

Abstract
An accurate diagnosis is an integral component of patient care for children with rare genetic disease. Recent advances in sequencing, in particular whole‐exome sequencing (WES), are identifying the genetic basis of disease for 25–40% of patients. The diagnostic rate is probably influenced by when in the diagnostic process WES is used. The Finding Of Rare Disease GEnes (FORGE) Canada project was a nation‐wide effort to identify mutations for childhood‐onset disorders using WES. Most children enrolled in the FORGE project were toward the end of the diagnostic odyssey. The two primary outcomes of FORGE were novel gene discovery and the identification of mutations in genes known to cause disease. In the latter instance, WES identified mutations in known disease genes for 105 of 362 families studied (29%), thereby informing the impact of WES in the setting of the diagnostic odyssey. Our analysis of this dataset showed that these known disease genes were not identified prior to WES enrollment for two key reasons: genetic heterogeneity associated with a clinical diagnosis and atypical presentation of known, clinically recognized diseases. What is becoming increasingly clear is that WES will be paradigm altering for patients and families with rare genetic diseases.
Keywords: clinical exome, FORGE Canada Consortium, rare diseases, whole‐exome sequencing
Rare genetic diseases affect at least 1 in 50 individuals (1, http://orphanet.net). The total number of these diseases is estimated to be 6000–7000 (2, http://www.omim.org) and while each is individually rare, together these genetic conditions contribute significantly to morbidity, mortality, and healthcare costs. Estimates suggest that up to 50% of patients with a rare genetic disease never receive a diagnosis 3. Patients without a diagnosis often embark on a diagnostic odyssey that includes multiple specialist consultations, imaging studies [e.g. magnetic resonance imaging (MRI), skeletal survey], invasive investigations (e.g. skin and muscle biopsy), and other laboratory and genetic tests. The diagnostic odyssey is by definition a slow, costly venture, and for many is ultimately disappointing when a diagnosis is not reached 4. A survey of patients with rare diseases demonstrated that for 25% of participants the time to diagnosis was extensive, ranging from 5 to 30 years, and during that time 40% received an incorrect diagnosis 5. As many as a third of these patients with rare diseases incurred inappropriate care for their eventual diagnosis, including those who underwent unnecessary surgical procedures 5. Providing a molecularly confirmed diagnosis, in a timely manner, for children and adults with rare diseases shortens the diagnostic odyssey, improves disease management, including targeted treatments and surveillance for later‐onset comorbidities for a subset of patients, and informs genetic counseling with respect to recurrence risks and prenatal diagnosis options for families.
Traditional diagnostic assessment in clinical genetics
Currently, an individual with a suspected rare genetic disease may have genetic testing performed on a gene‐by‐gene basis or by a gene‐panel (simultaneous analysis of two or more candidate genes) approach. Single gene testing is traditionally performed by Sanger sequencing; the gold standard that has been used for molecular diagnosis for two decades. This is often performed sequentially; if no mutations are identified in a proposed candidate gene, additional candidate gene(s) may subsequently be selected for testing by the physician. The sequencing of a single gene may take several weeks to months to complete and costs anywhere from one to a few thousand US dollars in a commercial diagnostic laboratory. Genetic testing performed using a gene‐panel approach is an advantageous strategy when more than one gene can cause the clinical presentation. Though the data is limited, the rate of molecular diagnosis for patients with rare diseases using traditional, comprehensive clinical genetics evaluation and targeted genetic testing (including karyotypes, chromosomal microarray analysis, single gene tests, gene‐panel tests, and specialized genetic biochemical laboratory tests) was 46% in one recent study that analyzed a cohort of 500 unselected consecutive patients referred to the genetics department at an American tertiary medical center 3. Of those diagnosed, 72% were diagnosed on their first visit, implying that subsequent genetic testing had diminishing returns using these traditional approaches and that next‐generation sequencing (NGS) (including WES and WGS) may be economically beneficial following the first clinical visit.
Impact of next‐generation sequencing on rare disease diagnosis
The advent of NGS technologies has created a paradigm shift in our approach to both the discovery of new disease genes and the timely diagnosis of genetic disease. Whole‐genome sequencing (WGS) refers to the sequencing of nearly the entire human genome simultaneously. WGS data can typically be generated in less than a month for approximately $2000 on the latest platforms. However, the assembly of the genome is computationally laborious and most of the non‐coding sequence is difficult to interpret. Whole‐exome sequencing (WES) can be completed in a similar timeframe and interrogates approximately 95% of the coding region of the genome, comprising ∼20,000 genes. Diseases that were previously intractable to gene identification given their rarity, clinical and genetic heterogeneity, and paucity of multiplex families have shown tremendous discovery success using WES. As a result, the pace of novel disease gene discoveries has increased dramatically over the last few years 6. However, it quickly became apparent that WES was also very useful for identifying mutations in known genes in these research patients 7. In one of the first studies to examine the success rate of WES in a clinical setting, a probable genetic diagnosis was established in 6 of the 12 (50%) patients recruited with a broad range of phenotypic presentations 8. A large‐scale research study of pediatric‐onset neurodevelopmental disease identified a known disease‐causing gene in 8% of patients who had the most relevant genes excluded prior to WES 9. A retrospective review of 28 families in the FORGE Canada research study with cerebellar ataxia showed a diagnostic success rate of 46% using WES 10, and a review of 9 families in FORGE with severe and early‐onset epilepsy demonstrated a diagnosis in 7, possibly 8, families 11. Similarly, there is literature to support the role of WES in patients with moderate‐severe intellectual disability or neurodevelopmental disorders, with success rates ranging from 16 to 45% 12, 13, 14, 15. Soden et al. undertook rapid WGS for patients with high‐acuity illness, defined as symptomatic at or shortly after birth requiring admittance to the neonatal or pediatric intensive care units 15. An impressive 73% (11 of 15) of families with acutely ill children were diagnosed using this approach, which may be a reflection of both the likelihood of a monogenic presentation with such an early and severe presentation as well as decreased prior testing. The success of WES and WGS in providing diagnoses to patients in a research setting is enabling the translation of this technology to the clinic, with the expectation of increasing the diagnostic yield over traditional genetic testing approaches and thereby providing answers to families who had previously gone without a molecular diagnosis.
