

Abstract
Patients with unresectable hepatocellular carcinoma (HCC) cannot generally be cured by systemic chemotherapy or radiotherapy due to their poor response to conventional therapeutic agents. The development of novel and efficient targeted therapies to increase their treatment options depends on the elucidation of the molecular mechanisms that underlie the pathogenesis of HCC. The DNA damage response (DDR) is a network of cell-signaling events that are triggered by DNA damage. Its dysregulation is thought to be one of the key mechanisms underlying the generation of HCC. Sphingosine-1-phosphate (S1P), a lipid mediator, has emerged as an important signaling molecule that has been found to be involved in many cellular functions. In the liver, the alteration of S1P signaling potentially affects the DDR pathways. In this review, we explore the role of the DDR in hepatocarcinogenesis of various etiologies, including hepatitis B and C infection and non-alcoholic steatohepatitis. Furthermore, we discuss the metabolism and functions of S1P that may affect the hepatic DDR. The elucidation of the pathogenic role of S1P may create new avenues of research into therapeutic strategies for patients with HCC.
Keywords: DNA damage response, Lipid mediator, Sphingosine-1-phosphate, Hepatocellular carcinoma
Introduction
The incidence of hepatocellular carcinoma (HCC), which is the most common type of primary liver cancer and which is associated with a very high mortality rate, is increasing [1]. The geographic distribution of HCC mortality is similar to that of its incidence; both are high in Asia and Africa [2]. Most HCC patients have severe liver dysfunction related to conditions such as chronic hepatitis B virus (HBV) or hepatitis C virus (HCV) viral infections, alcohol abuse, and metabolic disease [2]. Such conditions lead to 80 % of cases being ineligible for curative surgical treatment [3]. Patients with unresectable HCC cannot generally be cured by systemic chemotherapy or radiotherapy [3], because hepatoma cells respond poorly to conventional approaches [4–6]. Given the limited efficacy of existing treatments for hepatoma cells, the molecular mechanisms underlying the pathogenesis of HCC need to be understood so that novel and efficient targeted therapies may be developed for these patients.
One of the key mechanisms driving the generation of HCC is thought to be dysregulation of the DNA damage response (DDR). The DDR is a network of cellular signaling pathways that repairs the tens of thousands of DNA damage events that occur in the cells of the human body each day [7]. The DDR pathways are triggered by DNA damage to coordinate DNA repair, cell cycle arrest, and cell death or senescence [8]. If DNA damage is not properly repaired by the DDR system, disease-causing mutations may arise, including the mutations that underlie many types of cancer [9–14].
DNA damage can be caused by endogenous and exogenous agents. These include ultraviolet light, environmental chemicals such as tobacco, and reactive oxygen species (ROS), which are byproducts from either oxidative respiration or the redox cycling induced by environmental toxins [7, 15, 16]. ROS are also produced at sites of inflammation or infection by inflammatory cells such as macrophages and neutrophils [7]. Indeed, HCC often occurs after chronic inflammation or injury of the liver, which promotes DNA damage [17].
Recently, sphingolipids have emerged as important signaling molecules. They are reported to be involved in many cellular functions, including cell growth, death, senescence, adhesion, migration, angiogenesis, and inflammation [18]. Among the bioactive sphingolipids, sphingosine-1-phosphate (S1P) is an important mediator in multiple signaling pathways that affect many pathological conditions, including inflammation and cancer [19–22]. Notably, S1P is implicated in DNA damage and the DDR [23, 24]. Recently, we discovered that S1P and its kinase play an important role in hepatocytes, and that they regulate the gene transcription involved with lipid metabolism in the liver [25]. An increasing amount of evidence, including our own, suggests that the alteration of S1P signaling and gene regulation in the liver may affect the DDR and liver carcinogenesis [23–26].
In this review, we explore how the regulation of DNA damage influences hepatocarcinogenesis of various etiologies, such as that associated with hepatitis B and C viral infections or non-alcoholic steatohepatitis. Furthermore, we discuss the directions of future research based on the metabolism and functions of S1P, which may affect the hepatic DDR, with a focus on S1P regulation by enzymes such as sphingosine kinases (SphKs) and S1P lyase. Given that the DDR plays such a critical role in liver carcinogenesis, the elucidation of the role of S1P in pathogenesis has the potential to identify new avenues of research into therapeutic strategies to treat patients with HCC.
DNA damage and hepatocarcinogenesis
Current evidence suggests that the majority of HCC patients have underlying cirrhosis, and that HCC is driven by inflammation. While the molecular mechanisms linking chronic inflammation to HCC development remain unclear, the inflammatory stimuli that induce the production of ROS include superoxide, nitric oxide, hydrogen peroxide, and hypochloric acid [27]. These ROS are the primary causative agents of oxidative DNA damage. After DNA damage is induced by ROS, the DDR machinery is activated to repair DNA replication defects and reverse cell cycle arrest. Because DDR proteins are functionally deregulated in many types of cancer cells [28], the disruption of the balance between ROS-induced DNA damage and the DDR could contribute to carcinogenesis in the liver.
Five major mechanisms exist within the DDR system: (1) base excision repair (BER), (2) mismatch repair (MMR), (3) nucleotide excision repair (NER), (4) homologous recombination (HR), and (5) non-homologous end-joining (NHEJ) [29]. BER repairs the DNA damage induced by ROS or radiation, and is typically regulated by DNA glycosylase and X-ray repair cross-complementing protein 1 (XRCC1). MMR is involved in the repair of replication error-induced DNA mismatches [30], and is mediated by the DNA mismatch repair protein MutS and exonuclease 1. NER removes bulky DNA adducts, and is regulated by transcription factor II human (TFIIH), xeroderma pigmentosum type B (XPB), xeroderma pigmentosum group D (XPD), DNA repair protein complementing XP-A cells (XPA), replication protein A (RPA), and DNA repair protein complementing XP-G cells (XPG). HR is involved in the regeneration of damaged chromosomal regions using homologous DNA as a template and is regulated by the Mre11–Rad50–Nbs1 (MRN) complex, C-terminal-binding protein 1 (CtlP), RPA, Rad51, Rad52, breast cancer susceptibility gene (BRCA) 1, and BRCA2. NHEJ repairs damaged DNA through the direct rejoining of DNA double-strand breaks (DSBs), and is regulated by Ku70–Ku80, DNA-dependent protein kinase (DNA-PK), X-ray repair cross-complementing protein 4 (XRCC4)-DNA ligase IV, and XRCC4-like factor (XLF).
Multiple studies have suggested that HR and NHEJ play key roles in carcinogenesis through the maintenance of genomic instability and chromosomal rearrangement. These DSB repair pathways are typically initiated by the protein kinases ataxia telangiectasia mutated (ATM) and Rad3-related (ATR). ATM is recruited to DSBs by the MRN complex [31] and acts as a primary activator of HR and NHEJ [32]. ATR is involved with broader types of DNA damage, including DSBs and disrupted DNA replication forks [33]. ATM and ATR exhibit some crossover in function, with both proteins implicated in the phosphorylation of DDR-related proteins, including histone H2AX, mediator of DNA damage checkpoint 1 (MDC1), tumor suppressor p53-binding protein 1 (53BP1), BRCA1, and meiotic recombination 11 homolog A (MRE11). Among these signaling molecules, H2AX is the most sensitive marker of DSBs. The phosphorylated form of H2AX (γ-H2AX) specifically localizes at DSB sites, and the degree of DNA damage can be evaluated by γ-H2AX immunohistochemical staining. We recently assessed γ-H2AX staining in HCC and non-cancerous liver diseases and found that the number of γ-H2AX foci was significantly increased in HCC tissues in comparison to cirrhotic liver tissue without HCC [9]. Moreover, Xiao et al. [34] recently reported that increased γ-H2AX expression was associated with increased tumor size, vascular invasion, TNM stage, and reduced survival in HCC patients. They also found that γ-H2AX knockdown effectively decreased the expression of vascular endothelial growth factor (VEGF), leading to reduced tumorigenesis, and the inhibition of angiogenesis in HCC. Taken together, these findings suggest that the number of DSBs may be valuable for assessing tumorigenesis and angiogenesis in HCC.
