to the defined cell compartments (first- and second-level clustering). Left panel: illustration summarizing the location of the compartments.
(C) Mapping of basal cells to the t-SNE map defined in (B) according to the expression of SCMs, for each marker gene respectively.
(D) Percentage of basal cells that do not express any of the SCMs Lgr5, Cd34, Gli1, Lgr6, Lrig1, or Krt14 (in red).
(E) Density of basal cells with (gray) and without (red) SCM expression along the pseudotime axis.
(F) Projection of the basal cells that did not express any SCMs (red) onto the t-SNE map defined in (B).
(G) Heatmap of 44 genes that are differentially expressed between SCM+ and SCM− basal cells. Negative binomial regression was used to define specific SCM+ and SCM– gene expression signatures (i.e., the additional number of molecules expressed for each gene if a cell belongs to the SCM+ or SCM– group). For each gene, the group-specific expression in SCM+ and SCM– cells as well as the difference between both groups is shown (median number of molecules).
As most of the SCMs were predominantly expressed in basal cells (Figure 6A), we asked whether basal cells that expressed SCMs (73% of basal cells, Figure 6D) had distinct transcriptional programs in comparison to basal cells without SCM expression. SCM− basal cells were in general “less basal” than those cells expressing SCMs, as evident from projecting these two groups of cells onto the differentiation axis (Figure 6E) and were enriched in the IFE and upper HF compartments (Figure 6F). Using negative binomial regression, we obtained a set of genes that was higher expressed in SCM+ compared to the SCM− cells. Interestingly, the SCM+-enriched genes did not constitute a “unique stem cell signature” and were instead mostly part of a pan-basal gene expression program including components that are involved in the extracellular matrix (ECM) and basement membrane formation, and cell adhesion (Figures 6G and S7G–S7J; Table S6). Some of these genes have been found to be expressed in SCM+ cell populations (Blanpain et al., 2004, Greco et al., 2009, Tumbar et al., 2004), and the recently reported importance of COL17A1 for counteracting HF stem cell aging underpins our findings (Matsumura et al., 2016).
Altogether, we did not observe a clearly delineated transcriptional state (i.e., a set of genes uniquely expressed in stem cells) that set SCM+ and SCM– basal cells apart. What was shared between all SCM+ basal cells was a stronger pan-basal signature. Moreover, the gene expression signatures separating established SCM+ populations are mostly linked to the spatial axis (Figure S7K).
Comparison of Signaling Pathway, Cell Adhesion, and ECM Components across All Epidermal Subpopulations
The identification of 25 distinct (sub-) populations in telogen epidermis enabled direct comparisons of gene expression patterns across all these cell populations. For epidermal homeostasis, firm regulation of signaling pathway activation, niche-component expression, and epigenetic mechanisms are critically important (Hsu et al., 2014, Mesa et al., 2015, Rompolas and Greco, 2014, Botchkarev et al., 2012, Botchkarev and Flores, 2014). Thus, we focused the comparison between subpopulations on six epidermal key pathways (Wnt, Hedgehog [Hh], NF-kB, Notch, Bmp, and Tgf-b), cell adhesion and ECM components (Figures 7A–7C), and components of the epigenetic machinery (data not shown). Unlike the expression of signaling pathway and ECM-related genes, the analysis of epigenetic components did not reveal distinctive expression patterns and these genes were generally expressed at relatively low levels throughout the epidermis.
Figure 7.

Functional Signatures Expressed in Epidermal Subpopulations
(A–C) Expression of genes linked to signaling pathways (A), cell adhesion (B), and extracellular matrix and basement membrane constituents (C) in each epidermal population (defined in either first- or second-level clustering). Shown is the median number of molecules expressed in each cell population (negative binomial regression model).
Markedly, in the Wnt, Hh, Bmp, and Tgf-b signaling pathways we observed most heterogeneity in the expression of ligands, receptors, and their corresponding modulators, whereas their intracellular pathway components were expressed relatively evenly across all subpopulations with a few exceptions such as Gli1 expression indicating active Hedgehog signaling in outer bulge subpopulations (Brownell et al., 2011). Notch pathway components were generally expressed in all subpopulations, with exception of Jag2, which was detected over baseline only in the most basal layers of the IFE and the bulge. Interestingly, there seemed to be a trend of a receptor-ligand division between IFE and HF, most evident in the Wnt and Tgf-b pathways. Wnt ligands for example showed higher expression in the IFE basal layer while Wnt receptors were predominantly expressed in HF populations.
While the expression of signaling pathway genes diverged primarily along the spatial axis, genes linked to different types of cell-cell and cell-ECM junctions showed a strong heterogeneity along the differentiation axis. As expected, genes linked to focal adhesion and hemidesmosome formation were highest expressed in basal populations irrespective of location, while the formation of tight junctions, adherens junctions, gap junctions, and desmosomes was increased in all suprabasal populations.
Among ECM genes, we observed functional division between gene sets linked to a pan-basal state and niche/location related gene signatures. While collagen Col17a1, a subset of glycoproteins (Agrn, Fcgbp) and most laminins (Lama3, Lama5, Lamb2, Lamc2) were expressed at equally high levels across all basal keratinocytes, the majority of ECM genes exhibited a spatial expression corresponding to the pseudospace-related expression patterns identified in Figure 4C.
Overall, these comparisons demonstrated the utility of the transcriptional data of murine epidermis generated within this study, and with the accompanying online tool (http://kasperlab.org/tools or http://linnarssonlab.org/epidermis/) we hope to inspire and enable additional studies in skin biology by using this in-depth single-cell resource.
Discussion
We generated a large resource of single-cell gene expression profiles from murine keratinocytes and used it to dissect epidermal heterogeneity. Four major novelties and highlights of this study are discussed in the following sections.
Identification of Previously Unidentified Epidermal Subpopulations in IFE and the HF
Two cycles of unsupervised clustering, using all cells or subsets of cells, revealed an apparent transcriptional hierarchy between populations (main clusters) and their subpopulations in the epidermis. The 13 main clusters reflected the major IFE differentiation stages and three broad spatial compartments of the HF (upper HF, outer bulge, and inner bulge) and were grouped according to their compartments and functions supporting compartmentalized HF maintenance (Schepeler et al., 2014). Surprisingly, our unbiased clustering (first and second level) failed to demarcate several previously described cell populations, such as Gli1+ or Lgr5+ cells in the lower bulge, Lgr6+ cells of the isthmus, and the Lrig1+ cells in the infundibulum (Table S3) (Brownell et al., 2011, Füllgrabe et al., 2015, Jaks et al., 2008, Jensen et al., 2009, Snippert et al., 2010). Instead, we found that each of these marker-based populations encompassed several subpopulations that were defined in this study. In consequence, although expression of these marker genes has been very useful as genetic tools to study general cell and lineage dynamics during HF maintenance (Jaks et al., 2010, Kretzschmar and Watt, 2014), these markers are not well suited for defining transcriptionally homogenous populations.
Many of the subpopulations we identified have been previously described using immunostaining, lineage tracing or cell-sorting based transcriptional profiling (e.g., Blanpain et al., 2004, Brownell et al., 2011, Füllgrabe et al., 2015, Jaks et al., 2008, Jensen et al., 2009, Snippert et al., 2010, Veniaminova et al., 2013). However, the clustered single-cell transcriptomes of this study yielded more “pure” transcriptional signatures compared to marker-based sorting strategies and thus allowed for a more precise molecular characterization of subpopulations. In addition, we describe several populations that have not been previously identified, have not been described in molecular terms or were only assumed to exist (Table S3). For example, we found two basal subpopulations in the IFE that neither represented the previously described Ivl+ or Lgr6+ populations (Füllgrabe et al., 2015, Mascré et al., 2012). Future studies are needed to resolve whether these two IFE populations represent coexisting cell populations of closed lineages or reflect certain stromal microenvironments or different differentiation stages. Moreover, we found a group of cells in the HF with simultaneous expression of outer bulge (OB) and inner bulge (IB) signatures, which could be placed in the OB. IB cells have the important role to keep OB cells quiescent, until inductive hair growth signals from the dermal papilla stimulate proliferation of lower bulge and hair germ cells in a gradient fashion (Greco et al., 2009, Hsu et al., 2011). Given that in principle all OB cells are competent to enter cell cycle upon damage (Hsu et al., 2011) yet only a subset does during homeostatic hair growth, some cells may have an extra safety mechanism to counteract cell-cycle entry during early anagen by autocrine expression of inhibitory IB signals such as Fgf18.
We also identified two populations lining the opening of the SG with a remarkably high expression of the defensin Defb6. Defensins are small cysteine-rich cationic proteins and function as host defense peptides (Gallo and Nakatsuji, 2011 and references therein). The strategic placement of these two populations at the SG opening, where sebum is released to grease the entire epidermis, indicates DEFB6 as critical in protecting the HF bulge against microorganisms (Chronnell et al., 2001). Elucidating the function of these cells in the context of epidermal physiology will be an interesting topic for future studies.
