

Abstract
Intracerebral levels of Transforming Growth Factor beta (TGFβ) rise rapidly during the onset of experimental autoimmune encephalomyelitis (EAE), a mouse model of Multiple Sclerosis (MS). We addressed the role of TGFβ responsiveness in EAE by targeting the TGFβ receptor in myeloid cells, determining that Tgfbr2 was specifically targeted in monocyte‐derived dendritic cells (moDCs) but not in CNS resident microglia by using bone‐marrow chimeric mice. TGFβ responsiveness in moDCs was necessary for the remission phase since LysMCreTgfbr2fl/fl mice developed a chronic form of EAE characterized by severe demyelination and extensive infiltration of activated moDCs in the CNS. Tgfbr2 deficiency resulted in increased moDC IL‐12 secretion that skewed T cells to produce IFN‐γ, which in turn enhanced the production of moDC‐derived reactive oxygen species that promote oxidative damage and demyelination. We identified SNPs in the human NOX2 (CYBB) gene that associated with the severity of MS, and significantly increased CYBB expression was recorded in PBMCs from both MS patients and from MS severity risk allele rs72619425‐A carrying individuals. We thus identify a novel myeloid cell‐T cell activation loop active in the CNS during chronic disease that could be therapeutically targeted. GLIA 2016;64:1925–1937
Keywords: TGFβ, MOG‐EAE, gene deletion, reactive oxygen species, monocyte‐derived dendritic cells
Abbreviations
- APCs
antigen presenting cells
- BMDC
bone marrow‐derived dendritic cells
- BMDM
bone marrow‐derived macrophages
- EAE
experimental autoimmune encephalomyelitis
- iLN
inguinal lymph nodes.
- moDCs
monocyte‐derived dendritic cells
- PBMCs
peripheral blood mononuclear cells
- ROS
reactive oxygen species
- SNPs
single nucleotide polymorphisms
- TGFβ
transforming growth factor beta
- Tregs
regulatory T cells
Introduction
It is a generally accepted concept that the development of demyelinating lesions in multiple sclerosis (MS) and its mouse model experimental autoimmune encephalomyelitis (EAE) is initiated by autoreactive T cells that become reactivated in the CNS (Sie et al., 2014). Once inside the brain, myelin‐reactive CD4+ T cells secrete factors that open the blood‐brain barrier and recruit a heterogeneous population of myeloid cells that drive demyelination. The strong genetic association of MHC class II and MS implies that myeloid cells are of prime importance (Sawcer et al., 2011), but as both infiltrating monocyte‐derived macrophages and dendritic cells as well as resident microglia can all express MHC class II when activated, the relative contributions of these populations during the different phases of diseases needs to be better understood. Compared with infiltrating DCs, microglia are normally considered poor APCs (Ford, 1996; Suter et al., 2003; Greter et al., 2005), a function requiring MHC II expression, but they can also contribute to EAE progression by secreting cytokines/chemokines and producing ROS (Becher, 2001; Heppner et al., 2005; Goldmann et al., 2013). Human genome‐wide association study data also indicate that redox regulation figures in disease susceptibility (Sawcer et al., 2011). A recent study demonstrated that microglia appeared to exhibit debris scavenging functions during EAE while infiltrating monocytes initiated demyelination at nodes of Ranvier (Yamasaki et al., 2014), again indicating microenvironmental and myeloid subset‐specific functionality during disease promotion and resolution (Harris, 2014).
EAE severity correlates very well with the number of spinal cord infiltrating monocytes, and mice in which monocytes are inhibited from entering the CNS are protected against EAE development (Fife et al., 2000; Mildner et al., 2009; Ajami et al., 2011). The most abundant myeloid cell population in the CNS during EAE are the inflammatory Ly6Chi monocyte‐derived DCs (moDCs; King et al., 2009; Hesske et al., 2010). They are characterized by the coexpression of F4/80, CD11b, CD11c, and MHC class II (Hesske et al., 2010) in accordance with a recently proposed classification (Guilliams et al., 2014). Once in the CNS, monocytes differentiate and become activated under the influence of the Th1‐ and Th17‐ derived cytokines IFN‐γ and GM‐CSF, respectively. IFN‐γ activates APCs by upregulating MHC class II and induces the production of IL‐12. The exact action of GM‐CSF is disputed but the main target of GM‐CSF during CNS inflammation is considered to be monocytes, this process being absolutely required for induction of EAE (Hesske et al., 2010; Codarri et al., 2013, 2011; Greter et al., 2012; Croxford et al., 2015). The presence of moDCs in the CNS is transient and their numbers decrease drastically during EAE remission (Hesske et al., 2010; Ajami et al., 2011). However, the factors that control inflammation and regulate this contraction of the monocyte compartment during remission are not currently defined.
