

Abstract
Interim analysis of especially sizeable trials keeps the decision process free of conflict of interest while considering cost, resources, and meaningfulness of the project. Whenever necessary, such interim analysis can also call for potential termination or appropriate modification in sample size, study design, and even an early declaration of success. Given the extraordinary size and complexity today, this rational approach helps to analyze and predict the outcomes of a clinical trial that incorporate what is learned during the course of a study or a clinical development program. Such approach can also fill the gap by directing the resources toward relevant and optimized clinical trials between unmet medical needs and interventions being tested currently rather than fulfilling only business and profit goals.
Key words: Clinical trial operation method, decision making, interim analysis, rational approach
INTRODUCTION
Interim analysis is one of the reliable rational approaches to clinical trials that incorporate what is learned during the course of a clinical study and how it is completed, without compromising the validity or integrity. This method may encompass the potential changes in all program-related resources and activities, including changes in logistical, monitoring, and recruitment procedures.
On a realistic level, the study not only requires the ability to measure the outcomes of interest continuously but also to make data and summarized information about those measurements available in a timely manner to different audiences according to the study role. In a clinical context, this means not just continuously tracking trial data collected on case report forms but also generating performance metrics that enable refinements in operations. Interest in this approach has mounted as a result of the soaring cost of clinical research and numerous trial failures, including, particularly, costly and well-publicized failures of major late-stage trials.
The simplest result of such an interim analysis is early stopping for futility or continuation of the study. This rational approach also allows clinical researchers to employ the same basic management principles as typical modern businesses, using real-time data and analysis to inform decisions that continually optimize operations.
INTERIM ANALYSIS AND STOPPING RULE
There are a number of practical and theoretical justifications for the implementation of this approach in clinical trials via a variety of group sequential designs that allow a limited number of planned analyses while maintaining a prespecified overall type I error rate and the blind of the study. It is highly desirable that the conduct of the interim analyses be done by a body independent of the one charged with the day-to-day activities of the clinical trial.
There are a number of prospective statistical strategies for positive stopping of a clinical trial early.[1,2] Flexible strategy and other statistical procedures such as stochastic limitation or conditional power approaches consider negative stopping.[3,4] These include stochastic limitation or conditional power procedures which allow for the early termination of a clinical trial; if given the available trial information so far, the probability of reaching statistical significance in favor of the new treatment is small. There are also Bayesian or semi-Bayesian counterparts for each of these frequentist approaches.[5,6,7] Stopping rules for interim analyses based on limited data requires more stringent P values for stopping than later analyses, which can have stopping P values somewhat near to the nominal levels of significance. The use of these practices for which there are no documented statistical strategies creates serious problems during the review process.
The Guidance for Industry on Adaptive Design Clinical Trials for Drugs and Biologics was released in 2010 by the Food and Drug Administration (FDA). Where definition included for an adaptive design, which was similar to that of the Adaptive Design Scientific Working Group: A study that includes a prospectively planned opportunity for modification of one or more specified aspects of the study design and hypotheses based on the analysis of data (usually interim data) from participants in the study. Thus, both the Adaptive Design Scientific Working Group and FDA support the notion that changes are based on prespecified decision rules. However, FDA defines this more generally: “The term prospective here means that the adaptation was planned before data were examined in an unblinded manner by any personnel involved in planning the revision. This can include plans that are introduced or made final after the study has started if the blinded state of the personnel involved is unequivocally maintained when the modification plan is proposed.”[8]
PLANNED AND UNPLANNED INTERIM ANALYSES
There is a need to adjust the nominal P values after the conduct of such planned or unplanned interim analyses because it should not be mollified by the fact that such interim analyses were made on the basis of information external to the clinical trial operations. These are perhaps the most difficult to handle and yet, the most common interim analysis issues to which statistical reviewers are faced with during the review process.