Transcriptional Resolution of the Differentiation and Proximal-Distal Axis
While our reconstruction of IFE differentiation did not challenge the accepted three-tier model, which postulates a differentiation trajectory from the basal layer over maturation in the spinous layer toward terminal differentiation in the granular layer, we found transient cell states, which are nearly unresolvable with bulk cell methods. Intriguingly, we observed a dramatic transcriptional change along the differentiation axis between gene groups I and III (Figure 3C). It is tempting to speculate whether this change indicates a point of no return along the differentiation trajectory, so that all basal cells—before reaching this point—are to some extent plastic and can provide long-term renewal capacity, although their likelihood to give rise to a long-term surviving clone declines as they move further along the differentiation axis.
Most of the HF subpopulations expressed large sets of genes associated with a distinct differentiation stage and could be positioned along the IFE differentiation axis. To what extent HF and IFE subpopulations share differentiation programs needs further analysis, but these results are indicative of a general pan-differentiation program for keratinocytes with only a few exceptions: SG-related cells and one inner bulge cell cluster (IB I). Most interesting in this regard are the IB I cells. These cells originate from one of the outer bulge populations, relocate during anagen to the lower part of the growing HF, and home back to the bulge in the following catagen-telogen transition to function as proliferation-inhibitory bulge-niche cells (Hsu et al., 2011). The fact that IB I cells could not be placed along the axis of the pan-differentiation program raises the question of whether anagen growth uses an entirely different differentiation program compared to keratinocytes of the non-cycling part of the HF.
Applying a similar strategy as for the reconstruction of the differentiation trajectory (Trapnell et al., 2014), we observed that the basal cells can be aligned along a continuous trajectory reflecting the proximal-distal HF axis. Recent lineage-tracing studies suggest compartmentalized maintenance of the HF, implying that “invisible” borders keep cells within their compartments and compartments separated (Schepeler et al., 2014). The reconstruction of a continuous profile along the spatial axis, however, requires that cells have gradually overlapping sets of genes along the entire HF axis. Thus, it is tempting to speculate whether this feature is important for the extraordinary plasticity of HF cells, reflected in their ability to replace each other upon damage, and take over the role and functions of the replaced cells (Donati and Watt, 2015). For example, isthmus as well as hair germ cells can directly repair bulge cell damage (Rompolas et al., 2013). During wound repair, HF cells are recruited to the IFE and can even convert to permanent progenitors of the IFE epidermis (Ito et al., 2005, Kasper et al., 2011, Levy et al., 2005, Page et al., 2013), but contribution in the opposite direction to damaged existing HFs has, to our best knowledge, never been reported. In concordance, all HF cells expressed typical IFE signature genes, but IFE cells did not express HF-specific genes. The overlapping expression signatures along the spatial axis do not exclude the existence of compartmental borders during homeostasis, established, for example, by a few critical proteins, but may explain the rapid cellular adaptability of epidermal cells upon damage (Rompolas and Greco, 2014, Takeo et al., 2015), because only a small number of additional genes is necessary for a cell to adjust to a new environment.
A Quantitative Model to Explain Tissue Heterogeneity
The transcriptional differences between most subpopulations of keratinocytes could be quantitatively modeled and reconstructed using only the differentiation and spatial signatures. The only exceptions were Defb6+ cells around the SG opening (uHF I and uHF II), which exhibited a unique signature and gene expression patterns of their spatial niche but no pattern of pan-keratinocyte differentiation, and mature SG cells, T cells, and Langerhans cells that only expressed cell-type-associated gene expression signatures. That keratinocyte populations and cellular heterogeneity can effectively be modeled using only two continuous signatures represents unique quantitative insights into cellular heterogeneity, and it will be interesting to investigate the universality of this model for other cell types in other tissues.
Comparison of Epidermal Stem Cell Populations
Finally, we compared basal cells with and without expression of reported stem and progenitor cell markers in an effort to identify a “stemness” gene expression signature. Interestingly, no unique gene expression signature was found in cells expressing these markers. Instead, our results suggest that long-term self-renewing cells in the IFE and the HF do not have a distinct stemness signature other than having a strong basal signature in common, whereas they differ in expression of spatial signatures relating to their location. Altogether, the capacity for long-term self-renewal in the IFE and HF might not require a stemness gene expression signature (Clevers, 2015), but stem cell function might rather coincide with the ability of cells to maintain or occupy certain spatial positions within a tissue and the ability to attach to the basement membrane.
In summary, our reference atlas of transcriptionally distinct cells in the murine epidermis and online tools for custom data visualization and querying will enable deeper inquiries into the physiology of the skin.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-CD3 | BioLegend | Cat#100201 |
| Rat monoclonal anti-CD34 | eBioscience | Cat#14-0341 |
| Rat monoclonal anti-CD207 | eBioscience | Cat#14-2073 |
| Goat polyclonal anti-COX-1 (PTGS1) | Santa Cruz | Cat#sc-1754; RRID: AB_2245319 |
| Rabbit polyclonal anti-EGFP | Thermo Fisher | Cat#A-11122; RRID: AB_2576216 |
| Rabbit polyclonal anti-KI67 | Novocastra | Cat#NCL-Ki67p |
| Goat polyclonal anti-KLK10 | Santa Cruz | Cat#sc-20386 |
| Rabbit polyclonal anti-KRT6 | Covance | Cat#PRB-169P; RRID: AB_10063923 |
| Rabbit polyclonal anti-KRT10 | Covance | Cat#PRB-159P; RRID: AB_291580 |
| Rabbit polyclonal anti-KRT14 | Covance | Cat#PRB-155P; RRID: AB_292096 |
| Mouse monoclonal anti-KRT15 | Abcam | Cat#ab2414 |
| Rabbit monoclonal anti-KRT17 | Cell Signaling | Cat#4543 |
| Goat polyclonal anti-KRT79 | Santa Cruz | Cat#sc-243156 |
| Rabbit polyclonal anti-LOR | Covance | Cat#PRB-145P |
| Goat polyclonal anti-MGST1 | Santa Cruz | Cat#sc-17003; RRID: AB_2143472 |
| FISH probes | ||
| Cd34 | Advanced Cell Diagnostics | Cat#319161-C2 |
| Cst6 | Advanced Cell Diagnostics | Cat#436181 |
| Flg2 | Advanced Cell Diagnostics | Cat#430131 |
| Gli1 | Advanced Cell Diagnostics | Cat#311001 |
| Krt10 | Advanced Cell Diagnostics | Cat#457901 |
| Krt79 | Advanced Cell Diagnostics | Cat#436201-C2 |
| Lgr5 | Advanced Cell Diagnostics | Cat#312171-C2 |
| Lgr6 | Advanced Cell Diagnostics | Cat#404961 / Cat#404961-C2 |
| Lrig1 | Advanced Cell Diagnostics | Cat#310521 |
| Thbs1 | Advanced Cell Diagnostics | Cat#457891 |
| Postn | Advanced Cell Diagnostics | Cat#418581 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Agencourt AMPure XP | Beckman Coulter | Cat#A63880 |
| Defined Keratinocyte-SFM (1X) | Thermo Fisher | Cat#10744019 |
| DNase I Solution (1 mg/ml) | Stem Cell Technologies | Cat#07900 |
| Dynabeads MyOne Streptavidin C1 | Thermo Fisher | Cat#65001 |
| Minimum Essential Medium Eagle -Spinner modification |
Sigma-Aldrich | Cat#M8167 |
| PvuI-HF | NEB | Cat#R3150S |
| Qiaquick Buffer PB | QIAGEN | Cat#19066 |
| Trypsin solution from porcine pancreas | Sigma-Aldrich | Cat#T4424 |
| Critical Commercial Assays | ||
| Anti-Sca-1 MicroBead Kit (FITC), mouse | Miltenyi Biotec | Cat#130-092-529 |
| C1 Single-Cell Auto Prep IFC for mRNA Seq (10 – 17 μm) | Fluidigm | Cat#100-6041 |
| KAPA Library Quantification Kit | KAPA Biosystems | Cat#07960140001 |
| RNAscope Fluorescent Multiplex Reagent Kit | Advanced Cell Diagnostics | Cat#320850 |
| Deposited Data | ||
| Raw data files for RNA sequencing | NCBI GEO | GSE67602 |
| Scripts and computational analysis workflow | Kasper Lab | https://github.com/kasperlab |
| Online tool for visualization of single-cell data | Kasper Lab Linnarsson Lab |
http://kasperlab.org/tools http://linnarssonlab.org/epidermis/ |
| Systematic staining catalog | Kasper Lab | http://kasperlab.org/data |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Charles River | JAX: 000664 |
| Mouse: Lgr5-EGFP-Ires-CreERT2 | Jackson Laboratory | JAX: 008875 |
| Software and Algorithms | ||
| MSigDB | Subramanian et al., 2005 | http://www.broadinstitute.org/gsea/msigdb/index.jsp |
| NetworkX | Schult and Swart, 2008 | https://networkx.github.io/ |
| scikit-learn | Pedregosa et al., 2011 | http://scikit-learn.org/ |
| VGAM | Yee, 2010 | https://cran.r-project.org/web/packages/VGAM/index.html |
| μManager | Edelstein et al., 2014 | http://micro-manager.org/ |
Contact for Reagent and Resource Sharing
Further information and requests for reagents or computational resources may be directed to, and will be fulfilled by the corresponding author Maria Kasper (maria.kasper@ki.se).