Over the past 2–3 years, several large studies have demonstrated the diagnostic utility of WES for a large number of patients analyzed in clinical diagnostic laboratories 16, 17, 18. The first of these was reported by the Baylor College of Medical Genetics Laboratory which demonstrated a 25% molecular diagnostic rate for 250 probands with varying indications (although 80% had a neurologic phenotype) 16. Interestingly, when they increased the number of patients to 2000, the diagnostic rate remained 25% 17. The Lee et al study of 814 patients who underwent WES in a clinical diagnostic laboratory reported a similar diagnostic rate of 22–31% 18. This range probably reflects a number of factors including analysis strategy [affected singleton versus trio (proband and parents)], family history, type of disorder, age at presentation, and acuity of the clinical presentation, which speaks to the likelihood the disorder is caused by a single gene. Lastly, where in the diagnostic evaluation the WES was utilized also likely plays a role in the success rate.
In the setting of limited resources, the question of singleton vs trio sequencing is an important one. The analysis of the largest reported cohort of 2000 patients was primarily as singleton WES 17. Approximately 400 patients in the cohort of 814 underwent trio analysis and this was associated with the higher diagnostic rate of 31% 18. The higher diagnostic rate for trios is perhaps not surprising given that de novo mutations are only readily detected by this approach and appear to be enriched in neurodevelopmental cohorts, a common indication for referral to these clinical laboratories. However, the reported diagnostic rates of 22–25% for singleton analysis speaks to the considerable diagnostic utility of WES even in this sequencing paradigm 16, 17, 18. In addition, other approaches may be just as successful. Analysis of sibling pairs and one parent for autosomal recessive conditions thought to be secondary to compound heterozygous mutations will enable rapid analysis which includes phasing of the variants to determine if the variants are in cis or trans. For consanguineous families, analysis of three family members is probably not necessary, although the addition of a second affected sibling is useful for decreasing shared variants when the inbreeding coefficient is high. Considerations regarding study design must also include cost and the urgency of the diagnosis, which may be related to possible treatment or management implications that might be missed or delayed if secondary analysis of parental data is required to reach a conclusive answer.
There is also now evidence that supports the diagnostic advantage of WES over single‐gene and panel‐based approaches for several referral indications 19. In the study by Neveling et al. the diagnostic rate of patients who received traditional Sanger‐based testing based on candidate genes in 2011 was compared with patients who instead underwent WES the following year with a filtered analysis of large gene panels based on the indication for referral. The diagnostic rates were higher for patients after WES for clinical presentations such as blindness (25% vs 52%), deafness (10% vs 44%), movement disorders (5% vs 20%) and mitochondrial disorders (11% vs 15%) 19. This success of WES over traditional approaches may reflect extensive genetic heterogeneity for some of these clinical presentations and thus the number of genes assayed using the respective approaches. For disease presentations, where a limited number of genes are known to cause the disorder, targeted panel NGS approaches may provide a more comprehensive assessment.
The range of diagnostic success is also influenced by when in the diagnostic evaluation WES occurs. For the large studies reported to date 16, 17, 18, such detailed information is not available; however, it is very likely that at a minimum first line testing (karyotype, chromosomal microarray, some single gene testing as indicated) had been completed for these patients prior to WES. Whereas Shashi et al. have hypothesized that use of WES and WGS may be economically beneficial when used early in the diagnostic evaluation of a patient 3, it has not yet been determined how the diagnostic rate will be influenced by the timing at which WES is performed. The recent completion of Canada's nation‐wide FORGE (Finding Of Rare disease Genes) project allowed us to retrospectively investigate the diagnostic utility of WES after standard‐of‐care genetic testing in a cohort of children from Canada with unexplained rare diseases.