Hepatitis B virus and DNA damage
Hepatitis B virus (HBV) is a DNA virus with four main coding regions: HBV X (HBX), surface (HBs), core (HBc), and pol. HBX strongly promotes hepatocarcinogenesis, and the forced expression of HBX induces liver cancer without any inflammatory features in vivo [35], suggesting that this region of the viral genome directly causes HCC. The HBX gene is frequently integrated into the human genome, and its gene product transactivates various signal transduction pathways, including the Ras/mitogen-activated protein kinase (MAPK) pathway [36].
HBX promotes DNA damage through several different mechanisms (Fig. 1). HBX can inactivate the p53 tumor suppressor, which increases the susceptibility of host cells to DNA damage [37–39]. HBX also induces endoplasmic reticulum (ER) stress [40], suggesting that ER stress might contribute to oxidative stress in the liver. Moreover, the HBX-mediated oscillation of oxidative stress levels activates pro-survival factors, including nuclear factor-kappa B (NF-kappa B), signal transducer and activator of transcription 3 (STAT3) [41], and Akt/protein kinase B [42]. Additionally, HBX functionally inhibits several DNA damage-binding (DDB) proteins, including X-associated protein 1 (XAP-1)/UV-damaged DNA-binding protein (UV-DDB), XPB and XPD, which are involved with NER-mediated DNA repair [43, 44]. HBX also activates nuclear factor erythroid 2-related factor (Nrf2), a regulator of the anti-oxidant/electrophile response element (ARE) present in the promoters of phase II detoxifying and antioxidant factors such as heme oxygenase 1 (HO-1) and NAD(P)H:quinone oxidoreductase 1 (Nqo1) [45, 46]. Taken together, these findings reveal a unique role for HBX as an oncogene that can enhance cell survival in a context of increased DNA damage.
Fig. 1.

The various functions of hepatitis B virus X (HBX), HCV core protein and non-alcoholic steatohepatitis (NASH). HBX, HCV core protein, and NASH promote DNA damage through several different mechanisms, as shown. ATM ataxia telangiectasia mutated, ATR ataxia telangiectasia and Rad3-related, Chk1 checkpoint kinase 1, Chk2 checkpoint kinase 2, ER endoplasmic reticulum, HO1 heme oxygenase 1, Keap1 Kelch-like ECH-associated protein 1, MAPK mitogen-activated protein kinase, MRN Mre11-Rad50-Nbs1, NF-kappa B nuclear factor kappa-light-chain-enhancer of activated B cells, Nqo1 NAD(P)H: quinone oxidoreductase 1, Nrf-2 nuclear factor erythroid 2-related factor 2, PKCδ protein kinase cδ, STAT3 signal transducer and activator of transcription 3, UV-DDB UV-damaged DNA-binding protein, XAP-1 HBV X-associated protein 1, XPB xeroderma pigmentosum group B, XPD xeroderma pigmentosum group D, γH2AX phosphorylated histone H2AX
More recently, a close relationship between HBX and ATR- and ATM-mediated pathways has been described. Because both ATM and ATR are important regulators of the initial steps of the DDR, it is plausible that HBX regulates multiple down-stream DDR signaling molecules [47]. HBX stimulates the ATR-checkpoint kinase 1 (Chk1) pathway, leading to the inhibition of the intra-S-phase checkpoint and the prevention of DNA damage-induced cell apoptosis [48]. HBX also activates ATR through the MAPK signaling pathway, leading to the phosphorylation of H2AX [49]. However, the relationship between HBX and ATR may be complicated, as others have reported activation of p38 MAPK through ATR [50].
There are comparatively few studies of the relationship between HBX and ATM. ATM regulates the DDR and stimulates redox signaling [3, 51]. In addition, we have recently reported that ATM can be activated by HBX both in vitro and in vivo [52]. Our study revealed that the increased levels of phosphorylated ATM induced by HBX are significantly decreased in the presence of antioxidants, suggesting that HBX activates ATM through oxidative stress. We also identified an increased level of protein kinase C-delta (PKC-δ) in HBX-expressing cells, which could be attenuated by an ATM inhibitor. PKC-δ is involved in redox signaling, and induces the dissociation of Nrf2 from the adaptor protein Keap1 (INrf2) [53, 54]. Therefore, HBX-mediated ATM activation may disrupt the DNA damage defense mechanisms by regulating the DDR systems and/or the PKC-δ/Nrf2-mediated redox pathway. The development of an ATM kinase inhibitor could help to prevent HBX-induced carcinogenesis.
Hepatitis C virus and DNA damage
Hepatitis C virus (HCV), another causative agent of HCC, is a small enveloped single-stranded RNA virus that is transmitted by blood-to-blood contact. Currently, more than 185 million individuals worldwide are infected with HCV. Twenty percent of individuals with chronic HCV infection suffer liver cirrhosis [55]. Combination therapy with pegylated interferon (immunomodulator) and ribavirin (an inhibitor of viral RNA synthesis) has been used in an attempt to eradicate HCV infection; however, this treatment is limited by its low efficacy and side effects. Very recently, two newly developed oral direct-acting antiviral agents, daclatasvir and asunaprevir, have emerged as promising interferon-free agents for the treatment of HCV genotype 1 [56]. Long-term observation will be required to determine whether daclatasvir/asunaprevir therapy can effectively prevent HCC.
Chronic HCV infection promotes double-stranded DNA breaks that lead to genetic mutations in host cells. Furthermore, multiple studies have demonstrated increased levels of oxidative stress markers in urine samples or liver tissue specimens from patients with HCV. The DNA damage marker 8-hydroxy-2′-deoxyguanosine (8-OHdG) is detectable in the nuclei of hepatocytes in HCV-infected liver tissue [57]. Moreover, both 8-nitroguanine and 8-OHdG are present in the liver of patients with HCV-positive chronic hepatitis, but not in the liver of non-infected controls [58]. Researchers examined the relationship between 8-OHdG levels and clinical and biochemical parameters that hepatic 8-OHdG levels were significantly correlated with serum transaminase levels and tumor histological grade [59]. Intriguingly, the number of 8-OHdG-positive hepatocytes was significantly higher in patients with HCV-positive chronic hepatitis than in HBV-positive patients [60]. Hepatic DNA damage was strongly correlated with iron overload, suggesting that HCV-induced iron overload might be a critical mediator of oxidative stress in the liver of HCV-positive patients. Consistent with this, 8-OHdG levels in HCV-positive liver tissues are positively correlated with the disease stage and telomerase activity [61]. Importantly, a recent report shows that the cumulative HCC incidence rate in HCV-positive patients with high levels of liver 8-OHdG expression is significantly greater than that in patients with low 8-OHdG levels [62]. Together, these data strongly implicate HCV as a causative agent of oxidative DNA damage in the liver.
The HCV genome encodes core, E1, E2, and non-structural (NS2, NS3, NS4A, NS4B, NS5, NS5A, and NS5B) proteins, all of which are involved in HCV-associated pathogenesis. Several studies have implicated the HCV core protein as a mediator of oxidative DNA damage (Fig. 1). HCV core inhibits the formation of the MRN complex, leading to inhibition of ATM-mediated DNA repair [63]. Through interactions with mitochondria, the HCV core protein redistributes cytochrome c from the mitochondria to the cytoplasm, leading to the oxidation of the glutathione pool and a decrease in nicotinamide adenine dinucleotide phosphate (NADPH) content [64, 65]. HCV core decreases the level of hepcidin, a negative regulator of iron absorption and hepatic iron content [66]. These data strongly suggest that HCV core can induce ROS-mediated DNA damage. Non-structural HCV proteins can also promote oxidative stress, and the overproduction of ROS has been observed in NS5A-transgenic mice [67]. Meanwhile, HCV NS3 and NS4A protein can induce the translocation of ATM to the cytoplasm, impairing ATM-mediated DNA repair [68]. These data demonstrate multiple mechanisms by which HCV proteins can promote oxidative stress and DNA damage in infected hepatocytes.