Experimental Model and Subject Details
Mice
All experiments were performed on female C57BL/6 mice. The mice were fed ad libitum, and handled and housed under standard conditions in the animal facility of Karolinska University Hospital Huddinge. All mouse experiments were performed in accordance to Swedish legislation and approved by the Stockholm South Animal Ethics Committee. Mice were sacrificed in the second telogen and hair cycle stages were determined by staining dorsal skin sections for Ki67 as described previously (Greco et al., 2009, Müller-Röver et al., 2001). Mice that showed signs of early anagen were excluded from this analysis. Cells from n = 19 mice were included in the final dataset.
Method Details
Cell Isolation
Full epidermal cells were isolated as described previously (Jaks et al., 2008). In brief, clipped and disinfected dorsal skin was isolated, dermal and adipose tissue was removed, and stripes of skin were floated on trypsin for 2 hr at 32°C. Epidermal tissue was subsequently scraped into S-MEM / 1% BSA and single cells were isolated by magnetic stirring at 120 rpm for 25 min / RT. The resulting cell suspension was filtered through 70 μm and 40 μm cell strainers, resuspended in Defined Keratinocyte Serum-free Medium without supplement (DK-SFM), and SCA-1+ and SCA-1− cells were separated using Anti-SCA-1-FITC magnetic beads according to the manufacturer’s instructions. Cells were stored on ice in DK-SFM with 0.1 mg/ml DNase I until capturing. Before capturing, the cell suspension was carefully resuspended and two times passed through a 20 μm cell strainer.
From each experimental mouse, mid-dorsal skin pieces (ca. 0.5 × 0.5 cm) were paraffin-embedded for hair cycle staging and remapping of marker genes.
Cell Capturing, Quality Control, and Single-Cell cDNA Synthesis
Epidermal cells were captured on a medium microfluidic chip (designed for cells from 10 μm – 17 μm) using the Fluidigm C1 Autoprep System. 14 μl filtered cell suspension (∼750 cells / μl in DK-SFM with DNase I) was mixed with 6 μl C1 Suspension Reagent and 14 μl were loaded onto the chip. Single-cells were then captured for 30 min at 4°C using the “Cell Load (1772x/1773x)” script. Capturing efficiency was evaluated on a Nikon TE2000E automated microscope and both bright field and SCA1-FITC images of every capturing position were taken using μManager. Before proceeding with the tagmentation step, each capture site was manually inspected and only capture sites containing single, healthy cells were processed.
Following the image acquisition, STRT-C1 Lysis, RT and PCR mix was added as previously described (Islam et al., 2014), and the “RT + AMP (1772x/1773x)” script was executed. After the cDNA synthesis had been finished (∼8.5 h), the amplified cDNA was harvested with 13 μl Harvest Reagent and cDNA quality was measured on an Agilent BioAnalyzer.
Tagmentation and Isolation of 5′ fragments
The amplified cDNA was fragmented and barcoded using Tn5 DNA transposase (‘tagmentation’) as described previously (Islam et al., 2014). 100 μl Dynabeads MyOne Streptavidin C1 beads were washed in 2x BWT, resuspended in 2 ml 2x BWT, and 20 μl washed beads were added to each well. After 15 min incubation at room temperature, all wells were pooled, the beads were immobilized on a magnet, and the supernatant (containing all internal cDNA fragments) was removed. The beads were resuspended in 100 μl Tris-NaCl-Tween (TNT), washed once in 100 μl Qiaquick PB, and then washed twice in 100 μl TNT. The beads were subsequently incubated in 100 μl restriction mix (1x NEB CutSmart, 0.4 U/μl PvuI-HF enzyme) for 1 hr at 37°C to cleave 3′ fragments which carry a PvuI recognition site. Afterward, the beads were washed three times in TNT, then resuspended in 30 μl ddH2O and incubated for 10 min at 70°C to elute the DNA. To remove short fragments, AMPure beads were used at 1.8 x volume and eluted in 30 μl.
Illumina High-Throughput Sequencing and Processing of Sequencing Reads
The molar concentrations of the libraries were quantified with KAPA Library Quant qPCR and fragment lengths were determined using a reamplified (12 cycles) sample on a BioAnalyzer. Sequencing was performed on an Illumina HiSeq 2000 with C1-P1-PCR2 as read 1 primer and C1-TN5-U as index read primer. Reads of 50 bp as well as 8 bp index reads corresponding to the cell-specific barcodes were generated. Each read was expected to start with a 6 bp unique molecular identifier (UMI), followed by 3-5 guanines and the 5′ end of the mRNA. Reads were processed as described previously (Islam et al., 2014) except that we removed any mRNA molecule (i.e., UMI) supported by only a single read.
Yield and Quality of Sequencing
Sequencing yielded around 25 million mapped reads per C1 chip (793 million mapped reads and 26 million sequenced molecules in total) and around 0.55 million mapped reads per cell after quality control (Figures S1G – S1I). Each unique mRNA molecule was detected 18 times on average during the sequencing indicating sufficient sequencing depth (Figures S1J – S1K). Measurement of RNA spike-in standards indicates strong uniformity between experiments and a sequencing efficiency of 20 - 30 % (Figures S1L – S1N).
Systematic Staining of All Populations by Immunohistochemistry and Single Molecule FISH
The existence and spatial location of the 25 populations and subpopulations defined during 1st and 2nd level clustering were confirmed and determined by antibody staining and/or single-molecule mRNA FISH (FISH) (see Table S7). One subpopulation (uHF III) could not be shown via positive marker staining because this population did not express unique genes in comparison to the other populations, but it formed its own cluster due to the lack of genes. Since all other 24 clusters of cells could be verified, we expect that this population represents a true population and is likely positioned in the SG canal (placed by staining exclusion). The following antibody dilutions were used: CD3 (1:100), CD34 (1:50), CD207 (1:50), COX-1 (PTGS1) (1:50), EGFP (1:500), Ki67 (1:2000), KLK10 (1:50), KRT6 (1:250), KRT10 (1:250), KRT14 (1:250), KRT15 (1:50), KRT17 (1:100), KRT79 (1:50), LOR (1:200), MGST1 (1:50). Cd34, Cst6, Flg2, Gli1, Krt10, Krt79, Lgr5, Lgr6, Lrig1, Thbs1, and Postn mRNA were visualized by FISH using the RNAscope Fluorescent Multiplex Kit (Advanced Cell Diagnostics, Inc.) according to the manufacturers instructions. Please note that the used FISH protocol was in our hands less sensitive compared to our single-cell RNA-seq data and thus for lower expressed genes only few dots can be expected. According to our negative controls, and the manufacturers description, approx. one false positive signal can occur in one out of 10 cells.
Both, antibody and FISH stainings were performed on formalin-fixed, paraffin-embedded (FFPE) sections of dorsal skin isolated from the same animals that were used for the single-cell sequencing. The only exception was staining for anti-EGFP, which was performed on dorsal skin of 8 week old Lgr5-EGFP-Ires-CreERT2 mice using horizontal whole mount staining (Füllgrabe et al., 2015). Images were acquired on either a LSM710-NLO confocal microscope (Zeiss) or a Nikon A1R confocal microscope.
Quantification and Statistical Analysis
Analysis and Visualization of Processed Sequencing Data
The following section describes the data analysis approach employed in this study both in general terms (1-7) and with specific details referring to distinct steps in the analysis process (8). To ensure complete transparency and facilitate reproduction, the complete code used in this study is available online (see Key Resources Table).
(1). Implementation
Analysis and visualization of data were performed in a Python environment built on the NumPy, SciPy, matplotlib, and pandas libraries. Affinity propagation and t-SNE used implementations available in the scikit-learn package (Pedregosa et al., 2011). Graphs were drawn using the NetworkX package (Schult and Swart, 2008). Cubic spline smoothing and likelihood ratio tests were performed using the VGAM package (Yee, 2010), which was accessed via Rpy2. The custom made scripts used for this analysis are available online (see Key Resources Table).
(2). Unsupervised Clustering Using Affinity Propagation
(a). Feature Selection
To filter out genes before affinity propagation (AP) clustering, all genes with an average expression below a specified cut-off and/or those with less than five highly correlated neighbors were excluded. Two genes were defined as highly correlated if their correlation value (Pearson r) was within the top 5% of all gene-gene correlation values within the whole dataset. The remaining genes were used to fit a noise model as
where CV is a gene’s coefficient of variation and mean its average. The 2,500 genes that showed the largest difference between observed CV and CV as predicted by the noise model were used as features for AP clustering.