Evaluation of the utility of WES for patients near the end of the diagnostic odyssey; FORGE Canada Consortium
We investigated the diagnostic yield of WES for a cohort of patients who had already received standard‐of‐care genetics evaluation and diagnostic testing in Canada and were suspected to have a genetic disorder by the referring physician. Initial evaluation included single gene testing, occasional small gene panels, and chromosome microarray. In addition, we examined why these patients were not diagnosed after standard‐of‐care assessment. To do this we performed a retrospective study of the patients enrolled in the FORGE Canada project who received a molecular diagnosis in a known disease gene. FORGE was a 2‐year, pan‐Canadian initiative to rapidly identify novel genes responsible for a wide spectrum of pediatric‐onset disorders present in the Canadian population 20. The project ascertained >500 children from 362 families with rare diseases from across Canada and utilized WES to provide a definitive molecular diagnosis in either a known disease‐causing gene (105 families) or in a gene that represented a novel discovery (83 families) (Fig. 1); the FORGE experience with novel gene discovery has been reviewed elsewhere 20. The study was approved by Research Ethics Boards from all participating sites and informed consent was obtained from all families. Our consent process informed families that we would not systematically look at WES data for incidental findings, but that if we observed a convincing mutation in a gene that was deemed to be medically actionable in childhood we would return the result to the family.
Figure 1.
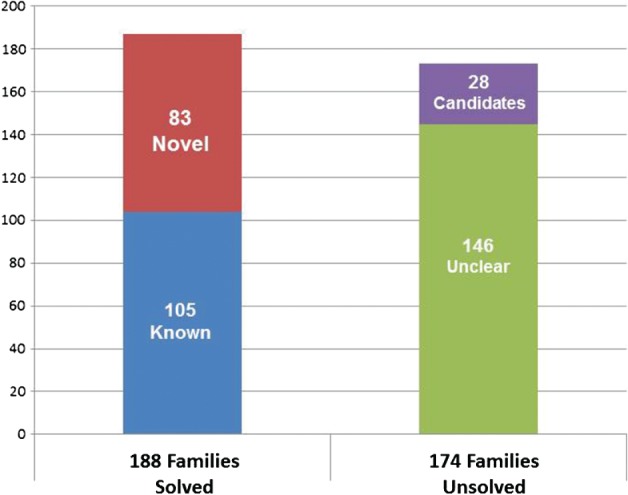
Outcomes of the 362 families studied using whole‐exome sequencing (WES) as part of the Finding Of Rare Disease GEnes (FORGE) Canada research study. Overall, 188 of the 362 (51.9%) families received a definitive molecular diagnosis, either in a gene previously known to cause a similar disease (known) or in a gene that had not been previously associated with the family's disease (novel). Of the 174 (48.1%) of families that remain unsolved, 28 have single‐surviving candidates for which we are gathering additional evidence to support pathogenicity.
Patient selection
Patients enrolled in the FORGE project were ascertained by physicians, mainly Medical Geneticists, from 21 participating academic centers across the country. In almost all patients, appropriate clinical and molecular investigations had been completed prior to enrollment to exclude known causes of the patient's clinical presentation (standard‐of‐care for the respective province, including chromosomal microarray and targeted gene testing when available). Access to clinical testing of specific single genes or panels of genes is dependent on the tests available within a province and varies considerably (e.g. few to 200 tests). There is also marked variability across Canada as to the resources available to fund out‐of‐country genetic testing. However, the vast majority of patients had already experienced a protracted diagnostic odyssey (several years to more than two decades) prior to entering FORGE. Patients were accepted either with a recognized clinical diagnosis (e.g. Dubowitz syndrome, etc.) or with a description of their clinical presentation if the diagnosis was unknown (e.g. microcephaly, short stature, sparse hair, intellectual disability).
WES data generation and analysis
Patients underwent WES at one of three centralized Genome Canada Science and Technology Innovation Centres (STICs): McGill University and Genome Quebec Innovation Centre (Montreal), The Centre for Applied Genomics (Toronto), or The Genome Sciences Centre (Vancouver), with over half of the samples run at the McGill site. Exome target enrichment was performed with the Agilent SureSelect 50 Mb (V3) All Exon Kit; samples were sequenced on the Illumina HiSeq 2000 platform, multiplexing three samples per lane. After removal of duplicate reads, the mean coverage of coding sequence regions ranged from ×70 to ×200. Alignment and variant annotation were performed by the FORGE informatics team at each STIC, using comparable analytical pipelines with publicly available tools and custom scripts as described previously 20. Annotations used included variant functional effect, gene OMIM associations, and metrics for missense single‐nucleotide‐variant pathogenicity (e.g. SIFT (http://sift.jcvi.org), Poly‐Phen (http://genetics.bwh.harvard.edu/pph2), Mutation Taster (http://www.mutationtaster.org/)). Filtering of variants was performed with our internal exome database to identify rare variants (<1%). Potentially deleterious rare variants in known disease genes associated with a phenotype similar to the patient were communicated to the referring physician for genotype‐phenotype evaluation. A definitive outcome for a patient occurred when the variant under consideration was in a gene previously known to cause disease (known gene) and the referring clinician provided feedback that this gene explained the affected individual's phenotype, rendering the patient ‘solved’. Genetic diagnoses were confirmed in clinically certified molecular diagnostic laboratories before the results were used to change patient management. We then reviewed why WES successfully identified these mutations in known genes for patients who had already had a standard‐of‐care diagnostic assessment.
