

Abstract
Chromatin immunoprecipitation, followed by quantification of immunoprecipitated DNA, can be used to measure RNA polymerase binding to any DNA segment in Escherichia coli. By calibrating measurements against the signal from a single RNA polymerase bound at a single promoter, we can calculate both promoter occupancy levels and the flux of transcribing RNA polymerase through transcription units. Here, we have applied the methodology to the E. coli lactose operon promoter. We confirm that promoter occupancy is limited by recruitment and that the supply of RNA polymerase to the lactose operon promoter depends on its location in the E. coli chromosome. Measurements of RNA polymerase binding to DNA segments within the lactose operon show that flux of RNA polymerase through the operon is low, with, on average, over 18 s elapsing between the passage of transcribing polymerases. Similar low levels of flux were found when semi-synthetic promoters were used to drive transcript initiation, even when the promoter elements were changed to ensure full occupancy of the promoter by RNA polymerase.
This article is part of the themed issue ‘The new bacteriology’.
Keywords: RNA polymerase, chromatin immunoprecipitation, promoter occupation, rifampicin, polymerase per second, polymerase density
1. Introduction
Many bacteria rely on transcription regulation in order to adapt to fluctuating environments. This often involves the interaction of regulatory activator proteins at or near promoters, which results in recruitment of the DNA-dependent RNA polymerase (RNAP) and subsequent transcript initiation and gene expression. In contrast, when the regulatory proteins are repressors, access to the promoter is blocked and, hence, expression of the corresponding transcription unit is silenced [1–3]. Most experimental studies of bacterial gene regulation have relied on measurements of fold-induction or fold-repression of measured levels of transcripts or gene products. However, few studies have addressed directly the issue of the number of RNAP molecules that engage with individual transcription units, and, to date, most calculations of RNAP flux through genes are based on estimates that work backwards from measured levels of RNA synthesis [4–6]. Here, we describe a new approach to direct quantification of RNAP bound to the Escherichia coli lac operon and its promoter, exploiting chromatin immunoprecipitation (ChIP). Recall that ChIP, in combination with analysis of immunoprecipitated DNA, permits us to detect protein binding at any chromosomal locus [7], independent of function, and many investigators have used it to measure the distribution of RNAP across bacterial chromosomes [8–11]. Here, we exploit the properties of the drug rifampicin, which blocks RNAP bound at promoters [9,12–14], to calibrate our ChIP measurements. This allows an absolute measure of promoter occupancy and RNAP flux through downstream genes.
2. Results
(a). Measurement of RNA polymerase flux though the lac operon
Formaldehyde treatment of cultures of E. coli efficiently cross-links RNAP to bound DNA targets [8,9]. Commercially available monoclonal mouse antibodies directed against the RNAP β subunit can then be used to immunoprecipitate RNAP from sonicated extracts of the cross-linked cells, and specific DNA targets can be quantified by PCR. We chose the well-characterized E. coli K-12 lactose (lac) operon to study RNA polymerase flux. Recall that the lac operon is expressed in a transcription unit from a promoter whose activity is repressed by the Lac repressor protein, and that induction requires a chemical inducer such as isopropyl β-d-1-thiogalactopyranoside (IPTG) [15,16]. To analyse RNAP flux through the lac operon, we used the lac0 pair of probes that samples the lac promoter, and the lac1–5 pairs of probes that sample approximately 300 base pair DNA sequences that are 518, 1421, 2308, 3691 and 4654 base pairs, respectively, downstream from the transcript start. Because each probe pair creates an amplicon that is a similar size, we can directly compare signal intensity between the different probes.
Escherichia coli K-12 strain MG1655, growing exponentially in medium, either with or without IPTG, was subject to our ChIP protocol (see Material and methods section for details, and electronic supplementary material, table S1 for probe sequences), and figure 1 shows quantification of the immunoprecipitated DNA at different loci in the lac operon (probed with the lac0–5 probes). The data show that the inclusion of IPTG in the bacterial growth media triggers a more than a 100-fold increase in levels of imunoprecipitated DNA, confirming that RNAP association with the lac operon is regulated by the Lac repressor. Accepting that the level of immunoprecipitated DNA corresponding to each probe reflects the amount of transcribing RNAP associated with the chromosomal DNA corresponding to each probe, the data argue that, at least for the first 2000 base pairs of the operon, RNAP levels remain constant, while they decline towards the end of the operon, presumably contributing to polarity effects [17]. Some quantitative differences seen with certain fragments, for example, with the lac3 probes, are likely owing to pause sequences [18,19].