(b). Affinity Propagation Clustering
Cell populations were defined using AP, a recently introduced approach for unsupervised clustering (Frey and Dueck, 2007). To ensure robustness toward differences in total gene expression between cells, Pearson correlation of log2-transformed data was used as distance metric for the clustering. To facilitate the visualization of clustered data as heatmaps and barplots, the cells / genes within the AP-defined clusters were brought into one-dimensional order based on Ward’s linkage. While mathematical aspects such as the highest possible reduction of variance within clusters were taken into consideration when selecting the clustering parameters preference and damping, parameter choice was mainly based on subjective measures of clustering performance.
(c). Evaluation of Clustering Robustness
To evaluate robustness of AP clustering, a resampling approach was used, where 25% of cells were removed from the dataset at random. The remaining cells were reclustered using the same parameters as for the main clustering and the percentage of cells in each defined group that remain clustered together was determined. In order to measure the background distribution (i.e., the percentage of cells which remain together by pure chance), the group labels were randomly permutated. Both the resampling and the label permutation were repeated 100 times.
(3). Nonlinear Dimensionality Reduction with t-Distributed Stochastic Neighbor Embedding
Dimensionality reduction to two dimensions for visualization purposes and as input for pseudotemporal/-spatial ordering was performed using t-distributed stochastic neighbor embedding (t-SNE) (Van der Maaten and Hinton, 2008). In most cases, a perplexity value between 20 and 25, an early exaggeration value of 2.0 – 3.0 and a learning rate of 1,000 were used.
(4). Negative Binomial Regression of Gene Expression
(a). Model Description
To assign expression of a gene to a cell population, a Bayesian general linear model (GLM) was used as described elsewhere (Zeisel et al., 2015). In such a model, it is assumed that the outcome (i.e., the measured expression of a gene in a population) is sampled from a distribution whose mean is determined by a linear combination of K predictors xi with coefficients βi. Therefore,
For each cell, the outcome and predictors are known and we aim to determine the values of the coefficients.
As predictors, we use a Baseline predictor and a binary Cell Type predictor. As we expect every gene to have a baseline expression proportional to the total number of expressed molecules within a particular cell, the Baseline predictor value is set as a cell’s molecule count normalized to the average molecule count of all cells. Meanwhile, the Cell Type predictor is set to 1 if a cell is included in a particular cell population cluster or a pseudospace / pseudotime bin. In consequence, the coefficient βk for a Cell Type predictor xk represents the additional number of molecules of a particular gene that are present if a cell is member of a particular cell type.
As real count data is usually overdispersed when compared to an ideal Poisson distribution, we used a negative binomial distribution, which can be represented as a Gamma distribution of Poisson distributions, for our model. Therefore, if y is the observed count,
with mean μ = ab and standard deviation .
As the standard deviation roughly scales as the square root of the mean, it can be described as with overdispersion factor r. Hence,
By attaching prior distributions to the overdispersion factor r and the coefficients βk, we acquire a full Bayesian negative binomial regression model, with
The model was implemented in STAN. A more detailed explanation of the model is provided elsewhere (Zeisel et al., 2015).
(b). Calling Genes That Are Specifically or Uniquely Expressed in Groups / Predictors
To define whether a gene can be considered specifically expressed in a particular cell population, we compared the posterior probability distributions of the Baseline coefficient and the Cell Type coefficient. A gene was considered activated in a cell population if its class-specific coefficient exceeded the Baseline coefficient with a specified posterior probability. In order to be defined as uniquely expressed in a particular cell population, a gene’s Cell Type coefficient had to exceed all other Cell Type coefficients as well as the Baseline coefficient with a specified posterior probability. The posterior probability cut-off at which genes were considered specifically or uniquely expressed was set at 99.9% for the regression model of the 1st level clustering and to 95% for all other regression models.
(c). Evaluating the Exploratory Quality of Regression Models
In order to evaluate how well a regression model explains the data, a simulated dataset was sampled from the model and compared to the observed data. In particular, for every gene and predictor xk in the model, values were randomly sampled one hundred times from the posterior probability distribution of each coefficient βk and subsequently multiplied with the predictor matrix used as input for the model. The resulting dataset contains the simulated expression data of g genes in m cells over K predictors. These data were subsequently summarized including either all or a subset of predictors and compared to the observed data. For each gene, the number of ‘explained’ (molecules both found in the observed and the simulated data), ‘underexplained’ (molecules found in the observed but not the simulated data) and ‘overexplained’ (molecules found in the simulated but not the observed data) molecules was determined. Data-model comparison occurred either on a single-cell level, a group level (for each gene, the number of molecules in the observed and simulated data were pooled between all cells within a group, thus averaging in-group noise) or a whole-dataset level (for each gene, the number of molecules in the observed and simulated data were pooled between all cells in the dataset).
(5). Pseudotemporal/-Spatial Ordering of Cells
(a). Bringing Cells into Pseudotemporal/-Spatial Order
Spatial and temporal ordering is based on the same analytical method and only distinguished by the input of cells (differentiating cells of the IFE for pseudotime; basal cells of HF and IFE for pseudospace). The pseudotemporal/-spatial ordering of IFE/basal cells is following a graph-based approach that was recently introduced by Magwene et al., 2003 and Trapnell et al., 2014. In brief, a minimum spanning tree (MST) is constructed between cells, which are defined by their position in – dimensionality-reduced – space. The longest path through the MST, called the diameter path, is subsequently defined and a PQ tree encoding all paths through the graph (or orderings of cells) under the constraints of the diameter path is constructed. The PQ tree is subsequently screened for orderings of cells that minimize the total traveling distance. While we generally follow the approach introduced by Trapnell et al., 2014 we diverge in several points. Since linear dimensionality reduction approaches such as PCA or ICA were insufficient to resolve and visualize the differentiation and spatial trajectories in the dataset, we used the nonlinear t-SNE method for dimensionality reduction and construction of the MST. Due to the high number of single cells included in our analysis (536 IFE cells and 486 basal cells) and due to a relative high level of noise, we furthermore did not consider all permutation emitted from the PQ. Instead, we restricted the number of orderings based on local optima derived from subsets of the graph.
(b). Testing the Robustness of Pseudotemporal or Pseudospatial Ordering
To test the robustness of the pseudotemporal/-spatial ordering, we (1) compared the results to orderings gained without any dimensionality reduction and (2) employed a resampling approach. During the resampling, we either compared the results of one hundred orderings gained from different initial t-SNE plots to our initial results to evaluate robustness against randomness in the dimensionality reduction or we randomly discarded 25% of cells from the dataset for one hundred times and compared the resulting ordering to our initial results to test for robustness against small changes in composition of the dataset. As negative control, we randomly shuffled cell labels.
(c). Modeling Gene Expression over Pseudospace/-Time and Calling Pseudospace/-Time-Dependent Genes
To model gene expression changes in dependency of pseudotime or pseudospace, a cubic smoothing spline with five effective degrees of freedom was fitted to the ordered expression data of all genes in the IFE or basal dataset which showed an average expression > 0.1 molecules. Pseudospace/-time dependency of gene expression was subsequently tested by comparing the spline-smoothed model to a pseudospace/-time-independent restricted model using the approximate likelihood ratio test. We considered all genes with a p-value below the Bonferroni-corrected significance level α = 0.001 to be pseudotime- or pseudospace-dependent. To visualize the expression patterns of all pseudotime- or pseudospace dependent genes and to perform gene set enrichment analysis, spline smoothed gene expression data was clustered using AP as described above. Genes within each cluster were ordered according to expression peak or onset of induction (defined as point in pseudospace/pseudotime where the expression of a gene exceeds 50% of the peak expression).
(d). Positioning Cells in Pseudospace/-Time
To link single cells not included in the model to a specific place in pseudotime or pseudospace, the expression data of g pseudospace/-time dependent genes in a particular cell M is correlated to all points in the fitted model (which contains the spline-fitted expression data of g pseudospace/-timespace-dependent genes over t points in pseudospace/-time) and the point with the highest Pearson r is returned.
To evaluate how well a particular cell or group of cells fits a pseudospace/-time model, we used several qualitative and quantitative approaches: on the one hand, we analyzed how many pseudospace/-time-dependent genes are expressed in a particular group of cells. We reasoned that a group of cells which exhibits e.g., features of a certain differentiation stage will express a high number of genes linked to this particular stage. On the other hand, we consider the p-value of the best fitting cell-to-point correlation a quantitative measure of fit. Furthermore, we employed a resampling approach to test the robustness of the correlation. In this approach, we randomly removed 75% of pseudotime- or pseudospace-dependent genes from the dataset for one hundred times and subsequently correlated each single cell to a specific point on the axis as described above. We then measured the average distance of the correlation points yielded from the reduced dataset to the correlation gained with the full dataset. We reasoned that cells which have a strong pseudotime-/pseudospace signature will be more robust against the resampling of the dataset and will thus show a narrower spread of correlation points.