Diagnostic rate; mutations in known disease genes
Of 362 families submitted to FORGE for WES, we identified disease‐causing mutations in known genes for 105 families (29%). The diseases studied were enriched for neurodevelopmental phenotypes and dysmorphic syndromes (Table 1). The success rate ranged from 12% (immunological disorders) to 44% (ciliopathies). When we considered the sequencing and data analysis strategy for each family who had undergone WES, the diagnostic rate in known disease genes ranged from 23% to 34%; 23% of 207 singletons, 32% of 72 sibling pairs, and 34% of 76 families who were either consanguineous or from isolated populations.
Table 1.
Broad phenotypes and associated diagnostic rates in known disease genes of the families studied using WES in FORGE
| Broad phenotype | Total families (N = 362) | Families with known genes (N = 105) | Diagnostic rate (%) |
|---|---|---|---|
| Neurodevelopmental | 98 | 31 | 31.6 |
| Dysmorphic syndromes | 80 | 18 | 22.5 |
| Ocular | 40 | 11 | 27.5 |
| Metabolic | 31 | 12 | 38.7 |
| Neuromuscular | 30 | 7 | 23.3 |
| Ciliopathy | 27 | 12 | 44.4 |
| Congenital malformation syndromes | 19 | 4 | 21.1 |
| Immunological | 17 | 2 | 11.8 |
| Other (isolated cardiac, endocrinology, skeletal dysplasia, connective tissue disorders, mental illness, lung disorder) | 20 | 8 | 40.0 |
FORGE, Finding Of Rare Disease GEnes; WES, whole‐exome sequencing.
Contributors to the diagnostic odyssey
The most common contributing factor for patients not receiving a molecular diagnosis prior to WES was that their disorder was associated with significant genetic heterogeneity (Table 2), which we defined as a disorder with more than three associated genes. It is a fiscal reality of our provincially funded healthcare systems that extensive gene‐by‐gene interrogation for a genetically heterogeneous condition is prohibitively expensive and thus generally not available. As a result, only the most common genes for a particular rare disease presentation tended to be evaluated prior to study enrollment. For example, several patients with a clinical diagnosis of Joubert syndrome were included in this study. There are currently over 20 genes known to be associated with Joubert syndrome 21. Prior to inclusion in this study, each patient had several of the more common genes associated with Joubert syndrome excluded, and for each of these patients a different, but previously known, Joubert syndrome‐associated gene was identified by WES. Given the vast number of clinical presentations associated with significant genetic heterogeneity, WES has remarkable advantages by providing a comprehensive approach.
Table 2.
Explanations for families who received a molecular diagnosis in FORGE using WES who had not been diagnosed during standard‐of‐care clinical genetics evaluation
| Explanation | Number of families (N = 105) |
|---|---|
| Genetic heterogeneity | 49 |
| Atypical presentation | 26 |
| Missed by another method | 9 |
| Gene identified while in the pipeline | 9 |
| Extremely rare condition | 5 |
| Conflation | 4 |
| Limited access to testing | 3 |
FORGE, Finding Of Rare Disease GEnes; WES, whole‐exome sequencing.
The next most common factor contributing to the diagnostic odyssey resolved by WES was that of phenotypic expansion (Table 2). There were 26 patients with an atypical presentation of a known disorder, such that the clinical diagnosis was unrecognizable to the clinician to inform genetic testing of the correct (as identified by WES) gene. This speaks to the challenges inherent in the diagnosis of rare diseases. The progressive nature of many rare diseases means that the clinician will often evaluate a patient early in the course of disease when ‘tell‐tale’ features may not yet be present (e.g. neonate in the NICU), making an early and accurate diagnosis difficult. The large number of atypical diagnoses also highlights how limited our understanding is of the phenotypic spectrum for many rare diseases. For example, mandibulofacial dysostosis and microcephaly (MFDM) syndrome was identified independently three times in the FORGE study. The initial cohort of patients with the classic presentation led to the discovery of the EFTUD2 gene as causative 22 and this was followed by the identification of patients on the mild and severe ends of this emerging syndrome. This suggests that we will not truly appreciate the variability of any rare disease until unbiased approaches, such as WES, have enabled the identification of the complete clinical spectrum.
In nine families a disease‐causing mutation was identified by WES that had been missed by another method of testing (Table 2). Two patients had undergone Sanger sequencing of the candidate gene in clinically accredited laboratories and the mutations had not been reported. In one patient the quality of the sequencing was poor, thus the mutation was not reported as it was presumed to be a sequencing error. In the other patient, the clinical lab re‐designed the test after communication regarding the WES result, and was subsequently able to identify the mutation. Most laboratories quote an error rate of approximately 1%, and our cases likely reflect this value. In two patients only common mutations in the gene were assessed by the clinical laboratory and were negative; the mutations identified by WES were not included in this targeted clinical test. Four patients had prior testing performed in a research laboratory and the results were conveyed as unrevealing. In one family the gene had been ruled out in some members of the family with a similar presentation but not in the patient who underwent WES. The affected members in this family were reasonably assumed to have the same underlying genetic basis for their pulmonary fibrosis; however, the findings by WES were in keeping with genetic heterogeneity in a large family with mutations in more than one gene causing the same phenotypic presentation. For another nine patients (Table 2) the causative gene was unknown at the outset of FORGE but was published while the project was underway, thus there was no clinical testing available prior to inclusion in this study.