Figure 1.
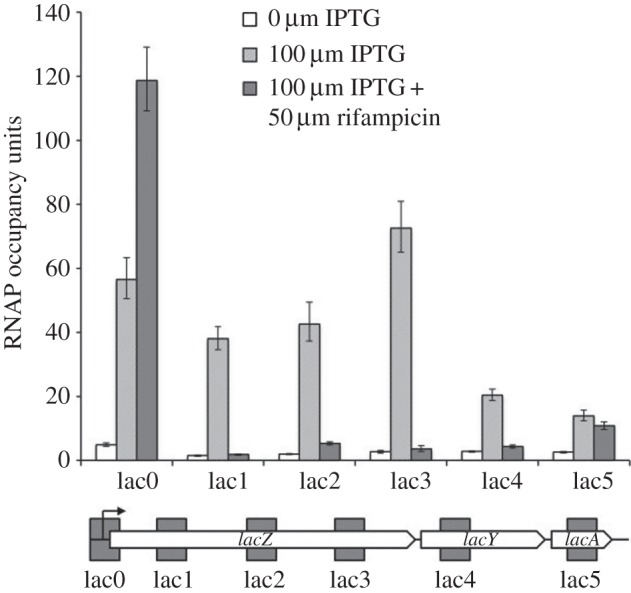
RNAP flux through the lac operon on the E. coli MG1655 chromosome. The figure shows experimentally measured RNAP occupancies at the lac promoter region (denoted lac0), or downstream regions (denoted lac1–5), illustrated in the sketch of the operon (approximately to scale). The probes are located from position −147 to +123 (lac0), position +518 to +781 (lac1), position +1421 to +1686 (lac2), position +2308 to +2575 (lac3), position +3691 to 3949 (lac4) and position +4654 to 4916 (lac5), all positions being with respect to the lac operon transcript start site. Cell cultures were grown and treated with formaldehyde, as described in the Material and methods section. Total DNA with cross-linked proteins was extracted and sonicated, and fragments cross-linked to RNAP were purified by immunoprecipitation. RNAP occupancy was measured by a ChIP–qPCR protocol. The figure illustrates measurements from cells grown with or without the inducer IPTG and with or without rifampicin, as indicated by the different shadings.
In order to calculate the absolute numbers of RNA polymerase molecules associated with the lac operon from the data in figure 1, we needed to measure the quantity of immunoprecipitated DNA that results from the binding of a single RNAP molecule. To do this, we exploited the property of rifampicin to block initiating RNAP at promoters and to inhibit transcript elongation [9]. Hence, rifampicin was added to MG1655 cells growing in the presence of IPTG, and figure 1 shows quantification of immunoprecipitated DNA at the different lac operon loci, probed with the lac0–5 probes. As expected, rifampicin causes a sharp decrease in the levels of immunoprecipitated DNA corresponding to the lac1–5 probes, but an increase with the lac0 probes. If we take the measured signal with the lac0 probe as indicative of a single promoter-bound RNAP, then we can deduce that, during induction in our growth conditions, the lac promoter is approximately 50% occupied, which is consistent with experimental data showing that the activity of the E. coli lac promoter is limited by the recruitment of RNAP [20,21]. Furthermore, the data permit an estimate of the flux of RNAP through the lac operon. Messenger elongation by RNAP in bacteria is known to proceed, on average, at 20–50 bases per second [22–24]. Because the DNA segment corresponding to each of the lac1–5 probes consists of approximately 300 base pairs, which would take at least 6 s to transcribe, approximately 33% observed occupancy by RNAP implies that a transcribing RNAP must arrive on average no more frequently than once every 18 s (3 × 6). This unexpected low level is likely owing to the time that individual RNAP molecules can take to escape from the promoter [25–27].