(6). Constructing Gene-Gene Neighbor Networks
To construct networks of pseudotime- and pseudospace-dependent genes, we used a shared nearest neighbor approach in combination with the previously described context likelihood of relatedness (CLR) algorithm (Faith et al., 2007). Specifically, we initially generated a gene-gene correlation matrix between all selected genes and subsequently used CLR to transform the correlation values based on their network context. For each gene, we then selected the n nearest neighbors. We considered two genes to be linked within the neighbor context if they shared a number ≥ k of nearest neighbors. Graphs were drawn using a force-directed spring layout with each node representing a gene and each edge connecting two interlinked genes.
In the pseudotime- and pseudospace-gene networks, two genes were considered linked if they shared at least 5 of 25 nearest neighbors. In the basal gene network, two genes were considered linked if they shared 10 or more of 25 nearest neighbors.
(7). Gene Set Enrichment Analysis
To link gene lists – for instance pseudotime- or pseudospace-dependent genes at particular stages – to potential biological roles, we queried the Molecular Signatures Database MSigDB using the ‘Investigate Gene Sets’ function (Subramanian et al., 2005). We only considered gene sets included in the CP, CP:BIOCARTA, CP:KEGG, CP:REACTOME, and BP categories of the dataset and excluded all matches with an FDR q-value . To avoid redundancies, the usually five reported gene sets were selected among the 20 most significant matches.
(8). Data Analysis Process
(a). Selection of Cells
Cells with less than 2,000 unique molecules were removed from the dataset, leaving 1,422 cells passing the quality criteria.
(b). 1st Level Clustering – AP Clustering
For the 1st level clustering, 2,500 features were selected as described in (2) using a mean expression cut-off of 0.05 molecules over the whole dataset (1,422 cells). Gene-gene and cell-cell Pearson distances were subsequently calculated and used as input for AP clustering. To achieve a better resolution of cell populations, gene clusters linked to ribosomal, housekeeping and intermediate early genes (IEGs) were removed after an initial round of clustering along the gene axis. In summary, 13 distinct cell populations could be defined during 1st level clustering. Clustering robustness was evaluated as described in (2). Additionally, the AP clustering approach was compared with unsupervised clustering by backSPIN (Zeisel et al., 2015) with good agreement. A t-SNE representation of the whole dataset was generated with the same features as used for the AP clustering.
(c). 1st Level Clustering – Negative Binomial Regression
A negative binomial regression model was generated as described in (4) using the 1st level clusters as predictors. The regression was performed on all genes with an average molecule count ≥ 0.25 over either the whole dataset or within at least one cluster (9,016 genes). Group-specific or –unique genes were called using a 99.9% posterior probability cut-off.
(d). 2nd Level Clustering – Cell Selection
2nd level clustering was performed separately on subsets of cells showing inner bulge (IB), outer bulge (OB), upper HF (uHF), or IFE basal (IFE B) signatures. Signature genes were identified from the 1st level clustering negative regression model: (1) as genes, which are only expressed over Baseline in either the IB, OB, uHF, or IFE B cluster(s), or (2) as genes, whose expression in one of these clusters exceeds the expression in all other clusters with 99.9% posterior probability. Following the identification of signature genes, the cumulative expression of the four different signatures was calculated for every cell in the dataset and cut-offs defining whether or not a single cell expresses a certain signature were specified. To avoid duplication of cells with more than one signature, cells were assigned to the four groups in the following order of primacy: IB > OB > uHF > IFE B. In this way, 87 IB, 273 OB, 364 uHF and 322 IFE B cells (from 630 IFE cells) were defined.
(e). 2nd Level Clustering – AP Clustering
From each of the four subsets of the data, features were selected as described in (2) using a mean expression cut-off of 0.1 molecules and genes linked to ribosomal, housekeeping and IEG clusters in the 1st level clustering were removed. Due to the considerably lower signal-to-noise ratios expected in the subpopulations, the selected genes were subjected to a first round of AP clustering and only clusters of genes that exhibited a strong and coordinated differential expression pattern were used as features for the final clustering of cells. Using this approach, three, seven, five, and three subclusters of cells were identified in the IB, uHF, OB, and IFE B data respectively. Clustering robustness was measured as described in (2).
(f). 2nd Level Clustering – Negative Binomial Regression
To perform negative binomial regression on the 2nd level clustering data while still considering the whole dataset, each cell assigned to the IB, OB, uHF or IFE B subset of the data was grouped according to its 2nd level cluster identity. All remaining cells (e.g., the immune cells or the cells of the IFE differentiation process which did not show an IFE B or IB/OB/uHF signature) were grouped according to 1st level cluster membership. The combination of the 2nd and 1st level clustering data allowed regression with 25 Cell Type predictors. The regression was performed on all genes with an average molecule count over either the whole dataset or within at least one cluster (9,784 genes). Group-specific or –unique genes were called using a 95% posterior probability cut-off.
(g). 1st and 2nd Level Clustering – Robustness towards Replication
To ensure that none of the cell populations defined during 1st and 2nd level clustering is the mere result of an experimental or technical artifact, the robustness of each cluster toward biological replication was analyzed. To this end, the number of cells in each cluster, the ratio of cells from SCA-1+ and SCA-1− fractions and the number of experimental mice from which the cells in each cluster were derived was calculated and compared to the number of mice expected by pure chance. To acquire the expected value of mice for a cell population, nSCA1+ / nSCA1- cells corresponding to the number of SCA-1+ and SCA-1− cells in the population were randomly sampled from the SCA-1+ and SCA-1− dataset and the total number of mice from which the sampled cells were derived was subsequently calculated. For each population, this sampling was repeated 10,000 times and a p-value was returned.
| Population | SCA-1+ Fraction | Number of Cells | Number of Mice | Number of Mice if Random | p-value |
|---|---|---|---|---|---|
| IFE B I | 91.5 % | 94 / 1422 | 10 / 19 | 13.26 | 0.0048 |
| IFE B II | 85.8 % | 134 / 1422 | 14 / 19 | 16.19 | 0.0703 |
| INFU B | 48.9 % | 94 / 1422 | 18 / 19 | 18.38 | 0.4925 |
| IFE D I | 45.0 % | 140 / 1422 | 19 / 19 | 18.83 | 1 |
| IFE D II | 30.9 % | 97 / 1422 | 19 / 19 | 18.65 | 1 |
| IFE K I | 21.1 % | 57 / 1422 | 15 / 19 | 17.63 | 0.0249 |
| IFE K II | 35.7 % | 14 / 1422 | 11 / 19 | 9.91 | 0.9014 |
| uHF I | 9.1 % | 33 / 1422 | 13 / 19 | 14.98 | 0.1343 |
| uHF II | 11.1 % | 36 / 1422 | 15 / 19 | 15.50 | 0.4892 |
| uHF III | 13.3 % | 45 / 1422 | 14 / 19 | 16.60 | 0.0438 |
| uHF IV | 23.4 % | 111 / 1422 | 19 / 19 | 18.76 | 1 |
| uHF V | 15.2 % | 79 / 1422 | 18 / 19 | 18.23 | 0.5875 |
| uHF VI | 10.8 % | 37 / 1422 | 13 / 19 | 15.63 | 0.053 |
| uHF VII | 13.0 % | 23 / 1422 | 11 / 19 | 13.01 | 0.1333 |
| SG | 5.3 % | 19 / 1422 | 8 / 19 | 11.54 | 0.0127 |
| OB I | 10.5 % | 105 / 1422 | 17 / 19 | 18.47 | 0.0583 |
| OB II | 9.8 % | 51 / 1422 | 16 / 19 | 16.91 | 0.3339 |
| OB III | 4.9 % | 41 / 1422 | 17 / 19 | 15.82 | 0.9194 |
| OB IV | 6.5 % | 46 / 1422 | 16 / 19 | 16.37 | 0.5234 |
| OB V | 6.7 % | 30 / 1422 | 15 / 19 | 14.34 | 0.7982 |
| IB I | 7.4 % | 54 / 1422 | 17 / 19 | 17.03 | 0.6533 |
| IB II | 15.8 % | 19 / 1422 | 9 / 19 | 11.84 | 0.0414 |
| IB III | 0.0 % | 14 / 1422 | 9 / 19 | 9.49 | 0.5027 |
| TC | 5.6 % | 18 / 1422 | 9 / 19 | 11.23 | 0.0952 |
| LH | 9.7 % | 31 / 1422 | 14 / 19 | 14.66 | 0.445 |
(h). Modeling of IFE Differentiation
To model IFE differentiation, all cells belonging to the non-infundibulum IFE basal clusters (IFE BI and IFE BII) or the remaining IFE cells identified in the 1st level clustering were considered (536 cells). Features were selected as described in (2) using a mean expression cut-off of 0.1 molecules and genes linked to ribosomal, housekeeping and IEG clusters in the 1st level clustering were removed. The remaining features were used as input for t-SNE (perplexity = 25, early exaggeration = 2.0) and the cells were brought into pseudotemporal order as described in (5). Cubic splines were fitted to the expression of 7,354 genes (mean expression molecules), 1,627 significantly pseudotime-dependent genes were identified and subsequently AP clustered into eight subgroups. All cells from the dataset were correlated to the differentiation trajectory and the robustness of the pseudotemporal ordering and the correlation was evaluated as described above.