Recent studies have revealed that HCV-infected cells are resistant to DNA damage-induced apoptosis. Several HCV components have been associated with p53 [69, 70] and HCV can inhibit the acetylation and activation of p53 in the nucleus by promoting cytoplasmic accumulation of the mouse double minute 2 homolog (MDM2)–p53 complex [71]. Therefore, HCV might be a critical mediator of cell survival in the face of increased oxidative DNA damage.
Non-alcoholic fatty liver disease and DNA damage
Non-alcoholic fatty liver disease (NAFLD) is a common metabolic disorder in developed countries. It is defined as hepatic steatosis which occurs without alcohol ingestion. Non-alcoholic steatohepatitis (NASH), a subtype of NAFLD, is characterized by hepatocytic injury and is well recognized as a risk factor for cryptogenic liver cirrhosis and HCC [72]. While several attempts have been made to explore the biological characteristics of NASH-associated HCC, the mechanism underlying carcinogenesis in NASH remains unclear because of limited patient numbers. However, we recently found that p27, a cell-cycle regulator that inhibits G1-S transition, is a marker of poor prognosis in patients with NASH-associated HCC [73]. The expression of p27 was positively correlated with tumor size and cell proliferation. The phosphorylation of p27 at serine 10 enables interaction with, and sequestration of, cyclin D-cyclin-dependent kinase 4/6. Furthermore, NASH-associated HCC patients with detectable phospho-p27 had reduced disease-free survival. These findings suggest that cell cycle regulator function is implicated in cancer progression in this disease.
Currently, the most accepted model for NAFLD progression centers on a ‘two-hit’ theory. Hepatic steatosis caused by insulin resistance is the first hit. The second hit is the hepatic injury induced by insults such as oxidative stress, lipid peroxidation, cytokine production, or mitochondrial dysfunction [74] (Fig. 1). Importantly, ROS-induced DNA damage is likely to be intricately involved in the pathogenesis and progression of NAFLD. Using a murine model, Daugherity et al. [75] found that mice which were fed a high-fat diet develop steatosis and fibrosis of the liver with increased ROS production and ATM phosphorylation. They also reported that ATM-knockout mice which were fed a high-fat diet showed less liver fibrosis, suggesting that ATM signaling could contribute to the pathogenesis and progression of NAFLD. A role of oxidative stress in the development of NAFLD is demonstrated by the significant increase in DNA damage and oxidative stress that is observed in mice that are fed methionine and choline-deficient diets [76]. Consistent with this, the treatment of these mice with an antioxidant reduced liver injury and steatosis. Of note, extensive studies have explored the possibility that extra-hepatic organs, including the gut and/or adipose tissue, might also be involved in the development of NASH. This novel mechanism is proposed to involve “multiple parallel hits,” and is mediated by molecules that are known to induce oxidative stress at the cellular level [77], such as hydroperoxide-detoxifying enzymes, the gut microbiota and adipose tissue-derived adipocytokines [78]. These findings suggest that oxidative stress plays a significant role in the progression of NAFLD.
Gentric et al. [79] identified increased numbers of highly polyploid mononuclear cells in the liver of NASH patients. Abnormal cell ploidy was accompanied by the oxidative stress-mediated activation of the G2/M DNA damage checkpoint and inefficient S/G2 cell cycle progression. They further demonstrated that antioxidant treatment prevented the development of polyploidy in murine models of NAFLD, supporting the notion that the manipulation of ROS levels could prevent disease progression. While the precise mechanism of ROS production in NAFLD remains elusive, one possible cause of oxidative stress is cytochrome P450 2E1 (CYP2E1), which plays a major role in oxidative stress-induced pathogenesis in alcoholic liver injury [80]. Previous studies have reported that CYP2E1 expression is increased in the liver of NASH patients [81, 82], suggesting that CYP2E1 is implicated in the production of ROS in the liver of patients with NAFLD. The reason why ethanol-inducible CYP2E1 is overexpressed in alcohol-abstinent individuals is unknown, and further studies are needed to elucidate the mechanism of oxidative stress induction in NASH.
The clinicopathological significance of oxidative stress in individuals with NAFLD/NASH has been recently reported. Seki et al. [83] reported that the levels of lipid peroxidation marker 4-hydroxy-2′-nonenal (HNE) adduct and 8-OHdG were frequently increased in patients with NAFLD, while they were rarely increased in the normal liver. Consistent with this, Nomoto et al. [84] reported that 8-OHdG was detected in approximately half of steatosis cases and 90 % of NASH patients. Tanaka et al. [85] reported that the ratio of 8-OHdG-positive hepatocytes was significantly increased in NASH-HCC in comparison to NASH without HCC. Together, these data indicate that oxidative DNA damage increases with NASH progression. Notably, Fujita et al. [86] reported that 8-OHdG levels were significantly decreased in the NASH liver following iron reduction therapy (phlebotomy plus an iron-restricted diet). Iron overload may therefore be an important cause of oxidative DNA damage in NASH patients.
S1P, a lipid mediator, works as a key regulatory molecule in cancer
Two decades ago, S1P was discovered as a signaling molecule that regulates cell growth, indicating that it may also play a role in cancer [87]. S1P is now known as an important regulatory molecule in cancer through its ability to promote cell proliferation, migration, invasion, angiogenesis and lymphangiogenesis (Fig. 2), as well as cell survival against ROS from chemotherapy or radiotherapy [19, 88–90]. S1P is generated intracellularly by two sphingosine kinases, SphK1 and SphK2 [91, 92]. SphK1 translocates from cytosol to the plasma membrane when activated by numerous agonists and stimuli, such as growth factors, hormones, pro-inflammatory cytokines, and many G protein-coupled receptor ligands [93, 94]. This translocation of SphK1 enables the localized production of S1P near its cell surface receptors and enables S1P “inside-out signaling” [20, 95]. In contrast, SphK2 is localized in the nucleus of many types of cells, and in the inner mitochondrial membrane [96, 97]. S1P is exported to the extracellular space where it regulates many functions by binding to, and signaling through, a family of five G protein-coupled receptors (S1PR1-5) [20]. The mechanism underlying the export of S1P out of cells involves the ATP binding cassette transporters, ABCA1 [96, 98], ABCC1, ABCG2 [95, 99, 100] and a newly found transporter, Spns2 [101–103].
Fig. 2.
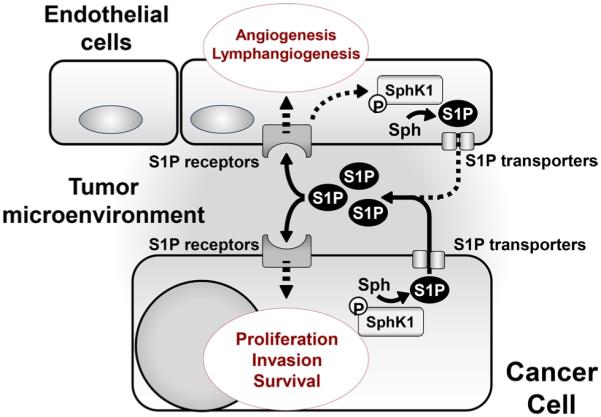
Sphingosine-1-phosphate (S1P) as an important regulatory molecule in cancer. S1P produced by SphK1 is exported from cancer cells via S1P transporters. It promotes cancer cell proliferation, invasion and survival by binding to specific G protein-coupled receptors (S1P receptors) in an autocrine and paracrine manner. S1P produced by cancer cells also stimulates S1P receptors on endothelial cells and promotes tumor-related angiogenesis and lymphangiogenesis
In contrast to the well-investigated extracellular actions of S1P through cell surface receptors, the intracellular mechanisms of S1P action have, until recently, been poorly understood. This situation has changed after discovery of the intracellular targets of S1P, including histone deacetylases (HDACs) [96] and TNF receptor-associated factor 2 (TRAF2) [93]. First, SphK2 forms a complex with histone H3 and HDACs, producing S1P, which regulates HDACs at specific lysine residues to affect gene transcription [96]. S1P binds to and inhibits both HDAC1 and HDAC2, indicating that the S1P produced in the nucleus by SphK2 influences the dynamic balance of histone acetylation, and thus the epigenetic regulation of specific target genes [96]. Second, TRAF2 is an adaptor protein, which contains a RING domain that is implicated in the regulatory ubiquitination of RIP1, a critical event in NF-κB activation in response to TNF-α. S1P is a missing cofactor that is required for the E3 ligase activity of TRAF2 and, consequently, the Lys-63-linked polyubiquitination of RIP1 and NF-κB activation [93], which explains the importance of SphK1 and S1P in cellular protection, inflammation, and the immune response.