(b). RNA polymerase supply at the lac promoter is location-dependent
In a previous study, we found that the measured activity of the lac promoter in E. coli strain MG1655 was dependent on its chromosomal location [28]. To show this, we constructed a portable lac promoter::green fluorescent protein (gfp) cassette that we inserted at different chromosomal locations. We found that expression varied by up to 200-fold according to location, and, using ChIP, we showed that the measured differences were due to different levels of RNAP associated with the gfp gene. Because lac promoter activity is limited by RNAP recruitment [20], we reasoned that the differences could be caused by the concentration of available RNAP differing from one chromosomal location to another. Hence, to test this, we replaced the lac promoter with the bacteriophage λ major leftward promoter (λPL), whose activity is known to be limited by RNAP escape rather than recruitment [29]. Figure 2 shows the results of an experiment where we compared gfp expression from either our lac promoter::gfp or λPL::gfp fusions inserted into the MG1655 chromosome at different specific locations. The data show that observed differences in expression are much smaller with the λPL::gfp fusions than with the lac promoter::gfp fusions, and this is consistent with the suggestion that the effective concentration of RNAP differs according to location along the E. coli chromosome.
Figure 2.

Chromosome position effects on activities of the lac (a) and λpL (b) promoters. The figure shows experimentally determined measurements of the expression of lac and λpL promoter::gfp fusions, at different locations on the E. coli chromosome. Fluorescent output from the reporter cassette was measured during growth in the presence of 100 µM IPTG, and is represented on the y-axis. Chromosomal positions of the reporter cassette are represented on the x-axis and denoted in the figure by the name of a neighbouring gene. Below each chart is a linear schematic of the E. coli genome, with the origin of replication (OriC), terminus (dif), macrodomains and non-structured regions (NSR, right, Ter, left, NSL, ori) shown as previously reported [30].
(c). Promoter determinants alter RNA polymerase recruitment and RNA polymerase escape
Although the E. coli lac operon promoter is often adopted as a paradigm, we wanted to compare results with an unrelated promoter, and so we used a previously constructed set of promoter::lac fusions [31], where the promoters carry different combinations of −35, extended −10 and −10 elements upstream of the galP1 transcript start region. In an initial experiment, we selected the KAB-TTTG promoter that carries the −35 element TAGACA (consensus is TTGACA), an extended −10 element of TTTG (consensus is TGTG) and a −10 hexamer of TATGGT (consensus is TATAAT). Figure 3 illustrates ChIP data from an experiment run either with or without rifampicin, from which, as before, RNAP occupancy can be calculated. Surprisingly, the data reveal occupancy and flux levels that are similar to the induced lac promoter. Hence, promoter occupation by RNAP, as judged by the ratio of signal without rifampicin to with rifampicin, is approximately 40%, whereas occupancy of the downstream DNA segments corresponding to the Lac1357 and Lac2720 probes, ranges from 10% to 20%, which would correspond to an RNAP flux of one every 30 s.
Figure 3.

RNAP flux through the lac operon controlled by synthetic promoters. The figure shows experimentally determined levels of RNAP occupancy at the promoter and downstream regions Lac1357 and Lac2720, measured by ChIP–qPCR, during exponential growth in the presence of absence of rifampicin, as indicated by the different shading. The positions of probe regions are shown on the schematic diagrams (approximately to scale). RNAP occupancy of each promoter in the presence of rifampicin is taken as 100% occupied and other figures normalized accordingly. Data are shown for the KAB-TTTG promoter and three mutant derivatives: p16G (an extended −10 element ‘up’ mutant), p34T (a −35 element ‘up’ mutant) and p12C (a −10 element ‘down’ mutant).
Because it is well established that promoter −35 and extended −10 elements contribute to the recruitment of RNAP at bacterial promoters [32,33], we repeated the experiment with derivatives of the KAB-TTTG promoter carrying the p34T point mutation that creates a consensus −35 element (TTGACA), or the p16G mutation that creates a consensus extended −10 element (TGTG). As a control, we also used a derivative of the KAB-TTTG promoter with the p12C mutation that creates a corrupted −10 element (CATGGT). The results, illustrated in figure 3, show that recruitment of RNAP is reduced by the −10 element mutation. In contrast, recruitment of RNAP to the promoter is increased to approximately 50% by the consensus −35 element, and to nearly 100% by the consensus extended −10 element. However, for both promoters, the increase in occupancy leads to only modest increases in RNAP flux through the downstream-transcribed DNA.