Five families were diagnosed with conditions that are exceedingly rare (<1/1,000,000) (Table 2), and the literature relatively sparse, such that most subspecialist physicians would not be expected to encounter a patient with that particular diagnosis in their career. For example, methylmalonate semialdehyde deficiency had only been molecularly diagnosed in two published individuals prior to our identification of mutations in the ALDH6A1 gene in a patient in this study 23.
In four individuals without a clinical diagnosis prior to WES, the patients were thought to have a novel syndrome due to a mutation in a single gene; however, they were in fact found to have a conflated clinical presentation comprising two or more known rare diseases (Table 2). This scenario has been reported previously by Yang et al. in which 4.6% (23 of 504) of those who received a molecular diagnosis had blended clinical presentations resulting from two single gene defects 17. Data analysis is challenging for these types of patients as the phenotype has to be dissected and attributed to mutations in different genes. For example, two siblings from consanguineous parents presented with severe congenital neutropenia and oculocutaneous albinism, and a novel disorder was suspected. WES identified homozygous mutations in G6PC3, which is known to cause severe congenital neutropenia type 4, in both siblings, and one sibling had homozygous mutations in SLC45A2 causing oculocutaneous albinism type 4 (OCA4). The second sibling had one mutation in SLC45A2, and manifested some features of OCA4 24. We expect that the subset of undiagnosed patients who are revealed by WES to have two distinct rare diseases will only rise as analysis of this data continues to improve over time.
Finally, for three patients (Table 2) limited availability of clinical testing anywhere in the world for ultra‐rare conditions at the time of the study meant that WES through FORGE was the best option for these clinicians to identify a molecular diagnosis; with the added potential for novel gene discovery if no pathogenic variant was found in the candidate gene of interest.
Impact on patient management
Rare diseases represent a valuable model for the implementation of personalized health. In addition to a clear diagnosis informing prognosis, recurrence risks, and providing prenatal options, there were six of the 105 families (26%) in this study whose medical management changed dramatically subsequent to a diagnosis through WES; three had their therapy adjusted and three had specific therapy initiated. For example, a child that was originally thought to have infantile myofibromatosis (IM) based on clinical suspicion and pathological findings was undergoing low‐dose chemotherapy to reduce the size of the myofibromas. WES was performed to identify the gene for IM, but instead identified the common causative mutation for fibrodysplasia ossificans progressiva (FOP) in ACVR1 25. Patients with FOP develop heterotopic ossifications secondary to mild trauma, such as intramuscular injections, contributing to poor outcomes. Once the diagnosis was clarified all interventions other than those that were life‐saving were stopped. In another family, two sisters with epilepsy and intellectual disability were diagnosed in their early 30s with autosomal recessive cerebral folate transport deficiency. Treatment for this rare disease with folinic acid has shown very encouraging outcomes if started early in the disease 26. Although the white matter damage was already extensive, folinic acid therapy initiated after diagnosis appears to be reducing seizure frequency and improving the quality of life for these patients. By providing a timely diagnosis we can halt the diagnostic odyssey and for a small number of conditions, prevent long term end‐organ damage when treatment options are available, the benefits of which cannot be overstated.
Clinical translation
Success rates for WES to provide a molecular diagnosis for patients have ranged from 22% to 46% in several large studies now summarizing outcomes for almost 3000 patients 15, 16, 17, 18. It is clear that this is a diagnostically useful test. The wide‐spread clinical translation of genome‐wide NGS approaches to enable personalized medicine for rare diseases requires guidance regarding clinical indications for testing using this approach; when it should occur in the diagnostic evaluation, how to approach incidental findings, and how these complex results should be reported to primary care clinicians and patients/families. Position statements and best practice guidelines are now available for many jurisdictions to address these issues 27, 28, 29, 30. Both the American College of Medical Genetics and Genomics (ACMG) (27; https://www.acmg.net) and the Canadian College of Medical Geneticists (CCMG) (28; http://www.ccmg‐ccgm.org) recommend the consideration of clinical genome‐wide sequencing for affected individuals in which either phenotype or family history raises suspicion of a monogenic rare disease.The ACMG and CCMG both recognize that this technology may be a more practical approach than standard‐of‐care genetic testing in certain scenarios, and both advocate for its use when it might be more efficient (with regards to both time and cost) than alternative approaches. The ACMG further recommends consideration of the use of this technology for the investigation of a fetus with a probably monogenic disorder and specific genetic tests have failed to arrive at a diagnosis 27.
Owing to the nature of genome‐wide sequencing in which genes unrelated to the primary indication of a patient are sequenced, careful consideration must be given to incidental findings. The ACMG 29, the European Society of Human Genetics (ESHG) 30, and most recently the CCMG 28, have all provided perspectives on how such findings might be handled in their jurisdictions. The ACMG has generated a list of genes that it recommends should be screened for deleterious mutations in clinical exome or genome sequencing studies, irrespective of the defined clinical phenotype of the proband 29. Adults would have the right to decline this information, but mutations that are medically actionable in childhood would be reported back to families. In contrast, the European guidelines present these as unsolicited findings, and do not advocate actively searching the data for deleterious variants in genes that are not associated with the primary reason for genetic testing 30. Similarly, the CCMG does not endorse the intentional clinical analysis of disease genes unrelated to the primary indication 28, but if one is inadvertently identified recommends that competent adults be given the option prior to testing to receive such findings. In children, incidental results that reveal a risk for a highly penetrant condition that is medically actionable during childhood should be reported back 28.