In summary, extracellular and intracellular S1P regulates multiple cellular functions that are involved in the pathogenesis of inflammation and cancer. Of note, S1P also regulates the DDR and survival of cancer cells in human patients.
S1P produced by SphK1 and DDR in the liver
There are increasing numbers of clinical and pathological reports on the importance of SphK1 in metastasis and in the prognosis of cancer patients [104–108]. Previous clinical studies have shown that SphK1 is overexpressed in several types of cancer and that its expression is correlated with poor patient outcomes [105]. Furthermore, an increasing number of reports suggest that SphK1 is involved in the response to DNA damage, and that it is related to chemotherapy and radiotherapy resistance. The mechanisms by which SphK1 and S1P regulate the DDR are now being investigated [23]. For instance, the loss of SphK1 in carcinoma cells increases the formation of ROS and sensitivity to doxorubicin-induced DNA damage [109].
The SphK1/S1P axis plays critical roles in cancer progression and is implicated in the repair of DNA damage. Using a mouse model of colitis-associated cancer [21, 110, 111], we recently demonstrated that S1P, produced by SphK1, is essential for the production of the multifunctional NF-kB-regulated cytokine IL-6, the persistent activation of the transcription factor STAT3 and the consequent upregulation of the S1P receptor, S1PR1. Importantly, it was reported that STAT3 interrupts ATR-to-Chk1 signaling by promoting the loss of Claspin, a protein that assists ATR in the phosphorylation of Chk1 [112]. Furthermore, it has been suggested that insulin-like growth factor binding protein-3 (IGFBP-3) promotes chemotherapy resistance via its upregulation of SphK1 and S1P [24]. The upregulation of this pathway by IGFBP-3 was demonstrated in estrogen receptor-negative breast epithelial cells and several triple-negative breast cancer cell lines [113, 114] in which IGFBP-3-induced S1P enhanced EGFR signaling. This is in agreement with a previous report, which indicated that SphK1 and S1P can transactivate EGFR [115]. Thus, it is possible that the activation of EGFR signaling by the upregulation of SphK1 and S1P promotes DNA damage repair, and that IGFBP-3-induced S1P signaling contributes to the chemo- and radio-resistance of cancer cells. Considering that the chronic inflammation and dysregulation of the DDR play critical roles in hepatocarcinogenesis, we cannot help but speculate that SphK1 and S1P may be linked with inflammation, DDR and hepatocarcinogenesis. Investigating the roles of SphK1 and S1P in the pathogenesis of human HCC from the standpoint of DNA damage will be an interesting direction for future study.
S1P produced by SphK2 and the DDR in the liver
We recently discovered that S1P produced in the nucleus of hepatocytes by SphK2, epigenetically regulates hepatic lipid metabolism, and that the dysregulation of SphK2 causes NAFLD in experimental models [25]. These findings suggest a previously overlooked possibility, that SphK2 and S1P may play a role in NASH-related carcinogenesis, which will also be an interesting direction of future research. As described earlier, the S1P produced by nuclear SphK2 acts intracellularly, as opposed to the S1P produced by SphK1, which acts extracellularly through cell surface receptors. The S1P produced by SphK2, in a complex with histone H3 and HDACs in the nucleus, works as an HDAC inhibitor to regulate histone acetylation and the epigenetic regulation of specific target genes [96]. Although it was reported that SphK2 is highly expressed in specific organs, including the liver and brain, the specific target for HDAC inhibition by S1P and SphK2 was, until recently, unclear. We reported that key genes encoding the nuclear receptors/enzymes involved in nutrient metabolism were significantly downregulated in the liver of SphK2−/− mice [25]. The nuclear levels of S1P, an endogenous inhibitor of HDAC 1/2, as well as the acetylation of H3K9, H4K5 and H2BK12, were significantly decreased in hepatocytes prepared from SphK2−/− mice [25]. That SphK2−/− mice rapidly developed fatty liver on a high-fat diet, suggests the importance of SphK2 in regulating hepatic lipid metabolism [25]. It has been reported that histone acetylation is essential, not only for the maintenance of chromatin structure, but also for genome stability [116]. S1P and SphK2 regulate histone acetylation and the epigenetic modification of key genes encoding the nuclear receptors/enzymes that are involved in nutrient metabolism, which may be closely related to the DDR and liver carcinogenesis. Further investigation is required to elucidate the involvement of nuclear S1P and SphK2 in the regulation of the DDR and liver carcinogenesis.
S1P regulation by phosphatases, lyases and the DDR
S1P can be dephosphorylated at the cell surface by a family of lipid phosphate phosphatases that demonstrates broad specificity. More specifically, intracellular S1P can be also dephosphorylated to form sphingosine by the action of two specific S1P phosphatases, Sgpp1 and Sgpp2, which reside in the endoplasmic reticulum. Furthermore, S1P may be irreversibly degraded by the S1P lyase to the nonsphingolipid substrates, hexadecenal and phosphoethanolamine [117]. Intracellular S1P levels are tightly maintained by the balance of synthesis and degradation. S1P lyase plays a major role in the generation of an S1P chemical gradient by degrading S1P in tissues [118]. S1P is enriched, in the submicromolar range, in the blood and lymph; in the interstitial fluids, it is present at much lower levels, creating a steep S1P gradient [118]. This vascular tissue S1P gradient regulates the traffic of immune cells such as lymphocytes, hematopoietic progenitor cells, and dendritic cells. Thus, S1P plays a critical role in immune functions and inflammation due to the S1P gradient produced by S1P phosphatases and lyase.
S1P lyase is activated by irradiation, and functions as a modulator of the DDR [117]. S1P lyase modulates the kinetics of DNA repair, the speed of recovery from G2 cell cycle arrest, and the extent of apoptosis after irradiation [119]. In these processes, S1P lyase affects the Cdk1–cyclin B complex by depleting cellular S1P and elevating ceramide levels, which delays the kinetics of DNA repair [119]. The depletion of S1P may therefore sensitize cancer cells to radiotherapy. Considering that hepatoma cells respond poorly to conventional chemotherapy and radiotherapy, the targeting of S1P signaling may be a more effective therapeutic approach for this disease.
Conclusion
Accumulating evidence indicates that perturbations of the DDR play critical roles in the liver carcinogenesis associated with HBV, HCV, and NASH. S1P, a bioactive sphingolipid, may be involved in the DDR and carcinogenesis; however, the detailed mechanisms of how S1P and its metabolizing enzymes are involved with HCC of varying etiologies remain unclear. After careful consideration of the connection between DDR and S1P, we hypothesize that S1P/SphKs may become established as one of the key regulators of hepatocarcinogenesis. However, we lack sufficient evidence at this time to make any certain conclusions. Further investigation of the role of DDR and lipid signaling in HCC is warranted. An increased understanding of the pathogenesis of HCC may lead to the development of treatment strategies that reduce radioresistance and chemoresistance, and thus improve patient survival.