3. Discussion
We have developed a simple method, based on ChIP with E. coli, for quantifying the binding of RNAP in vivo to any specific segment of DNA. For promoter regions, we can directly measure occupancy by RNAP, whereas for regions within transcription units, we can deduce the flux of RNAP. The method exploits rifampicin that specifically targets RNAP and blocks it in open complexes at promoters. Here, we make the assumption that formaldehyde equally efficiently cross-links rifampicin-blocked RNAP, RNAP that is bound and paused at promoters, and elongating RNAP, to cognate DNA targets. This appears reasonable as RNAP has a large molecular mass, and makes intimate contacts with the DNA template in all three situations.
Our results are consistent with previous observations that transcript initiation at the lac operon promoter is limited by RNAP recruitment [20,21]. We believe that this explains, at least in part, our previous observation that the expression of a lac promoter::gfp fusion differs according to its location on the E. coli chromosome [28], because the activity of a promoter that is limited by RNAP recruitment will depend on the local concentration of available RNAP. Hence, we suggest that, according to its position on the E. coli chromosome, the promoter will sample different locations, including locations where the effective concentration of RNAP is higher or lower. Consistent with this, a recent live-cell super-resolution microscopy study of RNAP in E. coli showed that the vast majority of non-transcribing RNAP molecules that were ‘searching’ for promoters were DNA-bound [34]. Interestingly, we found that Lac repressor-mediated repression of the lac operon promoter is not dependent on location [28]. From this, we deduce that diffusion of the Lac repressor ensures that its effective concentration is the same at all locations within the E. coli cell, whereas diffusion of larger RNAP molecules is constrained, and this is consistent with calculations of macromolecular mobility in bacteria [35].
The low measured flux of RNAP through the lac operon is consistent with previous observations, from both in vitro [26,27] and in vivo [36] studies, that the transition of RNAP from the transcriptionally competent open complexes to the elongating complex, via the promoter escape phase, is not simple and can be slow and rate-limiting. We observe low flux of one RNAP every 18–50 s, irrespective of whether transcription was being driven by the lac promoter or by genetically engineered promoters, even when the promoter is fully occupied. This underscores that promoters, as well as being drivers of transcription, are also bottlenecks, and delays to RNAP result in reduced flux through downstream-transcribed sequences [36,37]. Contributing reasons for delays include pauses owing to scrunching [38–40], pauses owing to disengagement of various RNAP determinants with promoter elements [32,33,41] and sigma factor-mediated pauses early in the elongation phase [42,43]. Additionally, it may well be that there are topological and mechanical reasons why transcribing RNAP molecules must be well separated, but, to date, these are speculative and poorly understood.
We are aware that the measured rates of RNAP flow that we report here are surprisingly low, and depend critically on estimates of RNA chain growth rates. Hence, it is worth underscoring that measured in vivo rates of RNA chain elongation [22–24] are corroborated by single molecule studies of RNA chain growth in vitro [44], and that previous estimates of RNAP flux, defined by synthetic biologists in terms of polymerase per second (PoPS) units, are consistent with our findings [5,45]. The prime motive for initiating this project was the need, perceived by the synthetic biologists, to provide robust characterization for ‘parts’ that could be used in novel circuits. Taken together, our results argue that full and robust characterization might not be possible, and, for many promoters, their ‘performance’ is context-dependent. Recent insights into transcript initiation and elongation, confirmed here by the low measured levels of RNAP flux through the lac operon, contradict the simple view that promoters are simply devices that ‘feed’ RNAP into transcription units [37]. Hence, while ‘parts’ such as the lac promoter have many uses in synthetic biology, their full exploitation will require considerable extension of our current knowledge base.
4. Material and methods
(a). Bacterial strains, plasmids and growth conditions
The experiments analysing the flux of RNAP through the chromosomal lac operon were completed using E. coli K-12 strain MG1655 [46], whereas the synthetic promoter experiments were conducted using a Δcrp derivative of strain M182 [47], with the promoter::lac fusion carried on the low copy number broad host range lac expression vector, pRW50 [31]. Fragments carrying the different KAB promoter derivatives were previously described [31,48]. The promoter derivatives are denoted pNX, where N is the position of the substitution upstream from the transcript start, and X is the substituted base on the non-template strand.