(i). Modeling of uHF Differentiation
To test whether the differentiation process follows similar lines in different compartments of the epidermis, pseudotemporal ordering of uHF cells was performed. For this, all non-SG (opening) uHF cells (uHF IV – VII, 250 cells) were used. Features were selected as described in (g). In contrast to (g), an initial round of dimensionality reduction (TruncatedSVD, 5 dimensions) was necessary to get a good t-SNE representation of the data (perplexity = 100, early exaggeration = 2.0). After pseudotemporal ordering and cubic spline fitting, 1,068 significantly pseudotime-dependent genes could be defined.
(j). Modeling of gene Expression Changes Along the Proximal-Distal Spatial Axis
In order to model spatial gene expression changes along the proximal-distal axis without interference from differentiation signatures, only cells from IFE and HF which show a clear basal signature were selected. Cells from the HF (uHF IV – VII, OB I – V, IB I – III∗) were considered basal if they were linked to a pseudotime position . Due to the early onset of differentiation in the IFE basal compartment, IFE cells were selected with a more stringent cut-off . In sum, 486 cells were classified as basal. Features were selected as described in (2) using a mean expression cut-off of 0.1 molecules and genes linked to ribosomal, housekeeping and IEG clusters in the 1st level clustering were removed. To make sure that no differentiation related modules of genes are included in the dataset, the genes were subjected to one round of AP clustering and only clusters not containing typical differentiation markers (e.g., Mt4 or Krt10) were included. Only the genes that passed this additional cycle of quality control were used as input for t-SNE (perplexity = 20, early exaggeration = 3.0) and the basal cells were subsequently brought into pseudospatial order as described in (5). Cubic splines were fitted to the expression of 6,788 genes (mean expression molecules), 547 significantly pseudospace-dependent genes were identified and subsequently AP clustered into eight subgroups. All cells from the dataset were correlated to the spatial axis and the robustness of the pseudospatial ordering and of the correlation was evaluated as described above.
∗ Although the cells of the inner bulge population IB I do not seem to show any distinct differentiation signatures, cells from IB I were considered in this model if under the set cut-off.
(k). Pseudospacetime – Creation
To link every cell to its position in two-dimensional space along the differentiation and spatial axes without interference from ambiguous genes, only genes, which were either uniquely pseudotime- (1,409 genes) or pseudospace-dependent (329 genes), were considered and correlation of all cells to both axes was recalculated using only the selected genes. Cells and cell populations which do not seem to fit to any position on either the pseudospace-, the pseudotime- or both axes (e.g., the immune or sebaceous gland cells, see (5)) were subsequently (partially) removed from the pseudospace.
(l). Pseudospacetime – Negative Binomial Regression
To perform negative binomial regression of the data under the constraints of the pseudospacetime model, both the pseudospace- and pseudotime-axis were divided into 15 equally sized bins and each pseudospace-/pseudotime-bin was considered a predictor in the regression model. Furthermore, additional predictors (sebaceous gland, sebaceous gland opening, pan-immune, T-cell and Langerhans cell) were generated for genetic signatures that cannot be explained by the pseudospacetime model. Regression was performed on the same set of 9,784 genes as selected in (f). Predictor-specific or –unique genes were called using a 95% posterior probability cut-off.
As a negative control, predictor identity was randomly shuffled between cells and the regression was performed as described above.
(m). Pseudospacetime – Model Comparison
To evaluate the explanatory quality of the pseudospacetime model, a simulated dataset was sampled from the traces of the negative regression model as described in (4) and subsequently compared to the observed data. To ensure comparability of the pseudospacetime model with the 1st and 2nd level clustering, only genes used consistently in the pseudospacetime, the 1st level, and the 2nd level regression were considered (6,949 genes).
(n). Stem Cell Analysis – Cell Selection
To select cells, which express the stem cell/progenitor markers Lgr5, Cd34, Gli1, Lgr6, Lrig1, and Krt14 above Baseline, the following cut-offs were chosen:
| Marker | Cut-off Selection | Cut-Off Value (Molecules) | Number of Positive Cells |
|---|---|---|---|
| Lgr5 | Maximal Baseline value predicted during 2nd level clustering regression multiplied by 10 | 0.34 | 138 |
| Cd34 | Maximal Baseline value predicted during 2nd level clustering regression multiplied by 10 | 0.85 | 297 |
| Gli1 | Maximal Baseline value predicted during 2nd level clustering regression multiplied by 10 | 0.23 | 84 |
| Lgr6 | Maximal Baseline value predicted during 2nd level clustering regression multiplied by 10 | 0.26 | 75 |
| Lrig1 | Maximal Baseline value predicted during 2nd level clustering regression multiplied by 5 | 1.95 | 207 |
| Krt14 | Maximal Baseline value predicted during 2nd level clustering regression multiplied by 10 | 35.18 | 149 |
(o). Stem Cell Analysis – AP Clustering
AP clustering was performed separately on all cells expressing a certain stem cell marker using the same approach as described for the 2nd level clustering in (e).
(p). Stem Cell Analysis – Basal Cell Clustering and t-SNE
To compare basal stem cells to each other and to basal cells, which do not express stem cell markers, all cells from IFE, uHF (uHF IV – VII) and OB with a pseudotime position were selected. In contrast to the cell selection described in (j), IFE cells were selected less stringently, inner bulge cells were not considered and ambiguous genes were removed before the pseudotime correlation (see (k)). In sum, 673 cells were considered as basal cells. Basal cells were subclustered into 7 groups using the same approach as described for the 2nd level clustering. The same features selected for the final clustering were used to generate a t-SNE representation of the basal dataset (perplexity = 20, early exaggeration = 2.0).
(q). Stem Cell Analysis – Negative Binomial Regression
To model genetic signatures which are either unique for each stem cell population or shared by all basal SCM+ or SCM− cells, we created two negative binomial regression models. (1) In the first model, gene expression in stem cells was modeled as a combination of Baseline expression, specific signatures unique to each stem cell population (e.g., all Lgr5+ cells) and signatures shared by SCM+ and SCM− cells. This model was used to determine stem cell population-specific gene expression signatures, which were called using a 95% posterior probability cut-off against Baseline. (2) The second approach modeled gene expression in stem cells as a combination of Baseline expression, two common signatures shared by all basal SCM+ and SCM− cells, and specific signatures unique to each compartment (IFE, uHF, upper OB and OB). As the second approach performed better in modeling SCM+ and SCM− signatures, it was used to define the SCM+ signature (90% posterior probability against Baseline; see Figure S7F) and to compare SCM+ to SCM− signatures. A gene was considered differentially expressed in SCM+ compared to SCM− cells (or vice versa) if it was represented with at least 0.25 molecules (median) in the SCM+ signature and if its SCM+ signature exceeds the SCM− signature with 90% posterior probability.
Data and Software Availability
Software
The computational analysis workflow and the scripts are available at https://github.com/kasperlab.
Data Resources
The accession number for the sequencing data reported in this paper is NCBI GEO: GSE67602.
Additional Resources
An online tool for the visualization of the single-cell dataset is available at http://kasperlab.org/tools or http://linnarssonlab.org/epidermis/.
A systematic staining catalog is provided at: http://kasperlab.org/data.
Author Contributions
S.J., S.L., and M.K. conceived and designed the study. S.J., A.Z., G.L.M., and P.L. performed sequencing experiments and computational analyses. S.J., T.J., and X.S. performed immunostaining experiments and microscopy analyses. S.J., A.Z., T.J., S.L., and M.K. interpreted data. S.J. and M.K. wrote the manuscript with input from all authors.
Acknowledgments
We thank Alexandra Are, Karl Annusver, and Åsa Bergström for technical help with immunohistochemistry and mice and Anna Juréus for help with RNA sequencing. We are grateful to Rickard Sandberg and Rune Toftgård for feedback and discussion on the manuscript. This work was supported by grants from the Swedish Cancer Society, Swedish Research Council (STARGET), Swedish Foundation for Strategic Research, Center for Innovative Medicine, and Ragnar Söderberg Foundation to M.K., European Research Council (261063, BRAINCELL), and Swedish Research Council (STARGET) to S.L., Human Frontier Science Program to A.Z., and Karolinska Institutet KID funding to S.J. and T.J. Parts of this study were performed at the Live Cell Imaging facility/Nikon Center of Excellence, Department of Biosciences and Nutrition, Karolinska Institutet, supported by grants from the Knut and Alice Wallenberg Foundation, the Swedish Research Council, the Center for Innovative Medicine, and the Jonasson donation to the School of Technology and Health, Royal Institute of Technology, Sweden.
Published: September 15, 2016
Footnotes
Supplemental Information includes seven figures and seven tables and can be found with this article online at http://dx.doi.org/10.1016/j.cels.2016.08.010.
Contributor Information
Sten Linnarsson, Email: sten.linnarsson@ki.se.