Acknowledgments
This work was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) [Grant Numbers 15H05676 and 15K15471 (for M.N.) and 15H04927 (for W.T.)]. M.N. was supported by the Uehara Memorial Foundation, Nakayama Cancer Research Institute, Takeda Science Foundation, and Tsukada Medical Foundation. K.T. was supported by an NIH/NCI grant (R01CA160688) and a Susan G. Komen Investigator Initiated Research Grant (IIR12222224). The authors thank Ingrid Woelfel for editing the English of the manuscript.
Footnotes
Compliance with ethical standards
Conflict of interest The authors declare no conflicts of interest in association with the present study.
References
- 1.Mittal S, El-Serag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol. 2013;47(Suppl):S2–6. doi: 10.1097/MCG.0b013e3182872f29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142:1264–73. doi: 10.1053/j.gastro.2011.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fujimaki S, Matsuda Y, Wakai T, Sanpei A, Kubota M, Takamura M, et al. Blockade of ataxia telangiectasia mutated sensitizes hepatoma cell lines to sorafenib by interfering with Akt signaling. Cancer Lett. 2012;319:98–108. doi: 10.1016/j.canlet.2011.12.043. [DOI] [PubMed] [Google Scholar]
- 4.Arii S, Yamaoka Y, Futagawa S, Inoue K, Kobayashi K, Kojiro M, et al. Results of surgical and nonsurgical treatment for small-sized hepatocellular carcinomas: a retrospective and nationwide survey in Japan. The Liver Cancer Study Group of Japan. Hepatology. 2000;32:1224–9. doi: 10.1053/jhep.2000.20456. [DOI] [PubMed] [Google Scholar]
- 5.Kuwahara Y, Li L, Baba T, Nakagawa H, Shimura T, Yamamoto Y, et al. Clinically relevant radioresistant cells efficiently repair DNA double-strand breaks induced by X-rays. Cancer Sci. 2009;100:747–52. doi: 10.1111/j.1349-7006.2009.01082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamphues C, Al-Abadi N, Durr A, Bova R, Klauschen F, Stenzinger A, et al. DNA index is a strong predictive marker in intra-hepatic cholangiocarcinoma: the results of a five-year prospective study. Surg Today. 2014;44:1336–42. doi: 10.1007/s00595-013-0701-7. [DOI] [PubMed] [Google Scholar]
- 7.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fortini P, Ferretti C, Dogliotti E. The response to DNA damage during differentiation: pathways and consequences. Mutat Res. 2013;743–744:160–8. doi: 10.1016/j.mrfmmm.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 9.Matsuda Y, Wakai T, Kubota M, Osawa M, Takamura M, Yamagiwa S, et al. DNA damage sensor gamma -H2AX is increased in preneoplastic lesions of hepatocellular carcinoma. Sci World J. 2013;2013:597095. doi: 10.1155/2013/597095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sai S, Wakai T, Vares G, Yamada S, Kamijo T, Kamada T, et al. Combination of carbon ion beam and gemcitabine causes irreparable DNA damage and death of radioresistant pancreatic cancer stem-like cells in vitro and in vivo. Oncotarget. 2015;6:5517–35. doi: 10.18632/oncotarget.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takabayashi H, Wakai T, Ajioka Y, Korita PV, Yamaguchi N. Alteration of the DNA damage response in colorectal tumor progression. Hum Pathol. 2013;44:1038–46. doi: 10.1016/j.humpath.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 12.Wakai T, Shirai Y, Sakata J, Korita PV, Ajioka Y, Hatakeyama K. Early DNA damage response in residual carcinoma in situ at ductal stumps and local recurrence in patients undergoing resection for extrahepatic cholangiocarcinoma. J Hepatobiliary Pancreat Sci. 2013;20:362–9. doi: 10.1007/s00534-012-0539-1. [DOI] [PubMed] [Google Scholar]
- 13.Wakai T, Shirai Y, Sakata J, Korita PV, Matsuda Y, Takamura M, et al. Alteration of p53-binding protein 1 expression as a risk factor for local recurrence in patients undergoing resection for extrahepatic cholangiocarcinoma. Int J Oncol. 2011;38:1227–36. doi: 10.3892/ijo.2011.959. [DOI] [PubMed] [Google Scholar]
- 14.Nagahashi M, Ajioka Y, Lang I, Szentirmay Z, Kasler M, Nakadaira H, et al. Genetic changes of p53, K-ras, and microsatellite instability in gallbladder carcinoma in high-incidence areas of Japan and Hungary. World J Gastroenterol. 2008;14:70–5. doi: 10.3748/wjg.14.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Curtin NJ. Inhibiting the DNA damage response as a therapeutic manoeuvre in cancer. Br J Pharmacol. 2013;169:1745–65. doi: 10.1111/bph.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawanishi S, Hiraku Y, Pinlaor S, Ma N. Oxidative and nitrative DNA damage in animals and patients with inflammatory diseases in relation to inflammation-related carcinogenesis. Biol Chem. 2006;387:365–72. doi: 10.1515/BC.2006.049. [DOI] [PubMed] [Google Scholar]
- 17.Yang SF, Chang CW, Wei RJ, Shiue YL, Wang SN, Yeh YT. Involvement of DNA damage response pathways in hepatocellular carcinoma. Biomed Res Int. 2014;2014:153867. doi: 10.1155/2014/153867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–50. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 19.Spiegel S, Milstien S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat Rev Immunol. 2011;11:403–15. doi: 10.1038/nri2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takabe K, Paugh SW, Milstien S, Spiegel S. “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev. 2008;60:181–95. doi: 10.1124/pr.107.07113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagahashi M, Hait NC, Maceyka M, Avni D, Takabe K, Milstien S, et al. Sphingosine-1-phosphate in chronic intestinal inflammation and cancer. Adv Biol Regul. 2014;54:112–20. doi: 10.1016/j.jbior.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagahashi M, Takabe K, Terracina KP, Soma D, Hirose Y, Kobayashi T, et al. Sphingosine-1-phosphate transporters as targets for cancer therapy. Biomed Res Int. 2014;2014:651727. doi: 10.1155/2014/651727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carroll B, Donaldson JC, Obeid L. Sphingolipids in the DNA damage response. Adv Biol Regul. 2015;58:38–52. doi: 10.1016/j.jbior.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chua MW, Lin MZ, Martin JL, Baxter RC. Involvement of the insulin-like growth factor binding proteins in the cancer cell response to DNA damage. J Cell Commun Signal. 2015;9:167–76. doi: 10.1007/s12079-015-0262-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagahashi M, Takabe K, Liu R, Peng K, Wang X, Wang Y, et al. Conjugated bile acid-activated S1P receptor 2 is a key regulator of sphingosine kinase 2 and hepatic gene expression. Hepatology. 2015;61:1216–26. doi: 10.1002/hep.27592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osawa Y, Nagaki M, Banno Y, Nozawa Y, Moriwaki H, Nakashima S. Sphingosine kinase regulates hepatoma cell differentiation: roles of hepatocyte nuclear factor and retinoid receptor. Biochem Biophys Res Commun. 2001;286:673–7. doi: 10.1006/bbrc.2001.5451. [DOI] [PubMed] [Google Scholar]
- 27.Bertram C, Hass R. Cellular responses to reactive oxygen species-induced DNA damage and aging. Biol Chem. 2008;389:211–20. doi: 10.1515/BC.2008.031. [DOI] [PubMed] [Google Scholar]
- 28.Löbrich M, Jeggo PA. The impact of a negligent G2/M check-point on genomic instability and cancer induction. Nat Rev Cancer. 2007;7:861–9. doi: 10.1038/nrc2248. [DOI] [PubMed] [Google Scholar]