The known works in this area are those of Geller, Pocock, Hughes, and Emerson. According to those, one approach is to assume that the accumulating data are continuously being looked at, but interim analyses are carried out only when they (the data) look interesting. This is equivalent to a continuous sequential design, and the repeated significance testing sequential designs of Armitage, McPherson, and Rowe may be appropriate. This ad hoc approach has been adopted by a number of statistical reviewers faced with problems of unplanned interim analyses during the review process of clinical trials. Besides this ad hoc approach, the more flexible alpha-spending function approach has also been suggested as a candidate for retrospective adjustment of P values due to unplanned interim analyses when the exact number of unplanned interim analyses actually carried out is known.
An example of multiple looks for a comparative trial in which two treatments are being compared for efficacy is as follows.
H0:p2 = p1
H1:p2 > p1
A standard design says that for 80% power with alpha of 0.05, we need about 100 patients per arm based on the assumption of p2 = 0.50 and p1 = 0.30 which results in 0.20 for the difference. Hence, what happens if we find P < 0.05 before all patients are enrolled? Why cannot we look at the data a few times in the middle of the trial and conclude that one treatment is better if we see P < 0.05?.
When we are looking to find a difference between 0.30 and 0.50, we would not expect to conclude that there is evidence for a difference. However, if we look after every four patients, we get the scenario where we would stop at 96 patients and conclude that there is a significant difference [Figures 1–3].[9]
Figure 1.
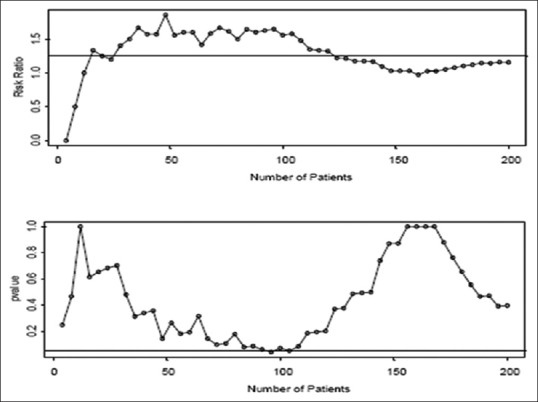
Plots above show simulated data where p1 = 0.40 and p2 = 0.50
Figure 3.
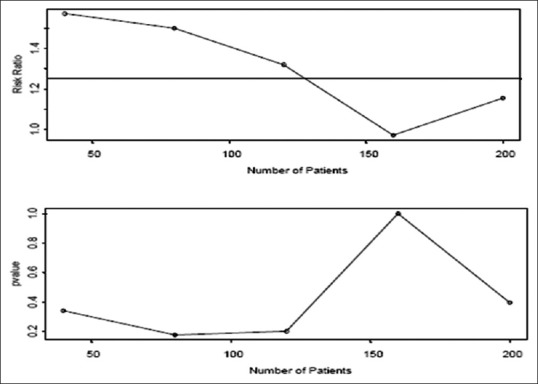
If we look after every forty patients, we get the scenario where we would not stop either. If we wait until the end of the trial (n = 200), we estimate p1 to be 0.45 and p2 to be 0.52. The P value used for testing shows a significant difference of 0.40
Figure 2.
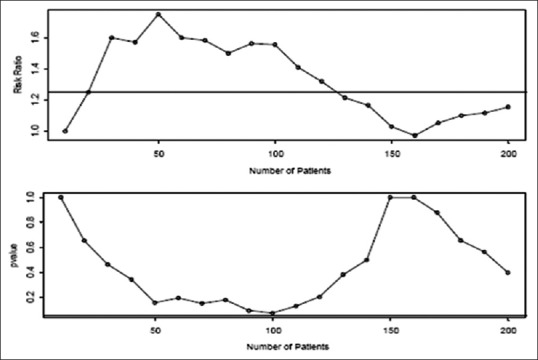
If we look after every ten patients, we get the scenario where we would not stop until all the 200 patients were observed and would conclude that there no significant difference (P = 0.40)
Would we have messed up if we looked early on?