Maria Kasper, Email: maria.kasper@ki.se.
Supporting Citations
The following references appear in the Supplemental Information: Collette et al., 2013, Fujiwara et al., 2011, Horsley et al., 2006, Magwene et al., 2003, Nijhof et al., 2006, Zeeuwen et al., 2002.
Supplemental Information
References
- Alcolea M.P., Jones P.H. Lineage analysis of epidermal stem cells. Cold Spring Harb. Perspect. Med. 2014;4:a015206. doi: 10.1101/cshperspect.a015206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi H., Li S., Qu X., Wang M., Bai X., Xu Z., Ao X., Jia Z., Jiang X., Yang Y., Wu H. DEC1 regulates breast cancer cell proliferation by stabilizing cyclin E protein and delays the progression of cell cycle S phase. Cell Death Dis. 2015;6:e1891. doi: 10.1038/cddis.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpain C., Lowry W.E., Geoghegan A., Polak L., Fuchs E. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell. 2004;118:635–648. doi: 10.1016/j.cell.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Botchkarev V.A., Flores E.R. p53/p63/p73 in the epidermis in health and disease. Cold Spring Harb. Perspect. Med. 2014;4 doi: 10.1101/cshperspect.a015248. a015248–a015248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botchkarev V.A., Gdula M.R., Mardaryev A.N., Sharov A.A., Fessing M.Y. Epigenetic regulation of gene expression in keratinocytes. J. Invest. Dermatol. 2012;132:2505–2521. doi: 10.1038/jid.2012.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownell I., Guevara E., Bai C.B., Loomis C.A., Joyner A.L. Nerve-derived sonic hedgehog defines a niche for hair follicle stem cells capable of becoming epidermal stem cells. Cell Stem Cell. 2011;8:552–565. doi: 10.1016/j.stem.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chronnell C.M., Ghali L.R., Ali R.S., Quinn A.G., Holland D.B., Bull J.J., Cunliffe W.J., McKay I.A., Philpott M.P., Müller-Röver S. Human beta defensin-1 and -2 expression in human pilosebaceous units: Upregulation in acne vulgaris lesions. J. Invest. Dermatol. 2001;117:1120–1125. doi: 10.1046/j.0022-202x.2001.01569.x. [DOI] [PubMed] [Google Scholar]
- Clevers H. STEM CELLS. What is an adult stem cell? Science. 2015;350:1319–1320. doi: 10.1126/science.aad7016. [DOI] [PubMed] [Google Scholar]
- Collette N.M., Yee C.S., Murugesh D., Sebastian A., Taher L., Gale N.W., Economides A.N., Harland R.M., Loots G.G. Sost and its paralog Sostdc1 coordinate digit number in a Gli3-dependent manner. Dev. Biol. 2013;383:90–105. doi: 10.1016/j.ydbio.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotsarelis G., Sun T.T., Lavker R.M. Label-retaining cells reside in the bulge area of pilosebaceous unit: Implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell. 1990;61:1329–1337. doi: 10.1016/0092-8674(90)90696-c. [DOI] [PubMed] [Google Scholar]
- Donati G., Watt F.M. Stem cell heterogeneity and plasticity in epithelia. Cell Stem Cell. 2015;16:465–476. doi: 10.1016/j.stem.2015.04.014. [DOI] [PubMed] [Google Scholar]
- Edelstein A.D., Tsuchida M.A., Amodaj N., Pinkard H., Vale R.D., Stuurman N. Advanced methods of microscope control using μManager software. J. Biol. Methods. 2014;1(2) doi: 10.14440/jbm.2014.36. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith J.J., Hayete B., Thaden J.T., Mogno I., Wierzbowski J., Cottarel G., Kasif S., Collins J.J., Gardner T.S. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5:e8. doi: 10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey B.J., Dueck D. Clustering by passing messages between data points. Science. 2007;315:972–976. doi: 10.1126/science.1136800. [DOI] [PubMed] [Google Scholar]
- Fuchs E. Epidermal differentiation: The bare essentials. J. Cell Biol. 1990;111:2807–2814. doi: 10.1083/jcb.111.6.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs E. Scratching the surface of skin development. Nature. 2007;445:834–842. doi: 10.1038/nature05659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H., Ferreira M., Donati G., Marciano D.K., Linton J.M., Sato Y., Hartner A., Sekiguchi K., Reichardt L.F., Watt F.M. The basement membrane of hair follicle stem cells is a muscle cell niche. Cell. 2011;144:577–589. doi: 10.1016/j.cell.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Füllgrabe A., Joost S., Are A., Jacob T., Sivan U., Haegebarth A., Linnarsson S., Simons B.D., Clevers H., Toftgård R., Kasper M. Dynamics of Lgr6+ progenitor cells in the hair follicle, sebaceous gland, and interfollicular epidermis. Stem Cell Reports. 2015;5:843–855. doi: 10.1016/j.stemcr.2015.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo R.L., Nakatsuji T. Microbial symbiosis with the innate immune defense system of the skin. J. Invest. Dermatol. 2011;131:1974–1980. doi: 10.1038/jid.2011.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco V., Chen T., Rendl M., Schober M., Pasolli H.A., Stokes N., Dela Cruz-Racelis J., Fuchs E. A two-step mechanism for stem cell activation during hair regeneration. Cell Stem Cell. 2009;4:155–169. doi: 10.1016/j.stem.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo N., Krutzsch H.C., Inman J.K., Roberts D.D. Thrombospondin 1 and type I repeat peptides of thrombospondin 1 specifically induce apoptosis of endothelial cells. Cancer Res. 1997;57:1735–1742. [PubMed] [Google Scholar]
- Hashimshony T., Wagner F., Sher N., Yanai I. CEL-Seq: Single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012;2:666–673. doi: 10.1016/j.celrep.2012.08.003. [DOI] [PubMed] [Google Scholar]
- Honma S., Kawamoto T., Takagi Y., Fujimoto K., Sato F., Noshiro M., Kato Y., Honma K. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature. 2002;419:841–844. doi: 10.1038/nature01123. [DOI] [PubMed] [Google Scholar]
- Horsley V., O’Carroll D., Tooze R., Ohinata Y., Saitou M., Obukhanych T., Nussenzweig M., Tarakhovsky A., Fuchs E. Blimp1 defines a progenitor population that governs cellular input to the sebaceous gland. Cell. 2006;126:597–609. doi: 10.1016/j.cell.2006.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu Y.-C., Pasolli H.A., Fuchs E. Dynamics between stem cells, niche, and progeny in the hair follicle. Cell. 2011;144:92–105. doi: 10.1016/j.cell.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu Y.-C., Li L., Fuchs E. Emerging interactions between skin stem cells and their niches. Nat. Med. 2014;20:847–856. doi: 10.1038/nm.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam S., Zeisel A., Joost S., La Manno G., Zajac P., Kasper M., Lönnerberg P., Linnarsson S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods. 2014;11:163–166. doi: 10.1038/nmeth.2772. [DOI] [PubMed] [Google Scholar]
- Ito M., Liu Y., Yang Z., Nguyen J., Liang F., Morris R.J., Cotsarelis G. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat. Med. 2005;11:1351–1354. doi: 10.1038/nm1328. [DOI] [PubMed] [Google Scholar]
- Jaks V., Barker N., Kasper M., van Es J.H., Snippert H.J., Clevers H., Toftgård R. Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nat. Genet. 2008;40:1291–1299. doi: 10.1038/ng.239. [DOI] [PubMed] [Google Scholar]
- Jaks V., Kasper M., Toftgård R. The hair follicle—a stem cell zoo. Exp. Cell Res. 2010;316:1422–1428. doi: 10.1016/j.yexcr.2010.03.014. [DOI] [PubMed] [Google Scholar]
- Janich P., Pascual G., Merlos-Suárez A., Batlle E., Ripperger J., Albrecht U., Cheng H.-Y.M., Obrietan K., Di Croce L., Benitah S.A. The circadian molecular clock creates epidermal stem cell heterogeneity. Nature. 2011;480:209–214. doi: 10.1038/nature10649. [DOI] [PubMed] [Google Scholar]