- 29.Slupphaug G, Kavli B, Krokan HE. The interacting pathways for prevention and repair of oxidative DNA damage. Mutat Res. 2003;531:231–51. doi: 10.1016/j.mrfmmm.2003.06.002. [DOI] [PubMed] [Google Scholar]
- 30.Yamaguchi S, Ogata H, Katsumata D, Nakajima M, Fujii T, Tsutsumi S, et al. MUTYH-associated colorectal cancer and adenomatous polyposis. Surg Today. 2014;44:593–600. doi: 10.1007/s00595-013-0592-7. [DOI] [PubMed] [Google Scholar]
- 31.Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11–Rad50–Nbs1 complex. Science. 2005;308:551–4. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 32.Bristow RG, Ozcelik H, Jalali F, Chan N, Vesprini D. Homologous recombination and prostate cancer: a model for novel DNA repair targets and therapies. Radiother Oncol. 2007;83:220–30. doi: 10.1016/j.radonc.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 33.Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–96. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 34.Xiao H, Tong R, Ding C, Lv Z, Du C, Peng C, et al. gamma-H2AX promotes hepatocellular carcinoma angiogenesis via EGFR/HIF-1alpha/VEGF pathways under hypoxic condition. Oncotarget. 2015;6:2180–92. doi: 10.18632/oncotarget.2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature. 1991;351:317–20. doi: 10.1038/351317a0. [DOI] [PubMed] [Google Scholar]
- 36.Koike K, Shirakata Y, Yaginuma K, Arii M, Takada S, Nakamura I, et al. Oncogenic potential of hepatitis B virus. Mol Biol Med. 1989;6:151–60. [PubMed] [Google Scholar]
- 37.Takada S, Kaneniwa N, Tsuchida N, Koike K. Cytoplasmic retention of the p53 tumor suppressor gene product is observed in the hepatitis B virus X gene-transfected cells. Oncogene. 1997;15:1895–901. doi: 10.1038/sj.onc.1201369. [DOI] [PubMed] [Google Scholar]
- 38.Prost S, Ford JM, Taylor C, Doig J, Harrison DJ. Hepatitis B x protein inhibits p53-dependent DNA repair in primary mouse hepatocytes. J Biol Chem. 1998;273:33327–32. doi: 10.1074/jbc.273.50.33327. [DOI] [PubMed] [Google Scholar]
- 39.Ogden SK, Lee KC, Barton MC. Hepatitis B viral transactivator HBx alleviates p53-mediated repression of alpha-fetoprotein gene expression. J Biol Chem. 2000;275:27806–14. doi: 10.1074/jbc.M004449200. [DOI] [PubMed] [Google Scholar]
- 40.Cho HK, Cheong KJ, Kim HY, Cheong J. Endoplasmic reticulum stress induced by hepatitis B virus X protein enhances cyclo-oxygenase 2 expression via activating transcription factor 4. Biochem J. 2011;435:431–9. doi: 10.1042/BJ20102071. [DOI] [PubMed] [Google Scholar]
- 41.Waris G, Huh KW, Siddiqui A. Mitochondrially associated hepatitis B virus X protein constitutively activates transcription factors STAT-3 and NF-kappa B via oxidative stress. Mol Cell Biol. 2001;21:7721–30. doi: 10.1128/MCB.21.22.7721-7730.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ha HL, Yu DY. HBx-induced reactive oxygen species activates hepatocellular carcinogenesis via dysregulation of PTEN/Akt pathway. World J Gastroenterol. 2010;16:4932–7. doi: 10.3748/wjg.v16.i39.4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Becker SA, Lee TH, Butel JS, Slagle BL. Hepatitis B virus X protein interferes with cellular DNA repair. J Virol. 1998;72:266–72. doi: 10.1128/jvi.72.1.266-272.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jaitovich-Groisman I, Benlimame N, Slagle BL, Perez MH, Alpert L, Song DJ, et al. Transcriptional regulation of the TFIIH transcription repair components XPB and XPD by the hepatitis B virus x protein in liver cells and transgenic liver tissue. J Biol Chem. 2001;276:14124–32. doi: 10.1074/jbc.M010852200. [DOI] [PubMed] [Google Scholar]
- 45.Schaedler S, Krause J, Himmelsbach K, Carvajal-Yepes M, Lieder F, Klingel K, et al. Hepatitis B virus induces expression of antioxidant response element-regulated genes by activation of Nrf2. J Biol Chem. 2010;285:41074–86. doi: 10.1074/jbc.M110.145862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Srisuttee R, Koh SS, Park EH, Cho IR, Min HJ, Jhun BH, et al. Up-regulation of Foxo4 mediated by hepatitis B virus X protein confers resistance to oxidative stress-induced cell death. Int J Mol Med. 2011;28:255–60. doi: 10.3892/ijmm.2011.699. [DOI] [PubMed] [Google Scholar]
- 47.Matsuda Y, Ichida T. Impact of hepatitis B virus X protein on the DNA damage response during hepatocarcinogenesis. Med Mol Morphol. 2009;42:138–42. doi: 10.1007/s00795-009-0457-8. [DOI] [PubMed] [Google Scholar]
- 48.Wu XY, Qian JJ, Lin Y, Zheng MH. Hepatitis B virus X protein disrupts DNA interstrand crosslinking agent mitomycin C induced ATR dependent intra-S-phase checkpoint. Eur J Cancer. 2008;44:1596–602. doi: 10.1016/j.ejca.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 49.Rakotomalala L, Studach L, Wang WH, Gregori G, Hullinger RL, Andrisani O. Hepatitis B virus X protein increases the Cdt1-to-geminin ratio inducing DNA re-replication and polyploidy. J Biol Chem. 2008;283:28729–40. doi: 10.1074/jbc.M802751200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang WH, Hullinger RL, Andrisani OM. Hepatitis B virus X protein via the p38MAPK pathway induces E2F1 release and ATR kinase activation mediating p53 apoptosis. J Biol Chem. 2008;283:25455–67. doi: 10.1074/jbc.M801934200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–90. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matsuda Y, Sanpei A, Wakai T, Kubota M, Osawa M, Hirose Y, et al. Hepatitis B virus X stimulates redox signaling through activation of ataxia telangiectasia mutated kinase. Int J Clin Exp Pathol. 2014;7:2032–43. [PMC free article] [PubMed] [Google Scholar]
- 53.Niture SK, Jain AK, Jaiswal AK. Antioxidant-induced modification of INrf2 cysteine 151 and PKC-delta-mediated phosphorylation of Nrf2 serine 40 are both required for stabilization and nuclear translocation of Nrf2 and increased drug resistance. J Cell Sci. 2009;122:4452–64. doi: 10.1242/jcs.058537. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54.Lee SE, Yang H, Jeong SI, Jin YH, Park CS, Park YS. Induction of heme oxygenase-1 inhibits cell death in crotonaldehyde-stimulated HepG2 cells via the PKC-delta-p38-Nrf2 pathway. PLoS One. 2012;7:e41676. doi: 10.1371/journal.pone.0041676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kohli A, Shaffer A, Sherman A, Kottilil S. Treatment of hepatitis C: a systematic review. JAMA. 2014;312:631–40. doi: 10.1001/jama.2014.7085. [DOI] [PubMed] [Google Scholar]
- 56.Schinazi R, Halfon P, Marcellin P, Asselah T. HCV direct-acting antiviral agents: the best interferon-free combinations. Liver Int. 2014;34(Suppl 1):69–78. doi: 10.1111/liv.12423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mahmood S, Kawanaka M, Kamei A, Izumi A, Nakata K, Niiyama G, et al. Immunohistochemical evaluation of oxidative stress markers in chronic hepatitis C. Antioxid Redox Signal. 2004;6:19–24. doi: 10.1089/152308604771978318. [DOI] [PubMed] [Google Scholar]