For the ChIP assays, triplicate single colonies were used to inoculate Luria Bertani (LB) media, supplemented with 35 µg ml−1 tetracycline, where appropriate, and incubated for 16 h at 37°C with aeration. Cells were then subcultured into fresh media to a final OD650 of 0.03, then incubated at 37°C with aeration until an OD650 of 0.4 was reached. The growth conditions used for fluorescence assays were the same, except M9 minimal media supplemented with 2 mM MgSO4, 0.1 mM CaCl2, 0.1% casamino acids, 0.3% fructose and 100 µM IPTG was used [28].
(b). Construction of plasmids, chromosomal recombination and green fluorescent protein measurements
To construct the gene doctoring donor plasmids required to insert the λPL::gfp fusion into the genome of E. coli MG1655, an oligodeoxynucleotide was synthesized to encode an EcoRI restriction target site and the λPL G-12 T up-mutant λ leftward promoter fused to the ribosome binding site of the lacZ gene. This ribosome binding site was used in order to make the fusion comparable to previously used lac promoter::gfp fusion [28]. This oligonucleotide primer was then used with a primer downstream of the HindIII site in the pJB plasmids to create an EcoRI–HindIII λPL promoter fragment, which was subsequently cloned into EcoRI–HindIII digested pJB plasmids containing the appropriate homology regions for insertion into the chromosomal targets used previously [28]. Insertion of the λPL promoter::gfp fusion into the target chromosomal loci was achieved exactly as described previously by the gene doctoring chromosome recombineering method [28,49], and fluorescence assays were run using the growth conditions described above.
(c). Chromatin immunoprecipitation and quantitative real-time PCR analysis
Chromatin immunoprecipitation (ChIP) and quantitative real-time PCR (qPCR) were performed as before, using commercial mouse monoclonal antibody for the RNAP β subunit (Neoclone no. W0002) to immunoprecipitate DNA cross-linked to RNAP [8,28,50]. Overnight cultures were used to subculture into fresh LB to a final OD650 of 0.025, and incubated at 37°C, with aeration, until an OD650 of 0.4 was reached. When appropriate, rifampicin was added to the culture to a final concentration of 50 µM, and incubated for 15 min, which is sufficient to trap RNAP molecules at promoters [8,9]. Cells were cross-linked by addition of formaldehyde to a final concentration of 1%, and incubated for a further 20 min at 37°C. After the ChIP protocol, immunoprecipitated DNA was quantified by qPCR, using the Agilent Technologies Stratagene Mx3005P machine and the Agilent Brilliant III Ultra-fast SYBR Green qPCR master mix, and this permitted calculation of RNAP occupancy units for each amplicon. Oligonucleotide primers were designed to amplify approximately 300 bp regions with the same PCR efficiency, either at promoter regions or within the lac operon (primer sequences are listed in electronic supplementary material, table S1). Control primers were used to amplify transcriptionally silent control regions. Enrichment of ChIP samples for RNAP binding was calculated relative to the transcriptionally silent control regions, as previously described [50], with the samples from rifampicin-treated cells used to define 100% occupation.
Supplementary Material
Acknowledgements
We are grateful to the EU Framework Programme 7 ST-FLOW Project for supporting this work.
Authors' contributions
The work was done by B.S. and J.B., who designed the experiments, together with D.L. and S.B. The manuscript was written by S.B. and J.B. We are grateful to Rita Godfrey for technical support, and to Victor de Lorenzo for stimulating discussions and advice throughout the work.
Competing interests
We have no competing interests.
Funding
This project was funded in part by the UK BBSRC and the Wellcome Trust.