The optimal timing for WES in the diagnostic process is still unclear, whether it should be at the first appointment if clinically indicated (e.g. disease presentation associated with significant genetic heterogeneity), second appointment after initial tests normal (e.g. first tier of genes ruled out), or towards the end of the diagnostic odyssey (e.g. extensive, possibly invasive tests have occurred). The reported literature shows diagnostic utility in all of these scenarios and the optimal timing is probably dependent on the particular patient and clinical circumstances. The data from FORGE Canada, which was focused on diagnostic outcomes from patients that were towards the end of the diagnostic odyssey, demonstrates yields as high as 29% for this patient group. These data also show that patients with genetically heterogeneous diseases, those with unknown disorders which may be due to an atypical presentation, and those with ultra‐rare diseases, will benefit immensely from relatively non‐biased clinical testing methods. The irony is that these patients with atypical and ultra‐rare diseases are not diagnosed as such until these technologies are used and mutations are identified, which underscores our current limited understanding of many phenotypic presentations of rare disease. Thus, improved phenotyping, reporting of atypical patients, and sharing of rare variants through databases is essential to improve the diagnosis and management going forward. Indeed, both our data and that of Soden et al. show that a significant proportion, 6% and 8% respectively, of patients will have therapies adjusted or initiated based on molecular results, highlighting the importance of providing every child with a diagnosis such that treatable conditions and windows of opportunity are not missed 15.
It is very likely that in the years to come, clinical genomic sequencing (a process used to determine the sequence of most, if not all, clinically significant genes and its associated interpretation, including bioinformatic analysis and clinical genotype–phenotype correlation) will be performed on an ever increasing number of patients with undiagnosed rare diseases. From the data accrued to date, we know that approximately 70% of those who undergo WES will remain without a molecular diagnosis in a known disease gene. We strongly believe that as clinical translation strategies are implemented in various jurisdictions, it is of utmost importance that the approach includes research opportunities for patients and families to consent to having coded genome‐wide and phenotypic data deposited and stored in national/international databases to assist in interpretation and solving the unsolved patients.
Solving the unsolved
There are a variety of reasons why up to 70% of patients might be unsolved after WES, including incomplete coverage of the exome and genetic mutations elusive to the technology itself [e.g. non‐coding variants, trinucleotide repeats, copy number variants, chromosome rearrangements, oligogenic inheritance, etc.; summarized in 31]. From our experience, however, there are a proportion of these cases in which the disease‐causing variant is in fact within the WES data but for a variety of reasons (not enough information on the variant, variant acting on gene function through novel molecular mechanism, unclear inheritance, etc.) there is insufficient evidence to support a definitive diagnosis. For some of these cases, performing WES on additional family members, including affected relatives or unaffected parents, may be useful. Analysis may also benefit from the use of large‐scale population variant control databases (such as ExAC at the Broad Institute; http://exac.broadinstitute.org); greatly reducing the number of rare variants identified and requiring interpretation.
In addition, there may be value in pursuing additional genomic technologies for those families that remain elusive to diagnosis. In a recent study by Gilissen et al., 50 patients with severe intellectual disability underwent trio WGS following uninformative WES and microarray analysis 32. To their surprise, 21 of the 50 (42%) received diagnoses in known ID genes through analysis for de novo and recessive coding mutations [both single nucleotide variants (SNVs) and copy number variants (CNVs)]. It is unclear if the SNV coding mutations that were identified (13 diagnoses) had been captured or missed in the previously uninformative WES data 32. It is well recognized that WGS offers a more complete coverage of the protein‐coding portion of the genome, but in theory one can expect the majority of these variants will be identified as technology to both capture and sequence the exome improves. Of interest, a total of eight de novo structural variants (five deletions, a tandem duplication, an inter‐chromosomal duplication and one complex inversion/duplication/deletion event), previously undetected by diagnostic microarray analysis, were identified, underlining that even as WES improves a proportion of cases will benefit from WGS.
In addition to diagnosing families with pathogenic mutations in known disease genes, the FORGE Canada project successfully demonstrated that a significant proportion of unexplained patients have pathogenic mutations in novel disease genes. An additional 23% of families studied in FORGE were shown to have pathogenic mutations in novel genes validated by cohort analysis and/or functional studies (reviewed in 20). Despite extensive efforts, this means that 48%, or 173 families, were not diagnosed by WES in FORGE, highlighting a need for both reanalysis of data for variants in recently identified disease‐causing genes and large‐scale data sharing to solve more of these intractable families.