- 58.Horiike S, Kawanishi S, Kaito M, Ma N, Tanaka H, Fujita N, et al. Accumulation of 8-nitroguanine in the liver of patients with chronic hepatitis C. J Hepatol. 2005;43:403–10. doi: 10.1016/j.jhep.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 59.Fujita N, Horiike S, Sugimoto R, Tanaka H, Iwasa M, Kobayashi Y, et al. Hepatic oxidative DNA damage correlates with iron overload in chronic hepatitis C patients. Free Radic Biol Med. 2007;42:353–62. doi: 10.1016/j.freeradbiomed.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 60.Fujita N, Sugimoto R, Ma N, Tanaka H, Iwasa M, Kobayashi Y, et al. Comparison of hepatic oxidative DNA damage in patients with chronic hepatitis B and C. J Viral Hepat. 2008;15:498–507. doi: 10.1111/j.1365-2893.2008.00972.x. [DOI] [PubMed] [Google Scholar]
- 61.Cardin R, Piciocchi M, Sinigaglia A, Lavezzo E, Bortolami M, Kotsafti A, et al. Oxidative DNA damage correlates with cell immortalization and mir-92 expression in hepatocellular carcinoma. BMC Cancer. 2012;12:177. doi: 10.1186/1471-2407-12-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chuma M, Hige S, Nakanishi M, Ogawa K, Natsuizaka M, Yamamoto Y, et al. 8-Hydroxy-2′-deoxy-guanosine is a risk factor for development of hepatocellular carcinoma in patients with chronic hepatitis C virus infection. J Gastroenterol Hepatol. 2008;23:1431–6. doi: 10.1111/j.1440-1746.2008.05502.x. [DOI] [PubMed] [Google Scholar]
- 63.Machida K, McNamara G, Cheng KT, Huang J, Wang CH, Comai L, et al. Hepatitis C virus inhibits DNA damage repair through reactive oxygen and nitrogen species and by interfering with the ATM-NBS1/Mre11/Rad50 DNA repair pathway in monocytes and hepatocytes. J Immunol. 2010;185:6985–98. doi: 10.4049/jimmunol.1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Okuda M, Li K, Beard MR, Showalter LA, Scholle F, Lemon SM, et al. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology. 2002;122:366–75. doi: 10.1053/gast.2002.30983. [DOI] [PubMed] [Google Scholar]
- 65.Korenaga M, Wang T, Li Y, Showalter LA, Chan T, Sun J, et al. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J Biol Chem. 2005;280:37481–8. doi: 10.1074/jbc.M506412200. [DOI] [PubMed] [Google Scholar]
- 66.Miura K, Taura K, Kodama Y, Schnabl B, Brenner DA. Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology. 2008;48:1420–9. doi: 10.1002/hep.22486. [DOI] [PubMed] [Google Scholar]
- 67.Wang AG, Lee DS, Moon HB, Kim JM, Cho KH, Choi SH, et al. Non-structural 5A protein of hepatitis C virus induces a range of liver pathology in transgenic mice. J Pathol. 2009;219:253–62. doi: 10.1002/path.2592. [DOI] [PubMed] [Google Scholar]
- 68.Lai CK, Jeng KS, Machida K, Cheng YS, Lai MM. Hepatitis C virus NS3/4A protein interacts with ATM, impairs DNA repair and enhances sensitivity to ionizing radiation. Virology. 2008;370:295–309. doi: 10.1016/j.virol.2007.08.037. [DOI] [PubMed] [Google Scholar]
- 69.Smirnova IS, Aksenov ND, Kashuba EV, Payakurel P, Grabovetsky VV, Zaberezhny AD, et al. Hepatitis C virus core protein transforms murine fibroblasts by promoting genomic instability. Cell Oncol. 2006;28:177–90. doi: 10.1155/2006/864648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Majumder M, Ghosh AK, Steele R, Ray R, Ray RB. Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J Virol. 2001;75:1401–7. doi: 10.1128/JVI.75.3.1401-1407.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nishimura T, Kohara M, Izumi K, Kasama Y, Hirata Y, Huang Y, et al. Hepatitis C virus impairs p53 via persistent overexpression of 3beta-hydroxysterol Delta24-reductase. J Biol Chem. 2009;284:36442–52. doi: 10.1074/jbc.M109.043232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 2010;51:1820–32. doi: 10.1002/hep.23594. [DOI] [PubMed] [Google Scholar]
- 73.Matsuda Y, Wakai T, Hirose Y, Osawa M, Fujimaki S, Kubota M. p27 Is a critical prognostic biomarker in non-alcoholic steatohepatitis-related hepatocellular carcinoma. Int J Mol Sci. 2013;14:23499–515. doi: 10.3390/ijms141223499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tessari P, Coracina A, Cosma A, Tiengo A. Hepatic lipid metabolism and non-alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. 2009;19:291–302. doi: 10.1016/j.numecd.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 75.Daugherity EK, Balmus G, Al Saei A, Moore ES, Abi Abdallah D, Rogers AB, et al. The DNA damage checkpoint protein ATM promotes hepatocellular apoptosis and fibrosis in a mouse model of non-alcoholic fatty liver disease. Cell Cycle. 2012;11:1918–28. doi: 10.4161/cc.20259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marcolin E, Forgiarini LF, Rodrigues G, Tieppo J, Borghetti GS, Bassani VL, et al. Quercetin decreases liver damage in mice with non-alcoholic steatohepatitis. Basic Clin Pharmacol Toxicol. 2013;112:385–91. doi: 10.1111/bcpt.12049. [DOI] [PubMed] [Google Scholar]
- 77.Takaki A, Kawai D, Yamamoto K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH) Int J Mol Sci. 2013;14:20704–28. doi: 10.3390/ijms141020704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–46. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- 79.Gentric G, Maillet V, Paradis V, Couton D, L’Hermitte A, Panasyuk G, et al. Oxidative stress promotes pathologic polyploidization in nonalcoholic fatty liver disease. J Clin Invest. 2015;125:981–92. doi: 10.1172/JCI73957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lieber CS. CYP2E1: from ASH to NASH. Hepatol Res. 2004;28:1–11. doi: 10.1016/j.hepres.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 81.Weltman MD, Farrell GC, Hall P, Ingelman-Sundberg M, Liddle C. Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology. 1998;27:128–33. doi: 10.1002/hep.510270121. [DOI] [PubMed] [Google Scholar]
- 82.Chalasani N, Gorski JC, Asghar MS, Asghar A, Foresman B, Hall SD, et al. Hepatic cytochrome P450 2E1 activity in non-diabetic patients with nonalcoholic steatohepatitis. Hepatology. 2003;37:544–50. doi: 10.1053/jhep.2003.50095. [DOI] [PubMed] [Google Scholar]
- 83.Seki S, Kitada T, Sakaguchi H. Clinicopathological significance of oxidative cellular damage in non-alcoholic fatty liver diseases. Hepatol Res. 2005;33:132–4. doi: 10.1016/j.hepres.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 84.Nomoto K, Tsuneyama K, Takahashi H, Murai Y, Takano Y. Cytoplasmic fine granular expression of 8-hydroxydeoxyguanosine reflects early mitochondrial oxidative DNA damage in non-alcoholic fatty liver disease. Appl Immunohistochem Mol Morphol. 2008;16:71–5. doi: 10.1097/PAI.0b013e31803156d5. [DOI] [PubMed] [Google Scholar]
- 85.Tanaka S, Miyanishi K, Kobune M, Kawano Y, Hoki T, Kubo T, et al. Increased hepatic oxidative DNA damage in patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. J Gastroenterol. 2013;48:1249–58. doi: 10.1007/s00535-012-0739-0. [DOI] [PubMed] [Google Scholar]
- 86.Fujita N, Miyachi H, Tanaka H, Takeo M, Nakagawa N, Kobayashi Y, et al. Iron overload is associated with hepatic oxidative damage to DNA in nonalcoholic steatohepatitis. Cancer Epidemiol Biomarkers Prev. 2009;18:424–32. doi: 10.1158/1055-9965.EPI-08-0725. [DOI] [PubMed] [Google Scholar]
- 87.Olivera A, Spiegel S. Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature. 1993;365:557–60. doi: 10.1038/365557a0. [DOI] [PubMed] [Google Scholar]