References
- 1.Ishihama A. 2000. Functional modulation of Escherichia coli RNA polymerase. Annu. Rev. Microbiol. 54, 499–518. ( 10.1146/annurev.micro.54.1.499) [DOI] [PubMed] [Google Scholar]
- 2.Browning DF, Busby SJ. 2004. The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2, 57–65. ( 10.1038/nrmicro787) [DOI] [PubMed] [Google Scholar]
- 3.Lee DJ, Minchin SD, Busby SJ. 2012. Activating transcription in bacteria. Annu. Rev. Microbiol. 66, 125–152. ( 10.1146/annurev-micro-092611-150012) [DOI] [PubMed] [Google Scholar]
- 4.Bon M, McGowan SJ, Cook PR. 2006. Many transcribed genes in bacteria and yeast are transcribed only once per cell cycle. FASEB J. 20, 1071–1074. ( 10.1096/fj.06-6087fje) [DOI] [PubMed] [Google Scholar]
- 5.Kelly JR et al. . 2009. Measuring the activity of BioBrick promoters using an in vivo reference standard. J. Biol. Eng. 3, 4 ( 10.1186/1754-1611-3-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu H, Sepulveda LA, Figard L, Sokac AM, Golding I. 2015. Combining protein and mRNA quantification to decipher transcriptional regulation. Nat. Methods 12, 739–742. ( 10.1038/nmeth.3446) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wade JT, Struhl K, Busby SJ, Grainger DC. 2007. Genomic analysis of protein-DNA interactions in bacteria: insights into transcription and chromosome organisation. Mol. Microbiol. 65, 21–26. ( 10.1111/j.1365-2958.2007.05781.x) [DOI] [PubMed] [Google Scholar]
- 8.Grainger DC, Hurd D, Harrison M, Holdstock J, Busby SJ. 2005. Studies of the distribution of Escherichia coli cAMP-receptor protein and RNA polymerase along the E. coli chromosome. Proc. Natl Acad. Sci. USA 102, 17 693–17 698. ( 10.1073/pnas.0506687102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herring CD, Raffaelle M, Allen TE, Kanin EI, Landick R, Ansari AZ, Palsson BO. 2005. Immobilisation of Escherichia coli RNA polymerase and location of binding sites by use of chromatin immunoprecipitation and microarrays. J. Bacteriol. 187, 6166–6174. ( 10.1128/JB.187.17.6166-6174.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho BK, Zengler K, Qiu Y, Park YS, Knight EM, Barrett CL, Gao Y, Palsson BO. 2009. The transcription unit architecture of the Escherichia coli genome. Nat. Biotechnol. 27, 1043–1049. ( 10.1038/nbt.1582) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis SE, Mooney RA, Kanin EI, Grass J, Landick R, Ansari AZ. 2011. Mapping E. coli RNA polymerase and associated transcription factors and identifying promoters genome-wide. Methods Enzymol. 498, 449–471. ( 10.1016/B978-0-12-385120-8.00020-6) [DOI] [PubMed] [Google Scholar]
- 12.Sippel A, Hartmann G. 1968. Mode of action of rafamycin on the RNA polymerase reaction. Biochem. Biophys. Acta 157, 218–219. ( 10.1016/0005-2787(68)90286-4) [DOI] [PubMed] [Google Scholar]
- 13.McClure WR, Cech CL. 1978. On the mechanism of rifampicin inhibition of RNA synthesis. J. Biol. Chem. 253, 8949–8956. [PubMed] [Google Scholar]
- 14.Campbell EA, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, Darst SA. 2001. Structural mechanism for rifampicin inhibition of RNA polymerase. Cell 104, 901–912. ( 10.1016/S0092-8674(01)00286-0) [DOI] [PubMed] [Google Scholar]
- 15.Reznikoff WS. 1992. The lactose operon-controlling elements: a complex paradigm. Mol. Microbiol. 6, 2419–2422. ( 10.1111/j.1365-2958.1992.tb01416.x) [DOI] [PubMed] [Google Scholar]