In 28 families studied in the FORGE project, single‐candidate genes were identified but little is known about the function of the gene, rendering cellular characterization and functional studies challenging. For such cases, finding just a single additional unrelated case with a deleterious‐appearing variant in the same gene and overlapping phenotype may provide sufficient evidence to causally implicate the gene or support further functional investigation. However, significant collaborative infrastructure, both within Canada and internationally, to enable sharing of phenotypic and genotypic datasets from patients with undiagnosed rare diseases will be needed to maximize the use of the data generated from such genome‐wide approaches, which up until now have been confined to the institutions that generated and/or analyzed the data. International efforts have made available a standard set of computer readable terms to describe the phenotypic features of a patient with a rare disease [e.g. Human Phenotype Ontology, HPO 33]. Clinical interface software, such as Phenotips (34; https://phenotips.org), have been developed to facilitate deep phenotyping for a patient using HPO terms in less than five minutes and are now being widely used in rare disease research projects such as the successor to FORGE, Care4Rare (http://care4rare.ca/). The associated web‐based matchmaking tool, PhenomeCentral (http://phenomecentral.org), shows contributors information about other phenotypically similar patients with deleterious‐appearing variants in the same candidate gene, and allows these researchers to contact each other when a match is made. Other similar ‘matchmaking’ databases have been developed, such as GeneMatcher (35, http://www.genematcher.com), which connects investigators based on their interest in the same gene, and DECIPHER (36; http://www.decipher.sanger.ac.uk), which allows investigators to search coded phenotypic and genotypic data to identify possible matches. Clinicians and scientists involved in these data sharing initiatives recognize the value in ever larger datasets to enable the identification of second families and thus MatchmakerExchange (http://matchmakerexchange.org) was launched this year to provide a more systematic approach to large‐scale data sharing for rare disease gene discovery through a federated network connecting databases through a common application programming interface (API). Thus far, the API connects PhenomeCentral, GeneMatcher and DECIPHER and thereby avoids the need for the same data to be entered into multiple databases. Such international sharing efforts will advance our knowledge of disease genes and their associated phenotypic spectrum, ultimately improving diagnostic accuracy.
Future perspectives
In the next decade we will witness a paradigm shift in the way we care for patients with rare genetic diseases, addressing a significant gap in the management of individuals with rare diseases. The diagnostic journey for patients will include clinical genomic sequencing, which in the not too distant future will be WGS as costs decrease with new platforms. The accessibility of WGS‐based diagnostics for patients will be central to understanding the complete compendium of human genetic pathology. Strategies and infrastructure should be put in place to facilitate discovery in clinical settings, and to further mine large phenotypic and genomic datasets for disease mechanisms. As we understand the etiology of more rare diseases, it is probable that we will increasingly implicate known genes and that the approach to the completion of the complete Mendelian dataset will be asymptotic. There will be a subset of rare diseases that are due to non‐coding mutations in regulatory regions that may be more readily identified with the imminent use of WGS and RNA sequencing, and to implicate and understand this type of variation will require large‐scale data sharing at an impressive level. Ultimately, such datasets will be instrumental in identifying modifiers of disease, providing insight into phenotypic variability, prognosis, and for a subset of diseases, identifying drug targets. Finally, NGS technologies are providing significant opportunities to implement personalized health strategies including prevention or early detection of disease, improved health maintenance, and development of tailored therapy for patients with rare genetic diseases.
Acknowledgements
The authors thank the FORGE Canada Consortium for providing this remarkable opportunity for rare disease research with such a spirit of national collaboration. We thank every participating family for making this study possible. Funding was provided by the Government of Canada through Genome Canada to the Ontario Genomics Institute (OGI‐049) and the Canadian Institutes of Health Research (CIHR). Additional funding was provided by Genome Quebec, Genome British Columbia and the University of Toronto McLaughlin Centre. KMB was supported by a CIHR Institute of Genetics Clinical Investigatorship Award.
Conflict of interest
None declared.
References
- 1.Orphanet. Prevalence of rare diseases: Bibliographic data. Orphanet Reports Series 2014; Rare Diseases collection.
- 2. McKusick VA. Mendelian Inheritance in Man and its online version, OMIM. Am J Hum Genet 2007: 80: 588–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shashi V, McConkie‐Rosell A, Rosell B et al. The utility of the traditional medical genetics diagnostic evaluation in the context of next‐generation sequencing for undiagnosed genetic disorders. Genet Med 2014: 16: 176–182. [DOI] [PubMed] [Google Scholar]
- 4. Graungaard AH, Skov L. Why do we need a diagnosis? A qualitative study of parents' experiences, coping and needs, when the newborn child is severely disabled. Child Care Health Dev 2007: 33: 296–307. [DOI] [PubMed] [Google Scholar]
- 5.Eurordis. EurordisCare2: survey of diagnostic delays, 8 diseases, Europe.