Every time we look at the data and consider stopping, we introduce the chance of falsely rejecting the null hypothesis. In other words, every time we look at the data, we have the chance of a type 1 error. If we look at the data multiple times, and we use alpha of 0.05 as our criterion for significance, we have a 5% chance of stopping each time. Under the true null hypothesis and just 2 looks at the data, we “approximate” the error rates as: Probability stop at the first look: 0.05, probability stop at the second look: 0.95 × 0.05 = 0.0475, and total probability of stopping is 0.0975.
We can obtain P < 0.05, but not declare statistical significance at the final look. O′Brien-Fleming bounds use more conservative stopping boundaries at early stages. These bounds spend little alpha at the time of the interim looks and lead to boundary values at the final stage that are close to those from the fixed sample design, avoiding the problem with the Pocock bounds. The classical Pocock and O′Brien-Fleming boundaries require a prespecified number of equally spaced looks. However, a Data Safety Monitoring Board (DSMB) may require more flexibility. Alternatively, one could specify an alpha-spending function that determines the rate at which the overall type I error is to be spent during the trial. At each interim look, the type I error is partitioned according to this alpha-spending function to derive the corresponding boundary values. Because the number of looks neither has to be prespecified nor equally spaced, an O-Brien-Fleming type alpha-spending function has become the most common approach to monitoring efficacy in clinical trials. Some investigators have suggested that using “P” to denote “statistical significance” as a way to denote the detection of an “effect” is inappropriate, and offer other solutions such as provision of effect size estimates and their precision from confidence intervals.[10,11]
Given the lack of standard statistical methods for retrospective adjustment of P values due to unplanned interim analyses, unplanned interim analyses should be avoided as they can flaw the results of a well-planned clinical trial. The performance of a clinical trial is only justified if the clinical investigators in advance consider ethical aspects and if an external Ethical Committee has approved the conduct of the study according to a defined protocol. A great deal of recent discussion in the clinical trials literature has focused on response-adaptive randomization in two-arm trials; however, this represents a fairly specific and relatively infrequently used type of adaptive clinical trial (ACT).[12,13,14,15,16,17,18,19]
OPERATIONAL REQUIREMENT WHILE CONDUCTING THE TRIAL
Trials are need to be carefully monitored so that decisions to stop early, whether based on trial data or external evidence, can be properly made and documented. What in practical terms can be done? First, make a realistic assessment of possible scenarios, using general experience from clinical trials. Rigorous assessment of directly relevant trials should be carried out, using techniques such as meta-analysis. Subjective beliefs about the likely relative efficacy of the treatments and the clinical benefits that would be required before a new treatment would be used routinely can also be documented at this stage, although these can be surprisingly variable, as illustrated by some work on a trial of treatment.
During an ongoing trial, different individuals become unblinded to data at different time points, and the regulatory document will be left open with some gray areas that merit further discussion. For instance, investigators typically remain blinded until the end of the study, whereas DSMB members may be partially or fully unblinded at the time of the first interim analysis. Suppose an investigator proposes a design change after the time of the first interim analysis based on external factors, such as the release of results from a similar trial, one could argue that the impetus for the proposed adaptation was not based on the results of unblinded data, which would fit the FDA definition for a valid adaptive design.[8] However, if the proposed adaptation has to be reviewed and approved by the DSMB, the fact they have seen unblinded data would seem to imply that the definition may not be met. The role of a blinded versus unblinded statistician in the process may also be important in determining whether the definition has been met. Further clarification of these types of areas is needed in the future to ensure that researchers and regulatory authorities agree on what constitutes a valid adaptive design. The implementation of these methods required the development of a structure to support DSMBs, which are relatively standard for modern clinical trials. This also required substantial training of clinical trialists to ensure that they understand the intricacies of the methods, as well as the potential pitfalls associated with the use of the methods.
During the design of a clinical trial, several important design decisions must be made. Although study success depends on their accuracy, there may be limited information to guide the decisions. This approach addresses the uncertainty by allowing a review of accumulating data during an ongoing trial, and modifying trial characteristics accordingly if the interim information suggests that some of the original decisions may not be valid. However, it is well known that implementing many of the proposed approaches will require the clinical trials community to address several statistical, logistical, and operational hurdles.