- 16.Wilson CJ, Zhan H, Swint-Kruse L, Matthews KS. 2007. The lactose repressor system: paradigms for regulation, allosteric behaviour and protein folding. Cell Mol. Life Sci. 64, 3–16. ( 10.1007/s00018-006-6296-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ullmann A, Joseph E, Danchin A. 1979. Cyclic AMP as a modulator of polarity in polycistronic transcriptional units. Proc. Natl Acad. Sci. USA 76, 3194–3197. ( 10.1073/pnas.76.7.3194) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larson MH, et al. 2014. A pause sequence enriched at translation start sites drives transcription dynamics in vivo. Science 344, 1042–1047. ( 10.1126/science.1251871) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vvedenskaya IO, Vahedian-Movahed H, Bird JG, Knoblauch JG, Goldman SR, Zhang Y, Ebright RH, Nickels BE. 2014. Interactions between RNA polymerase and the ‘core recognition element’ counteract pausing. Science 344, 1285–1289. ( 10.1126/science.1253458) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malan TP, Kolb A, Buc H, McClure WR. 1984. Mechanism of CRP-cAMP activation of lac operon transcription initiation. J. Mol. Biol. 180, 881–909. ( 10.1016/0022-2836(84)90262-6) [DOI] [PubMed] [Google Scholar]
- 21.Buckle M, Fritsch A, Roux P, Geiselmann J, Buc H. 1991. Kinetic studies on promoter-RNA polymerase complexes. Methods Enzymol. 208, 236–258. ( 10.1016/0076-6879(91)08016-B) [DOI] [PubMed] [Google Scholar]
- 22.Murakawa GJ, Kwan C, Yamashita J, Nierlich DP. 1991. Transcription and decay of the lac messenger: role of an intergenic terminator. J. Bacteriol. 173, 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vogel U, Jensen KF. 1994. The RNA chain elongation rate in Escherichia coli depends on the growth rate. J. Bacteriol. 176, 2807–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen H, Shiroguchi K, Ge H, Xie XS. 2015. Genome-wide study of mRNA degradation and transcript elongation in Escherichia coli. Mol. Syst. Biol. 11, 781–791. ( 10.15252/msb.20145794) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsu LM. 2008. Promoter escape by Escherichia coli RNA polymerase. EcoSal Plus 3 ( 10.1128/ecosalplus.4.5.2.2) [DOI] [PubMed] [Google Scholar]
- 26.Chander M, Lee A, Vallery TK, Thandar M, Jiang Y, Hsu LM. 2015. Mechanisms of very long abortive transcript release during promoter escape. Biochemistry 54, 7393–7408. ( 10.1021/acs.biochem.5b00712) [DOI] [PubMed] [Google Scholar]
- 27.Bauer DL, Duchi D, Kapanidis AN. 2016. E. coli RNA polymerase pauses during initial transcription. Biophys. J. 110, p21a. ( 10.1016/j.bpj.2015.11.170) [DOI] [Google Scholar]
- 28.Bryant JA, Sellars LE, Busby SJ, Lee DJ. 2014. Chromosome position effects on gene expression in Escherichia coli K-12. Nucleic Acids Res. 42, 11 383–11 392. ( 10.1093/nar/gku828) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kincade JM, deHaseth PL. 1991. Bacteriophage lambda promoters pL and pR: sequence determinants of in vivo activity and of sensitivity to the DNA gyrase inhibitor, coumermycin. Gene 97, 7–12. ( 10.1016/0378-1119(91)90003-T) [DOI] [PubMed] [Google Scholar]
- 30.Boccard F, Esnault E, Valens M. 2005. Spatial arrangement and macrodomain organization of bacterial chromosomes. Mol. Microbiol. 57, 9–16. ( 10.1111/j.1365-2958.2005.04651.x) [DOI] [PubMed] [Google Scholar]
- 31.Burr T, Mitchell J, Kolb A, Minchin S, Busby S. 2000. DNA sequence elements located immediately upstream of the −10 hexamer in Escherichia coli promoters: a systematic study. Nucleic Acids Res. 28, 1864–1870. ( 10.1093/nar/28.9.1864) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miroslavova NS, Busby SJ. 2006. Investigations of the modular structure of bacterial promoters. Biochem. Soc. Symp. 73, 1–10. ( 10.1042/bss0730001) [DOI] [PubMed] [Google Scholar]
- 33.Hook-Barnard IG, Hinton DM. 2007. Transcription initiation by mix and match elements: flexibility for polymerase binding to bacterial promoters. Gene Regul. Syst. Biol. 1, 275–293. [PMC free article] [PubMed] [Google Scholar]
