

Abstract
Introduction:
Prion diseases are protein conformation disorders and neither caused by viroid or virus but is a transmissible particle labeled a prion by Pruisner. Normal prion protein becomes infectious by a different folding, but the triggers are not known. Based on the characteristic brain pathology, they are grouped under spongiform encephalopathy affecting both man and animals. Estimated prevalence is one per million. Creutzfeldt–Jakob disease (CJD) registry from National Institute and Neurosciences (NIMHANS), Bengaluru, reported 69 cases in 30 years.
Patient and Methods:
Patients seen by our team from December 2011 to October 2015 who satisfied criteria for probable CJD were evaluated for clinical, electrophysiological, radiological, and demographic factors. None of them underwent histopathological examination of brain tissue or tonsils. Cerebrospinal fluid protein 14-3-3 was not done. All of them were followed up by telephonic inquiry for the course of the illness. All of them received symptomatic medications with anticonvulsants, flupirtine 200 mg orally daily, and other symptomatic medications.
Results:
Sporadic CJD is the most common form seen in India and is probably under reported. males seem to be more affected, and the mean duration for the bed bound state is 12 months. Drugs were only effective for a very brief period in controlling myoclonus and behavior.
Discussion:
Sporadic CJD is one of the most common and rapidly fatal forms of dementia in India. Cortical ribboning and periodic complexes are the most common laboratory findings. Familial CJD is a very rare occurrence and variant CJD is probably not prevalent.
Conclusion:
All patients with rapidly progressive dementia should be handled with biohazard precautions unless proved otherwise. Role of alcohol and smoking in the transformation of PrPc to PrPsc needs to be evaluated.
Key words: Cortical ribbon, Creutzfeldt–Jakob disease, periodic complexes, rapidly progressive dementia
INTRODUCTION
Prion diseases are otherwise called as protein conformation disorders as it is clear that it is neither a virus nor a viroid which is responsible but transmissible proteinaceous particle labeled by Pruisner as “PRION” in 2004.[1] It can be genetic, sporadic, and spontaneous as well as infective making this disease a very unique one. PRNP is the gene coding PrP protein and located in the short arm of chromosome 20. Creutzfeldt in 1920 and Jakob in 1921 reported dementing illness with spongiform change.[1,2] The first report of transmissibility in human prion disease was reported by Gajdusek and group by transmitting Kuru to Chimpanzees. The incubation period is very long, but the clinical course is very short. The infectious agent is a protease-resistant 27–30 kDa protein PrPsc, which is a conformer of PrPc. PrPsc has ability to bind to PrPc as a template for its own replication. The normal prion protein takes an infectious form by different folding spontaneously triggering a domino effect which misfolds prion protein throughout the brain based on the genetic variations of the individual. More than 50 prion protein mutations are reported in the inherited forms. This aggregates and cause proteinase resistance, decreased water solubility, and tendency to polymerize causing cell damage.[2] Based on characteristic brain pathology, they are grouped under spongiform encephalopathy affecting both humans and animals. The human forms are Kuru due to endocannibalism, familial fatal insomnia, Gerstmann–Straussler–Scheinker disease, and Creutzfeldt–Jakob disease (CJD). The animal forms are bovine spongiform encephalopathy, or mad cow disease, scrapie affecting sheep and goat, mink and feline encephalopathy, elk, deer forms, etc., Variant CJD is believed to be transmitted from bovine spongiform encephalopathy due to dietary and environmental exposure currently seen only in the UK.[3,4]
Epidemiology
Estimated incidence is 1/million. The sporadic variety is most common. Familial is suspected on family history following a dominant pattern and confirmed by genetic testing. It forms 5–10% in the USA. About 405 cases of infectious CJD are reported. The biggest cluster related to contaminated Dural grafts in Japan, human growth hormone therapy in France, and 189 variant CJD described.[5,6,7] From Bengaluru over 30 years, 69 cases are reported by Shankar et al. from 1968 to 1997.[8] Ten cases are reported from Delhi and seven cases from Mumbai.[7]
Clinical features
Sporadic CJD manifests between 55 and 75 years with rapidly progressing dementia and behavioral symptoms in the form of delusions, hallucinations, delirium, depression, apathy, agitation, confusion, disorientation, memory loss, pyramidal, extrapyramidal, and cerebellar features with myoclonus. The myoclonus is typically generalized, nonepileptic and occurs at a frequency of 1 Hz. The myoclonus can be elicited by sound, light, and touch. Patients also have severe asthenia, anxiety, weight loss, altered sleep-wake cycle, and some patients show features of anterior horn cell involvement. Both sexes are equally affected. Ataxia and abnormalities of vision occur in some cases. The visual symptoms may be loss of acuity or distortion of seen objects. Headache, vertigo, and sensory symptoms can be seen in some patients. Patients can develop eye signs in the form of paralysis of convergence and upgaze. The disease progresses very rapidly on a week to week basis resulting in mute state, contractures and death occurs in 1–3 years. Very rarely patients have survived up to 10 years. Familial ones have a variety of phenotypes and given different names. Variant forms affect younger people and have a slightly less catastrophic course.