Mechanisms for stopping the trial must be identified, and criteria for stopping a trial should be explicit. Mortality and excess toxicity are obvious end points to monitor, but more complex features such as quality of life are much more difficult to assess and analyze. A particular dilemma arises when considering which end points to monitor because only short-term results, such as tumour response, acute morbidity, and early deaths, are available quickly, whereas the real value of many trials is their potential to give information on long-term survival and late morbidity. By definition, decisions to stop have to be made primarily on the early information, and it is of importance to assess to what extent this can act as surrogate information for the long-term outcomes. Monitoring for toxicity is always worthwhile, but monitoring for efficacy is likely to be most beneficial when mature data are accruing fast relative to the entry of new patients.
If a trial does stop early, what are the priorities? The surviving trial patients should be informed of the position, which will be much easier if they gave genuine informed consent. The next priority should be the release of full results, quickly, via peer-reviewed journals, although this is difficult given the current constraints of most journals.
From the statistical viewpoint, monitoring methods can be classified according to whether the method is frequentist or Bayesian,[20] and comprehensive reviews of statistical aspects of monitoring can be found in studies by Whitehead,[21] Jennison and Turnbull,[22] and Piantadosi.[23] However, regardless of the specific method used, a key issue is that statistical rules are only a part of the question, as they tend to oversimplify the information relevant to the decision that must be taken. The decision to stop a trial before the prespecified final analysis should not only be guided by statistical considerations, but also by practical issues (toxicity, ease of administration, costs, etc.), as well as clinical considerations. For this reason, it is preferable to refer to statistical methods as guidelines, rather than rules.[24]
The recognized potential ethical benefits of ACTs include a higher probability of receiving an effective intervention for participants, optimizing resource utilization, and accelerating treatment discovery. Ethical challenges voiced include developing procedures, so trial participants can make informed decisions about taking part in ACTs and plausible, though unlikely risks of research personnel are altering the enrollment patterns.[25]
CONCLUSIONS
The decision to conduct an interim analysis should be based on sound scientific reasoning that is guided by clinical and statistical integrity, standard operating practices for interim analyses, and regulatory concerns. Such a decision must not and should not be based on natural tendencies toward operational or academic curiosity. Therefore, unplanned interim analyses should be avoided as they can flaw the results of a well-planned clinical trial. A good performance metrics enable greater understanding of the study progress, far tighter control, more effective allocation of resources such as monitoring time, faster enrollment, and in the larger scheme of things, shorter timelines and lower costs in operations, and decision-making process.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Montori VM, Devereaux PJ, Adhikari NK, Burns KE, Eggert CH, Briel M, et al. Randomized trials stopped early for benefit: A systematic review. JAMA. 2005;294:2203–9. doi: 10.1001/jama.294.17.2203. [DOI] [PubMed] [Google Scholar]
- 2.Ioannidis JP. Contradicted and initially stronger effects in highly cited clinical research. JAMA. 2005;294:218–28. doi: 10.1001/jama.294.2.218. [DOI] [PubMed] [Google Scholar]
- 3.Schulz KF, Grimes DA. Multiplicity in randomised trials II: Subgroup and interim analyses. Lancet. 2005;365:1657–61. doi: 10.1016/S0140-6736(05)66516-6. [DOI] [PubMed] [Google Scholar]
- 4.Pocock SJ. When (not) to stop a clinical trial for benefit. JAMA. 2005;294:2228–30. doi: 10.1001/jama.294.17.2228. [DOI] [PubMed] [Google Scholar]
- 5.Roger JL, Berry DA. Group sequential clinical trials: A classical evaluation of Bayesian decision-theoretic designs. J Am Stat Assoc. 1994;176:1528–34. [Google Scholar]
- 6.Emerson SS. Stopping a clinical trial very early based on unplanned interim analyses: A group sequential approach. Biometrics. 1995;51:1152–62. [PubMed] [Google Scholar]
- 7.Grossman J, Parmar MK, Spiegelhalter DJ, Freedman LS. A unified method for monitoring and analysing controlled trials. Stat Med. 1994;13:1815–26. doi: 10.1002/sim.4780131804. [DOI] [PubMed] [Google Scholar]
- 8.Food and Drug Administration. Guidance for Industry: Adaptive Design Clinical Trials for Drugs and Biologics. 2010. [Last accessed on 2016 Jun 15]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM201790.pdf .