- 34.Stracy M, Lesterlin C, Garza de Leon F, Uphoff S, Zawadzki P, Kapanidis AN. 2015. Live-cell superresolution microscopy reveals the organisation of RNA polymerase in the bacterial nucleoid. Proc. Natl Acad. Sci. USA 112, 4390–4399. ( 10.1073/pnas.1507592112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parry BR, Surovtsev IV, Cabeen MT, O'Hern CS, Dufresne ER, Jacobs-Wagner C. 2014. The bacterial cytoplasm has glass-like properties and is fluidised by metabolic activity. Cell 156, 183–194. ( 10.1016/j.cell.2013.11.028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reppas NB, Wade JT, Church GM, Struhl K. 2006. The transition between transcription initiation and elongation in E. coli is highly variable and often rate limiting. Mol. Cell 24, 747–757. ( 10.1016/j.molcel.2006.10.030) [DOI] [PubMed] [Google Scholar]
- 37.Browning DF, Busby SJ. 2016. Local and global regulation of transcription initiation in bacteria. Nat. Rev. Microbiol. ( 10.1038/nrmicro787) [DOI] [PubMed] [Google Scholar]
- 38.Revyakin A, Liu C, Ebright RH, Strick TR. 2006. Abortive initiation and productive initiation by RNA polymerase involve DNA scrunching. Science 314, 1139–1143. ( 10.1126/science.1131398) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kapanidis AN, Margeat E, Ho SO, Kortkhonjia E, Weiss S, Ebright RH. 2006. Initial transcription by RNA polymerase proceeds through a DNA scrunching mechanism. Science 314, 1144–1147. ( 10.1126/science.1131399) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Winkelman JT, Vvedenskaya IO, Zhang Y, Zhang Y, Bird JG, Taylor DM, Gourse RL, Ebright RH, Nickels BE. 2016. Multiplexed protein-DNA cross-linking: scrunching in transcription start site selection. Science 351, 1090–1093. ( 10.1126/science.aad6881) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsu LM. 2002. Promoter clearance and escape in prokaryotes. Biochim. Biophys. Acta 1577, 191–207. ( 10.1016/S0167-4781(02)00452-9) [DOI] [PubMed] [Google Scholar]
- 42.Perdue SA, Roberts JW. 2011. Sigma 70-dependent transcription pausing in Escherichia coli. J. Mol. Biol. 412, 782–792. ( 10.1016/j.jmb.2011.02.011) [DOI] [PubMed] [Google Scholar]
- 43.Strobel EJ, Roberts JW. 2015. Two transcription pause elements underlie a sigma 70-dependent pause cycle. Proc. Natl Acad. Sci. USA 112, 4374–4380. ( 10.1073/pnas.1512986112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Larson MH, Landick R, Block SM. 2011. Single-molecule studies of RNA polymerase: one singular sensation, every little step it takes. Mol. Cell 41, 249–262. ( 10.1016/j.molcel.2011.01.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nielsen AAK, Der BS, Shin J, Vaidyanathan P, Paralanov V, Strychalski E, Ross D, Densmore D, Voight CA. 2016. Genetic circuit design automation. Science 352, 6281 ( 10.1126/science.aac7341) [DOI] [PubMed] [Google Scholar]
- 46.Blattner FR, et al. 1997. The complete genome sequence of Escherichia coli K-12. Science 277, 1453–1462. ( 10.1126/science.277.5331.1453) [DOI] [PubMed] [Google Scholar]
- 47.Busby SJ, Kotlarz D, Buc H. 1983. Deletion mutagenesis of the Escherichia coli galactose operon promoter region. J. Mol. Biol. 167, 259–274. ( 10.1016/S0022-2836(83)80335-0) [DOI] [PubMed] [Google Scholar]
- 48.Miroslavova NS. 2005. Studies on the recognition of Escherichia coli promoters and their elements. PhD Thesis, University of Birmingham, UK.
- 49.Lee DJ, Bingle LE, Heurlier K, Pallen MJ, Penn CW, Busby SJ, Hobman JL. 2009. Gene doctoring: a method for recombineering in laboratory and pathogenic Escherichia coli strains. BMC Microbiol. 9, 252 ( 10.1186/1471-2180-9-252) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bonocora RP, Fitzgerald DM, Stringer AM, Wade JT. 2013. Non-canonical protein-DNA interactions identified by ChIP are not artefacts. BMC Genomics 14, 254 ( 10.1186/1471-2164-14-254) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.