- Jensen K.B., Watt F.M. Single-cell expression profiling of human epidermal stem and transit-amplifying cells: Lrig1 is a regulator of stem cell quiescence. Proc. Natl. Acad. Sci. USA. 2006;103:11958–11963. doi: 10.1073/pnas.0601886103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K.B., Collins C.A., Nascimento E., Tan D.W., Frye M., Itami S., Watt F.M. Lrig1 expression defines a distinct multipotent stem cell population in mammalian epidermis. Cell Stem Cell. 2009;4:427–439. doi: 10.1016/j.stem.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper M., Jaks V., Are A., Bergström Å., Schwäger A., Svärd J., Teglund S., Barker N., Toftgård R. Wounding enhances epidermal tumorigenesis by recruiting hair follicle keratinocytes. Proc. Natl. Acad. Sci. USA. 2011;108:4099–4104. doi: 10.1073/pnas.1014489108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman C.K., Zhou P., Pasolli H.A., Rendl M., Bolotin D., Lim K.-C., Dai X., Alegre M.-L., Fuchs E. GATA-3: An unexpected regulator of cell lineage determination in skin. Genes Dev. 2003;17:2108–2122. doi: 10.1101/gad.1115203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar K., Watt F.M. Markers of epidermal stem cell subpopulations in adult mammalian skin. Cold Spring Harb. Perspect. Med. 2014;4:a013631. doi: 10.1101/cshperspect.a013631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar K., Cottle D.L., Donati G., Chiang M.-F., Quist S.R., Gollnick H.P., Natsuga K., Lin K.-I., Watt F.M. BLIMP1 is required for postnatal epidermal homeostasis but does not define a sebaceous gland progenitor under steady-state conditions. Stem Cell Reports. 2014;3:620–633. doi: 10.1016/j.stemcr.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy V., Lindon C., Harfe B.D., Morgan B.A. Distinct stem cell populations regenerate the follicle and interfollicular epidermis. Dev. Cell. 2005;9:855–861. doi: 10.1016/j.devcel.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magwene P.M., Lizardi P., Kim J. Reconstructing the temporal ordering of biological samples using microarray data. Bioinformatics. 2003;19:842–850. doi: 10.1093/bioinformatics/btg081. [DOI] [PubMed] [Google Scholar]
- Mascré G., Dekoninck S., Drogat B., Youssef K.K., Broheé S., Sotiropoulou P.A., Simons B.D., Blanpain C. Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature. 2012;489:257–262. doi: 10.1038/nature11393. [DOI] [PubMed] [Google Scholar]
- Matsumura H., Mohri Y., Binh N.T., Morinaga H., Fukuda M., Ito M., Kurata S., Hoeijmakers J., Nishimura E.K. Hair follicle aging is driven by transepidermal elimination of stem cells via COL17A1 proteolysis. Science. 2016;351 doi: 10.1126/science.aad4395. aad4395–aad4395. [DOI] [PubMed] [Google Scholar]
- Mesa K.R., Rompolas P., Zito G., Myung P., Sun T.Y., Brown S., Gonzalez D.G., Blagoev K.B., Haberman A.M., Greco V. Niche-induced cell death and epithelial phagocytosis regulate hair follicle stem cell pool. Nature. 2015;522:94–97. doi: 10.1038/nature14306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlacki M., Darido C., Jane S.M., Wilanowski T. Loss of Grainy head-like 1 is associated with disruption of the epidermal barrier and squamous cell carcinoma of the skin. PLoS ONE. 2014;9:e89247. doi: 10.1371/journal.pone.0089247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R.J., Liu Y., Marles L., Yang Z., Trempus C., Li S., Lin J.S., Sawicki J.A., Cotsarelis G. Capturing and profiling adult hair follicle stem cells. Nat. Biotechnol. 2004;22:411–417. doi: 10.1038/nbt950. [DOI] [PubMed] [Google Scholar]
- Müller-Röver S., Handjiski B., van der Veen C., Eichmüller S., Foitzik K., McKay I.A., Stenn K.S., Paus R. A comprehensive guide for the accurate classification of murine hair follicles in distinct hair cycle stages. J. Invest. Dermatol. 2001;117:3–15. doi: 10.1046/j.0022-202x.2001.01377.x. [DOI] [PubMed] [Google Scholar]
- Niemann C., Horsley V. Development and homeostasis of the sebaceous gland. Semin. Cell Dev. Biol. 2012;23:928–936. doi: 10.1016/j.semcdb.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann C., Watt F.M. Designer skin: Lineage commitment in postnatal epidermis. Trends Cell Biol. 2002;12:185–192. doi: 10.1016/s0962-8924(02)02263-8. [DOI] [PubMed] [Google Scholar]
- Nijhof J.G.W., Braun K.M., Giangreco A., van Pelt C., Kawamoto H., Boyd R.L., Willemze R., Mullenders L.H., Watt F.M., de Gruijl F.R., van Ewijk W. The cell-surface marker MTS24 identifies a novel population of follicular keratinocytes with characteristics of progenitor cells. Development. 2006;133:3027–3037. doi: 10.1242/dev.02443. [DOI] [PubMed] [Google Scholar]
- Page M.E., Lombard P., Ng F., Göttgens B., Jensen K.B. The epidermis comprises autonomous compartments maintained by distinct stem cell populations. Cell Stem Cell. 2013;13:471–482. doi: 10.1016/j.stem.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011;12:2825–2830. [Google Scholar]
- Petersson M., Niemann C. Stem cell dynamics and heterogeneity: Implications for epidermal regeneration and skin cancer. Curr. Med. Chem. 2012;19:5984–5992. [PubMed] [Google Scholar]
- Picelli S., Björklund Å.K., Faridani O.R., Sagasser S., Winberg G., Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods. 2013;10:1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- Rompolas P., Greco V. Stem cell dynamics in the hair follicle niche. Semin. Cell Dev. Biol. 2014;25-26:34–42. doi: 10.1016/j.semcdb.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rompolas P., Mesa K.R., Greco V. Spatial organization within a niche as a determinant of stem-cell fate. Nature. 2013;502:513–518. doi: 10.1038/nature12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R. Entering the era of single-cell transcriptomics in biology and medicine. Nat. Methods. 2014;11:22–24. doi: 10.1038/nmeth.2764. [DOI] [PubMed] [Google Scholar]
- Sato F., Kawamoto T., Fujimoto K., Noshiro M., Honda K.K., Honma S., Honma K., Kato Y. Functional analysis of the basic helix-loop-helix transcription factor DEC1 in circadian regulation. Interaction with BMAL1. Eur. J. Biochem. 2004;271:4409–4419. doi: 10.1111/j.1432-1033.2004.04379.x. [DOI] [PubMed] [Google Scholar]
- Schepeler T., Page M.E., Jensen K.B. Heterogeneity and plasticity of epidermal stem cells. Development. 2014;141:2559–2567. doi: 10.1242/dev.104588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schult, D.A., and Swart, P.J. (2008). Exploring network structure, dynamics, and function using NetworkX. Proceedings of the 7th Python in Science Conference (SciPy2008).
- Snippert H.J., Haegebarth A., Kasper M., Jaks V., van Es J.H., Barker N., van de Wetering M., van den Born M., Begthel H., Vries R.G. Lgr6 marks stem cells in the hair follicle that generate all cell lineages of the skin. Science. 2010;327:1385–1389. doi: 10.1126/science.1184733. [DOI] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeo M., Lee W., Ito M. Wound healing and skin regeneration. Cold Spring Harb. Perspect. Med. 2015;5:a023267. doi: 10.1101/cshperspect.a023267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan D.W.M., Jensen K.B., Trotter M.W.B., Connelly J.T., Broad S., Watt F.M. Single-cell gene expression profiling reveals functional heterogeneity of undifferentiated human epidermal cells. Development. 2013;140:1433–1444. doi: 10.1242/dev.087551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toufighi K., Yang J.-S., Luis N.M., Aznar Benitah S., Lehner B., Serrano L., Kiel C. Dissecting the calcium-induced differentiation of human primary keratinocytes stem cells by integrative and structural network analyses. PLoS Comput. Biol. 2015;11:e1004256. doi: 10.1371/journal.pcbi.1004256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbar T., Guasch G., Greco V., Blanpain C., Lowry W.E., Rendl M., Fuchs E. Defining the epithelial stem cell niche in skin. Science. 2004;303:359–363. doi: 10.1126/science.1092436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Maaten L., Hinton G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008;9:2579–2605. [Google Scholar]
- Veniaminova N.A., Vagnozzi A.N., Kopinke D., Do T.T., Murtaugh L.C., Maillard I., Dlugosz A.A., Reiter J.F., Wong S.Y. Keratin 79 identifies a novel population of migratory epithelial cells that initiates hair canal morphogenesis and regeneration. Development. 2013;140:4870–4880. doi: 10.1242/dev.101725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Pasolli H.A., Williams T., Fuchs E. AP-2 factors act in concert with Notch to orchestrate terminal differentiation in skin epidermis. J. Cell Biol. 2008;183:37–48. doi: 10.1083/jcb.200804030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee T.W. The VGAM package for categorical data analysis. J. Stat. Softw. 2010;32:1–34. [Google Scholar]
- Zeeuwen P.L.J.M., van Vlijmen-Willems I.M.J.J., Hendriks W., Merkx G.F.M., Schalkwijk J. A null mutation in the cystatin M/E gene of ichq mice causes juvenile lethality and defects in epidermal cornification. Hum. Mol. Genet. 2002;11:2867–2875. doi: 10.1093/hmg/11.23.2867. [DOI] [PubMed] [Google Scholar]
- Zeisel A., Muñoz-Manchado A.B., Codeluppi S., Lönnerberg P., La Manno G., Juréus A., Marques S., Munguba H., He L., Betsholtz C. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347:1138–1142. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.