- 6. Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare‐disease genetics in the era of next‐generation sequencing: discovery to translation. Nat Rev Genet 2013: 14: 681–691. [DOI] [PubMed] [Google Scholar]
- 7. Polychronakos C, Seng KC. Exome diagnostics: already a reality? J Med Genet 2011: 48: 579. [DOI] [PubMed] [Google Scholar]
- 8. Need AC, Shashi V, Hitomi Y et al. Clinical application of exome sequencing in undiagnosed genetic conditions. J Med Genet 2012: 49: 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dixon‐Salazar TJ, Silhavy JL, Udpa N et al. Exome sequencing can improve diagnosis and alter patient management. Sci Transl Med 2012: 4: 138ra178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sawyer SL, Schwartzentruber J, Beaulieu CL et al. Exome sequencing as a diagnostic tool for pediatric‐onset ataxia. Hum Mutat 2014: 35: 45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dyment D, Tetreault M, Beaulieu C et al. Whole‐exome sequencing broadens the phenotypic spectrum of rare pediatric epilepsy: a retrospective study. Clin Genet 2015: 88: 34–40. [DOI] [PubMed] [Google Scholar]
- 12. Rauch A, Wieczorek D, Graf E et al. Range of genetic mutations associated with severe non‐syndromic sporadic intellectual disability: an exome sequencing study. Lancet 2012: 380: 1674–1682. [DOI] [PubMed] [Google Scholar]
- 13. Vissers LE, de Ligt J, Gilissen C et al. A de novo paradigm for mental retardation. Nat Genet 2010: 42: 1109–1112. [DOI] [PubMed] [Google Scholar]
- 14. de Ligt J, Willemsen MH, van Bon BW et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 2012: 367: 1921–1929. [DOI] [PubMed] [Google Scholar]
- 15. Soden SE, Saunders CJ, Willig LK et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med 2014: 6: 265ra168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang Y, Muzny DM, Reid JG et al. Clinical whole‐exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med 2013: 369: 1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang Y, Muzny DM, Xia F et al. Molecular findings among patients referred for clinical whole‐exome sequencing. JAMA 2014: 312: 1870–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee H, Deignan JL, Dorrani N et al. Clinical exome sequencing for genetic identification of rare mendelian disorders. JAMA 2014: 312: 1880–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Neveling K, Feenstra I, Gilissen C et al. A post‐hoc comparison of the utility of Sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat 2013: 34: 1721–1726. [DOI] [PubMed] [Google Scholar]
- 20. Beaulieu CL, Majewski J, Schwartzentruber J et al. FORGE Canada Consortium: outcomes of a 2‐year national rare disease gene discovery project. Am J Hum Genet 2014: 94: 809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Srour M, Hamdan FF, Schwartzentruber JA et al. Mutations in TMEM231 cause Joubert syndrome in French Canadians. J Med Genet 2012: 49: 636–641. [DOI] [PubMed] [Google Scholar]
- 22. Lines MA, Huang L, Schwartzentruber J et al. Haploinsufficiency of a spliceosomal GTPase encoded by EFTUD2 causes mandibulofacial dysostosis with microcephaly. Am J Hum Genet 2012: 90: 369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marcadier J, Smith A, Pohl D et al. Mutations in ALDH6A1 encoding methylmalonate semialdehyde dehydrogenase are associated with dysmyelination and transient methylmalonic aciduria. Orphanet J Rare Dis 2013: 8: 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fernandez BA, Green JS, Bursey F et al. Adult siblings with homozygous G6PC3 mutations expand our understanding of the severe congenital neutropenia type 4 (SCN4) phenotype. BMC Med Genet 2012: 13: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu H, Sawyer SL, Gos M et al. Atypical fibrodysplasia ossificans progressiva diagnosed by whole‐exome sequencing. Am J Med Genet A 2015: 167: 1337–1341. [DOI] [PubMed] [Google Scholar]
- 26. Grapp M, Just I, Linnankivi T et al. Molecular characterization of folate receptor 1 mutations delineates cerebral folate transport deficiency. Brain 2012: 135: 2022–2031. [DOI] [PubMed] [Google Scholar]
- 27. ACMG Board of Directors . Points to consider in the clinical application of genomic sequencing. Genet Med 2012: 14: 759–761. [DOI] [PubMed] [Google Scholar]
- 28. Boycott K, Hartley T, Adam S et al. The clinical application of genome‐wide sequencing for monogenic diseases in Canada: position statement of the Canadian College of Medical Geneticists. J Med Genet 2015: 52: 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Green RC, Berg JS, Grody WW et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 2013: 15: 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van El CG, Cornel MC, Borry P et al. Whole‐genome sequencing in health care. Recommendations of the European Society of Human Genetics. Eur J Hum Genet 2013: 21: S1–S5. [PMC free article] [PubMed] [Google Scholar]
- 31. Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. N Engl J Med 2014: 370: 2418–2425. [DOI] [PubMed] [Google Scholar]
- 32. Gilissen C, Hehir‐Kwa JY, Thung DT et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014: 511: 344–347. [DOI] [PubMed] [Google Scholar]
- 33. Robinson PN, Köhler S, Bauer S, Seelow D, Horn D, Mundlos S. The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am J Hum Genet 2008: 83: 610–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Girdea M, Dumitriu S, Fiume M et al. PhenoTips: patient phenotyping software for clinical and research use. Hum Mutat 2013: 34: 1057–1065. [DOI] [PubMed] [Google Scholar]
- 35. Sobreira N, Schiettecatte F, Boehm C, Valle D, Hamosh A. New tools for Mendelian disease gene identification: PhenoDB variant analysis module; and GeneMatcher, a web‐based tool for linking investigators with an interest in the same gene. Hum Mutat 2015: 36: 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bragin E, Chatzimichali EA, Wright CF et al. DECIPHER: database for the interpretation of phenotype‐linked plausibly pathogenic sequence and copy‐number variation. Nucleic Acids Res 2014: 42: D993–D1000. [DOI] [PMC free article] [PubMed] [Google Scholar]