Diagnosis
It is based on clinical features, course, and electroencephalogram (EEG) changes which are classical in the sporadic ones and not present in variant forms. EEG shows typically 1/s spikes or mid-positive triphasic waves which are symmetrical and synchronous with a normal background in the early stages and background becomes altered as disease advances. Magnetic resonance imaging (MRI) is mandatory criteria for the diagnosis of probable CJD. Diffusion-weighted images have a sensitivity of up to 100%. This is believed to be due the decrease in the isotropic water diffusibility which corresponds to spongiform changes and nerve cell loss as well as accumulation of the pathological form of PrPSc. The typical features are cortical ribboning, signal intensity changes in the thalamus which are bilateral and symmetrical and more often seen in variant CJD but can also be seen in sporadic CJD and called as “Pulvinar sign.” Among other basal ganglia structures, caudate is affected in 85–90% of patients and Putamen in 84% of the patients and only about 7% show globus pallidus changes. The combination of caudate and putaminal involvement is seen in 67% and called as “inverted Hockey stick sign.”[9] Tonsillar biopsy is helpful in variant form. Detection of 14-3-3 protein in the cerebrospinal fluid (CSF) improves the diagnostic accuracy by 98% in cases of sporadic CJD. Revised WHO definition (given below) is applied for general diagnosis and pathology when possible [Table 1].[10]
Table 1.
Revised WHO definition of Creutzfeldt–Jakob disease subtypes (incorporating the new definition of probable sporadic Creutzfeldt–Jakob disease as per section 3.1)

Precautions in handling tissues
During surgery, nonbreakable gloves should be used. No frozen sections should be taken. In autopsy, all universal precautions should be followed. Masks, eye shields, and cut resistant gloves are used. Better to remove calvarium with a hand saw. Use restricted area and should not contaminate the outer surface of containers and the funeral home should be notified. All the areas should be washed with freshly opened undiluted sodium hypochlorite. This is washed off after 10–15 min with water. All used materials should be put in a biohazard bag, autoclaved at 132 degree Celsius and discarded. Any liquids should be disinfected with equal volume of 2 N Na OH before disposing. Moreover, all surfaces should be wiped with the same for 15–60 min. Tissues are formalin fixed for 10 days before cutting. Agitate the blocks with 50–100 ml of 100% formic acid for 1 h and return to formalin for 2 days before embedding. Use double gloves while processing and treat all disposables before disposing for 1 h with undiluted bleach. It is better to use a disposable microtome or keep a separate microtome for these cases. Covered slides do not need any precaution, but the blocks need to be labeled properly.[10,11]
PATIENTS AND METHODS
Patients admitted to our team from December 2011 to October 2015, who satisfied criteria for probable CJD by the WHO criteria were included for the study.[11] All demographic details, habits, diet, exposure to toxins, human products such as hormones, immunoglobulins, vaccines, surgery, trauma, organ transplant, and family history were recorded. All patients underwent all mandatory investigations for dementia including CSF for chronic infections, B12 levels, thyroid function tests, serology as well as MRI and EEG. However, histopathology, Western blot, and tonsillar biopsy not done in any case.