- 88.Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat Rev Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- 89.Nagahashi M, Ramachandran S, Kim EY, Allegood JC, Rashid OM, Yamada A, et al. Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res. 2012;72:726–35. doi: 10.1158/0008-5472.CAN-11-2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aoyagi T, Nagahashi M, Yamada A, Takabe K. The role of sphingosine-1-phosphate in breast cancer tumor-induced lymphangiogenesis. Lymphat Res Biol. 2012;10:97–106. doi: 10.1089/lrb.2012.0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lacana E, Maceyka M, Milstien S, Spiegel S. Cloning and characterization of a protein kinase A anchoring protein (AKAP)-related protein that interacts with and regulates sphingosine kinase 1 activity. J Biol Chem. 2002;277:32947–53. doi: 10.1074/jbc.M202841200. [DOI] [PubMed] [Google Scholar]
- 92.Liu H, Toman RE, Goparaju SK, Maceyka M, Nava VE, Sankala H, et al. Sphingosine kinase type 2 is a putative BH3-only protein that induces apoptosis. J Biol Chem. 2003;278:40330–6. doi: 10.1074/jbc.M304455200. [DOI] [PubMed] [Google Scholar]
- 93.Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465:1084–8. doi: 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012;22:50–60. doi: 10.1016/j.tcb.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Takabe K, Kim RH, Allegood JC, Mitra P, Ramachandran S, Nagahashi M, et al. Estradiol induces export of sphingosine 1-phosphate from breast cancer cells via ABCC1 and ABCG2. J Biol Chem. 2010;285:10477–86. doi: 10.1074/jbc.M109.064162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009;325:1254–7. doi: 10.1126/science.1176709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Strub GM, Paillard M, Liang J, Gomez L, Allegood JC, Hait NC, et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2011;25:600–12. doi: 10.1096/fj.10-167502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sato K, Malchinkhuu E, Horiuchi Y, Mogi C, Tomura H, Tosaka M, et al. Critical role of ABCA1 transporter in sphingosine 1-phosphate release from astrocytes. J Neurochem. 2007;103:2610–9. doi: 10.1111/j.1471-4159.2007.04958.x. [DOI] [PubMed] [Google Scholar]
- 99.Mitra P, Oskeritzian CA, Payne SG, Beaven MA, Milstien S, Spiegel S. Role of ABCC1 in export of sphingosine-1-phosphate from mast cells. Proc Natl Acad Sci USA. 2006;103:16394–9. doi: 10.1073/pnas.0603734103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim RH, Takabe K, Milstien S, Spiegel S. Export and functions of sphingosine-1-phosphate. Biochim Biophys Acta. 2009;1791:692–6. doi: 10.1016/j.bbalip.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kawahara A, Nishi T, Hisano Y, Fukui H, Yamaguchi A, Mochizuki N. The sphingolipid transporter spns2 functions in migration of zebrafish myocardial precursors. Science. 2009;323:524–7. doi: 10.1126/science.1167449. [DOI] [PubMed] [Google Scholar]
- 102.Hisano Y, Kobayashi N, Kawahara A, Yamaguchi A, Nishi T. The sphingosine 1-phosphate transporter, SPNS2, functions as a transporter of the phosphorylated form of the immunomodulating agent FTY720. J Biol Chem. 2011;286:1758–66. doi: 10.1074/jbc.M110.171116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nagahashi M, Kim EY, Yamada A, Ramachandran S, Allegood JC, Hait NC, et al. Spns2, a transporter of phosphorylated sphingoid bases, regulates their blood and lymph levels and the lymphatic network. FASEB J. 2013;27:1001–11. doi: 10.1096/fj.12-219618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Van Brocklyn JR, Jackson CA, Pearl DK, Kotur MS, Snyder PJ, Prior TW. Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J Neuropathol Exp Neurol. 2005;64:695–705. doi: 10.1097/01.jnen.0000175329.59092.2c. [DOI] [PubMed] [Google Scholar]
- 105.Ruckhaberle E, Rody A, Engels K, Gaetje R, von Minckwitz G, Schiffmann S, et al. Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res Treat. 2008;112:41–52. doi: 10.1007/s10549-007-9836-9. [DOI] [PubMed] [Google Scholar]
- 106.Li W, Yu CP, Xia JT, Zhang L, Weng GX, Zheng HQ, et al. Sphingosine kinase 1 is associated with gastric cancer progression and poor survival of patients. Clin Cancer Res. 2009;15:1393–9. doi: 10.1158/1078-0432.CCR-08-1158. [DOI] [PubMed] [Google Scholar]
- 107.Liu SQ, Su YJ, Qin MB, Mao YB, Huang JA, Tang GD. Sphingosine kinase 1 promotes tumor progression and confers malignancy phenotypes of colon cancer by regulating the focal adhesion kinase pathway and adhesion molecules. Int J Oncol. 2013;42:617–26. doi: 10.3892/ijo.2012.1733. [DOI] [PubMed] [Google Scholar]
- 108.Pyne S, Edwards J, Ohotski J, Pyne NJ. Sphingosine 1-phosphate receptors and sphingosine kinase 1: novel biomarkers for clinical prognosis in breast, prostate, and hematological cancers. Front Oncol. 2012;2:168. doi: 10.3389/fonc.2012.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Taha TA, Osta W, Kozhaya L, Bielawski J, Johnson KR, Gillanders WE, et al. Down-regulation of sphingosine kinase-1 by DNA damage: dependence on proteases and p53. J Biol Chem. 2004;279:20546–54. doi: 10.1074/jbc.M401259200. [DOI] [PubMed] [Google Scholar]
- 110.Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell. 2013;23:107–20. doi: 10.1016/j.ccr.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Huang WC, Nagahashi M, Terracina KP, Takabe K. Emerging role of sphingosine-1-phosphate in inflammation, cancer, and lymphangiogenesis. Biomolecules. 2013:3. doi: 10.3390/biom3030408. doi:10.3390/biom3030408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Koganti S, Hui-Yuen J, McAllister S, Gardner B, Grasser F, Palendira U, et al. STAT3 interrupts ATR-Chk1 signaling to allow oncovirus-mediated cell proliferation. Proc Natl Acad Sci USA. 2014;111:4946–51. doi: 10.1073/pnas.1400683111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Martin JL, Lin MZ, McGowan EM, Baxter RC. Potentiation of growth factor signaling by insulin-like growth factor-binding protein-3 in breast epithelial cells requires sphingosine kinase activity. J Biol Chem. 2009;284:25542–52. doi: 10.1074/jbc.M109.007120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Martin JL, de Silva HC, Lin MZ, Scott CD, Baxter RC. Inhibition of insulin-like growth factor-binding protein-3 signaling through sphingosine kinase-1 sensitizes triple-negative breast cancer cells to EGF receptor blockade. Mol Cancer Ther. 2014;13:316–28. doi: 10.1158/1535-7163.MCT-13-0367. [DOI] [PubMed] [Google Scholar]
- 115.Sukocheva O, Wadham C, Holmes A, Albanese N, Verrier E, Feng F, et al. Estrogen transactivates EGFR via the sphingosine 1-phosphate receptor Edg-3: the role of sphingosine kinase-1. J Cell Biol. 2006;173:301–10. doi: 10.1083/jcb.200506033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S, et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell. 2010;18:436–47. doi: 10.1016/j.ccr.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fyrst H, Saba JD. An update on sphingosine-1-phosphate and other sphingolipid mediators. Nat Chem Biol. 2010;6:489–97. doi: 10.1038/nchembio.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol. 2012;30:69–94. doi: 10.1146/annurev-immunol-020711-075011. [DOI] [PubMed] [Google Scholar]
- 119.Kumar A, Oskouian B, Fyrst H, Zhang M, Paris F, Saba JD. S1P lyase regulates DNA damage responses through a novel sphingolipid feedback mechanism. Cell Death Dis. 2011;2:e119. doi: 10.1038/cddis.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]