- 9.Sinha BK. Interim Analysis in Clinical Trial, ppt. [Last accessed on 2016 Feb 21]. Available from: http://www.uta.fi>dpph>materials>InterimAnalysis .
- 10.Colquhoun D. An investigation of the false discovery rate and the misinterpretation of P values. R Soc Open Sci. 2014;1:140216. doi: 10.1098/rsos.140216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halsey LG, Curran-Everett D, Vowler SL, Drummond GB. The fickle P value generates irreproducible results. Nat Methods. 2015;12:179–85. doi: 10.1038/nmeth.3288. [DOI] [PubMed] [Google Scholar]
- 12.Saxman SB. Ethical considerations for outcome-adaptive trial designs: A clinical researcher's perspective. Bioethics. 2015;29:59–65. doi: 10.1111/bioe.12084. [DOI] [PubMed] [Google Scholar]
- 13.Saxman SB. Commentary on Hey and Kimmelman. Clin Trials. 2015;12:113–5. doi: 10.1177/1740774514568874. [DOI] [PubMed] [Google Scholar]
- 14.Korn EL, Freidlin B. Commentary on Hey and Kimmelman. Clin Trials. 2015;12:122–4. doi: 10.1177/1740774515569611. [DOI] [PubMed] [Google Scholar]
- 15.Joffe S, Ellenberg SS. Commentary on Hey and Kimmelman. Clin Trials. 2015;12:116–8. doi: 10.1177/1740774515568917. [DOI] [PubMed] [Google Scholar]
- 16.Hey SP, Kimmelman J. Are outcome-adaptive allocation trials ethical? Clin Trials. 2015;12:102–6. doi: 10.1177/1740774514563583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buyse M. Commentary on Hey and Kimmelman. Clin Trials. 2015;12:119–21. doi: 10.1177/1740774515568916. [DOI] [PubMed] [Google Scholar]
- 18.Berry DA. Commentary on Hey and Kimmelman. Clin Trials. 2015;12:107–9. doi: 10.1177/1740774515569011. [DOI] [PubMed] [Google Scholar]
- 19.Parker RM, Browne WJ. The place of experimental design and statistics in the 3Rs. ILAR J. 2014;55:477–85. doi: 10.1093/ilar/ilu044. [DOI] [PubMed] [Google Scholar]
- 20.Freedman LS, Spiegelhalter DJ, Parmar MK. The what, why and how of Bayesian clinical trials monitoring. Stat Med. 1994;13:1371–83. doi: 10.1002/sim.4780131312. [DOI] [PubMed] [Google Scholar]
- 21.Whitehead J. The Design and Analysis of Sequential Clinical Trials. 2nd ed. Chichester: Ellis Horwood; 1992. [Google Scholar]
- 22.Jennison C, Turnbull BW. Statistical approaches to interim monitoring of medical trials: A review and commentary. Stat Sci. 1990;5:299–317. [Google Scholar]
- 23.Piantadosi S. Clinical Trials: A Methodologic Perspective. New York: John Wiley & Sons; 1997. [Google Scholar]
- 24.Souhami RL. The clinical importance of early stopping of randomized trials in cancer treatments. Stat Med. 1994;13:1293–5. doi: 10.1002/sim.4780131303. [DOI] [PubMed] [Google Scholar]
- 25.Legocki LJ, Meurer WJ, Frederiksen S, Lewis RJ, Durkalski VL, Berry DA, et al. Clinical trialist perspectives on the ethics of adaptive clinical trials: A mixed-methods analysis. BMC Med Ethics. 2015;16:27. doi: 10.1186/s12910-015-0022-z. [DOI] [PMC free article] [PubMed] [Google Scholar]