All patients were treated for all comorbidities. Sodium valproate and clonazepam were used for the treatment of myoclonus and seizures. Flupirtine 200 mg a day was tried in all cases. Behavioral problems were treated with quetiapine 25–100 mg a day. Counseling regarding illness and precautions to be undertaken were given to all caregivers. The course was followed up by telephonic conversation with family members.
RESULTS
We had a total of 15 patients, one familial and others sporadic. Out of the 14 patients with sporadic CJD, all showed cortical ribboning, the majority were males (92.8%) as compared to females (7.14%). Seven patients (42.85%) were from higher and middle socioeconomic class. Eight patients (57.14%) were chronic alcoholics and six patients (42.85%) were chronic smokers. The mean age was 55.2 ± 11.9 years. The mean Vitamin B12, folate, and homocysteine levels were 797 pg/ml, 8.11 pg/ml, and 13.43 µmol/L, respectively. The electrolyte and thyroid functions were normal. Liver function test was impaired in two patients. There were eight patients in the geriatric age group (≥60 years) which constituted 57.14% of the cases. About 14.8% patients had undergone major surgery in the past and two had received blood transfusions during those procedures. Two patients (14.28%) had a history of hypertension and five patients (35.71%) had a history of diabetes mellitus and were on regular oral medication for the same. None of the patients were having history of exposure to sexually transmitted diseases and confirmed by subsequent investigations as well. Symptoms of akathisia were seen in six, delirium in eight, depression and sleep problems in 11 patients, respectively. Clinical and neuropsychological assessment revealed features of frontal lobe dysfunction in 13 (92%), temporal lobe dysfunction in 4 (28.6%), parietal lobe dysfunction in 4 (28.6%), and occipital lobe involvement in 6 patients (42.9%), respectively. Four patients had features of cerebellar dysfunction. Myoclonic jerks were seen in 13 and generalized seizures in 1 patient. One patient was a 63-year-old female, who was in end stage disease, had a very strong family history with involvement of mother, aunt, and son. At the time of visit to our center, she was bed bound and comatose. She had rhythmic myoclonic jerks, rigidity, and extensor plantar response. This was the only familial case in our series. Her imaging showed pulvinar sign [Figure 1].
Figure 1.
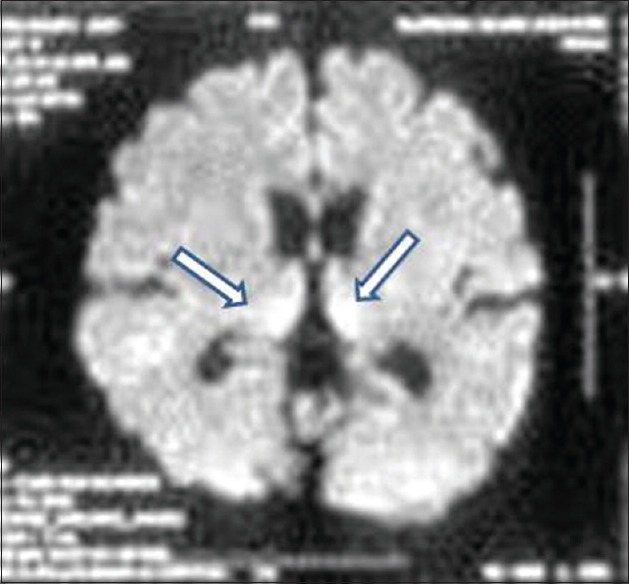
Case of familial Creutzfeldt–Jakob disease showing pulvinar sign
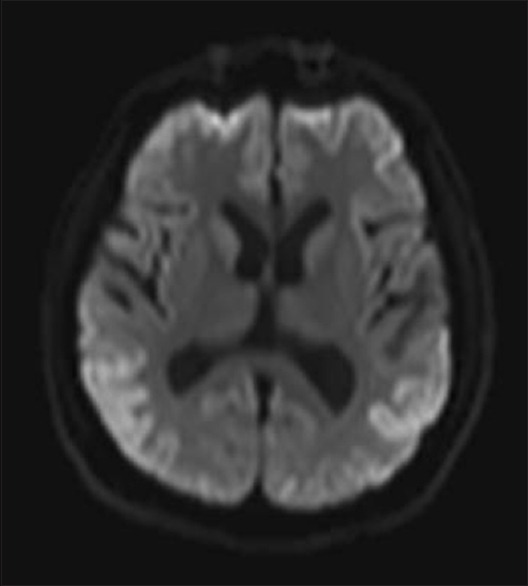
EEG showed periodic complexes in 11 patients and two patients showed mid-positive triphasic waves and one patient showed frontal intermittent rhythmic delta waves [Figures 2 and 3]. The patient with familial CJD showed very typical 1/s sharp waves as well as mid-positive triphasic waves CT scan revealed diffuse atrophy with caudate hypodensity in 5 (35.7%) patients. MRI showed cortical ribboning in all the patients, but characteristic pulvinar sign was seen in three patients [Figures 4 and 5]. Caudate nuclei and thalamic signal changes were seen in nine patients. Visual evoked potentials were prolonged in two patients, and antimeasles antibody along with anti-Jo antibody was positive in one patient. Otherwise vasculitic and paraneoplastic workup were noncontributory. All our patients were bed bound within 12 months of diagnosis and at 12 months, two patients died. Further, follow-up is not available.
Figure 2.
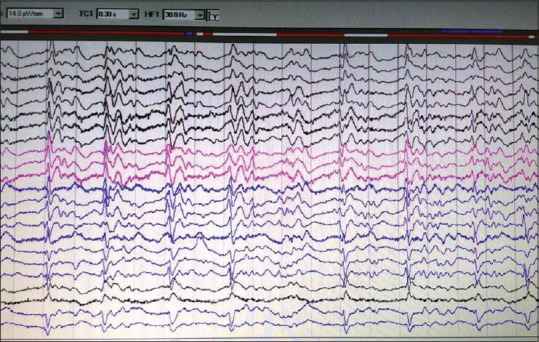
Electroencephalogram showing classical 1/s periodic sharp wave complex
Figure 3.
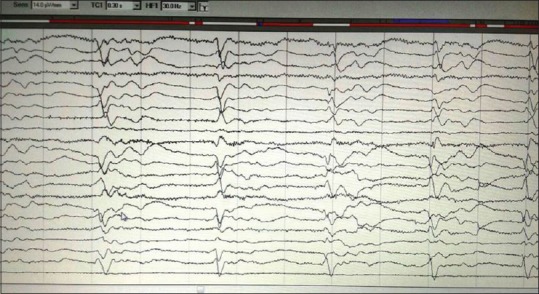
Electroencephalogram showing periodic mid-positive triphasic waves
Figure 4.
Diffusion-weighted imaging showing cortical ribboning
Figure 5.
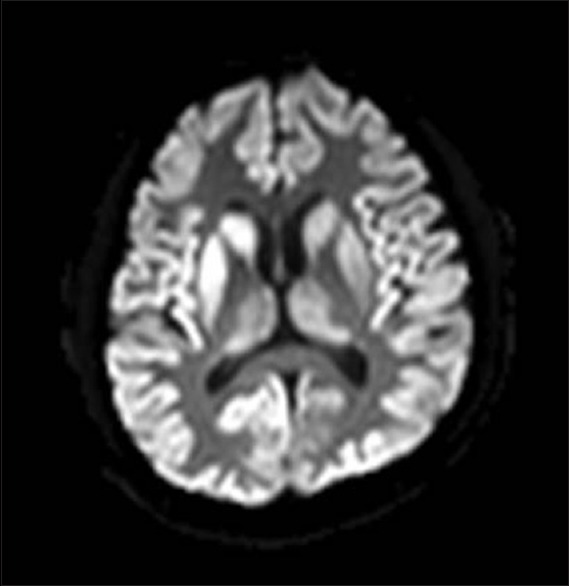
Diffusion-weighted imaging showing cortical ribboning, inverted hockey stick sign, and thalamic signal changes
DISCUSSION
Our experience reveals that spongiform encephalopathy is not uncommon in our country. Males in the fifth to sixth decade seem to be commonly affected. Sporadic variety is the most common. Common comorbidity is smoking and alcohol. The most common symptoms are sleep problems, delirium, and myoclonus. EEG and MRI showed changes in all cases. The average time to bed bound state from the first evaluation is around 12 months. Onset to peak is not clear as the exact onset could not be ascertained.
CONCLUSION
CJD is not uncommon in India. Sporadic form seems to be the most common variety. Whether alcohol and smoking have a role in misfolding the prion protein needs to be evaluated. Maximum affected are males and mean duration to bed bound state is about 12 months. Drugs are helpful for a brief period only in controlling behavior and myoclonus. All rapidly progressive dementias should be handled with care to avoid the biohazards unless spongiform encephalopathy is excluded.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Bradley WG, editor. Neurology in Clinical Practice: Principles of Diagnosis and Management. Vol. 2. Philadelphia, USA: Taylor and Francis; 2004. p. 1567. [Google Scholar]
- 2.Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature. 1996;383:685–90. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 3.Collinge J. Prion diseases of humans and animals: Their causes and molecular basis. Annu Rev Neurosci. 2001;24:519–50. doi: 10.1146/annurev.neuro.24.1.519. [DOI] [PubMed] [Google Scholar]
- 4.Harvey RJ, Fox NC, Rossor MN. Dementia handbook. Martin Dunitz. 1999:65. [Google Scholar]
- 5.Brown P, Brandel JP, Preece M, Sato T. Iatrogenic Creutzfeldt-Jakob disease: The waning of an era. Neurology. 2006;67:389–93. doi: 10.1212/01.wnl.0000231528.65069.3f. [DOI] [PubMed] [Google Scholar]
- 6.Collinge J, Whitfield J, McKintosh E, Beck J, Mead S, Thomas DJ, et al. Kuru in the 21st century – An acquired human prion disease with very long incubation periods. Lancet. 2006;367:2068–74. doi: 10.1016/S0140-6736(06)68930-7. [DOI] [PubMed] [Google Scholar]
- 7.Mehndiratta MM, Bajaj BK, Gupta M, Anand R, Tatke M, Seryam S, et al. Creutzfeldt-Jakob disease: Report of 10 cases from North India. Neurol India. 2001;49:338–41. [PubMed] [Google Scholar]
- 8.Shankar SK, Chandra P. Creutzfeld–Jakob disease in Western India in 30 years (1968–1997) Bangalore, India: CJD Registry NIMHANS; 1997. [Google Scholar]
- 9.Kallenberg K, Schulz-Schaeffer WJ, Jastrow U, Poser S, Meissner B, Tschampa HJ, et al. Creutzfeldt-Jakob disease: Comparative analysis of MR imaging sequences. AJNR Am J Neuroradiol. 2006;27:1459–62. [PMC free article] [PubMed] [Google Scholar]
- 10.Revised WHO Definition of CJD Subtypes (Incorporating the New Definition of Probable Sporadic CJD as Per Section 3.1. [Last accessed on 2015 Nov 05, 7:45 pm]. Available from: http://www.who.int/csr/resources/publications/bse/whoemczdi989 .
- 11.Brown P. Autopsy Performance and Reporting. Northfield, Ill: College of American Pathologists; 1990. “Guidelines for high risk autopsy cases: Special precautions for Creutzfeldt – Jakob disease.”; pp. 68–74. [Google Scholar]