TGFβ is a pleiotropic cytokine with important functions during embryogenesis, patterning, development, homeostasis, and repair. TGFβ activity increases dramatically in the CNS during EAE, as elegantly demonstrated by in vivo bioluminescence studies (Luo et al., 2007; Lanz et al., 2010; Speck et al., 2014). Attempts to define the role of TGFβ during EAE have yielded conflicting results. Administration of recombinant TGFβ prevented the development of EAE symptoms (Racke et al., 1991, 1993). In concordance, EAE was exacerbated by the administration of TGFβ1‐neutralizing antibodies (Johns and Sriram, 1993). Paradoxically, over‐production of TGFβ by astroglia also exacerbated EAE (Wyss‐Coray et al., 1997), and more recently the pharmacological inhibition of TGFβR signaling attenuated EAE (Luo et al., 2007). TGFβ can have multifunctional roles and the net effect on the EAE outcome appears to depend on the location, timing, and the nature of the cell that is targeted. In this respect TGFβ has differential effects on virtually every cell type in the CNS (Wyss‐Coray et al., 1997; Lalive et al., 2005; Lanz et al., 2010; He et al., 2014). It is also well established that in the presence of IL‐6 then TGFβ is critical for the induction of Th17 cells, while in the absence of IL‐6 then Tregs can be induced (Fu et al., 2004; Liu et al., 2006; Ostroukhova, 2006). We and others have demonstrated that TGFβ has strong immunoregulatory effects on APCs both in vitro and in vivo (Biollaz et al., 2009; Parsa et al., 2012; Zhang et al., 2014).
We thus hypothesized that loss of TGFβ responsiveness in the myeloid compartment would interfere with the de‐activation of inflammatory monocyte‐derived cells and affect EAE progression. To address this question we bred mice with Tgfbr2 targeted specifically in myeloid cells. We demonstrate that these mice are characterized by the retention of moDCs in the CNS and development of severe chronic EAE with increased demyelination and no remission. Examination of the T cell compartment during chronic EAE revealed a skewing of CNS‐infiltrating T cells towards IFN‐γ production. In vitro models provided evidence for a pathogenic Th1‐skewing APC‐T cell‐signaling loop whereby loss of TGFβ responsiveness in APCs led to increased levels of Th1‐polarizing IL‐12. IFN‐γ in turn sustained chronic activation of myeloid cells with increased ROS production that contributes to oxidative damage and demyelination. Furthermore, we identified SNPs in the human NOX2 (CYBB) gene to be associated with MS severity measured as a combination of switch to second‐line treatment and high Multiple Sclerosis Severity Score (MSSS). The presence of the associated allele of the SNPs correlated with increased expression in PBMCs, and the expression of the CYBB gene was increased in PBMC among MS cases compared with patients with other neurological diseases (OND).
Materials and Methods
Mice
All mice were bred and maintained under specific pathogen‐free conditions at Karolinska Institutet or at the University of Zurich. All animal experiments were approved and performed in accordance with national and cantonal animal care guidelines and ethical permits. We generated two lines of LysMCreTgfbr2fl/fl mice: Tgfbr2fl/fl mice were imported from Dr. Ming Li at Sloan Kettering Institute, or obtained from Dr. Per Levéen, Lund University. LysMCre mice were obtained from The Jackson Laboratory (Lyz2‐Cre) or directly from Dr. Irmgard Förster, University of Cologne, and bred to obtain LysMCreTgfbr2fl/fl mice (LysMCre/+Tgfbr2fl/fl) and littermate controls (LysMCre/+TGFbR2fl/wt or LysM+/+TGFbR2fl/fl). The animals were crossed for >10 generations to C57BL6.
Induction of EAE
To induce EAE mice were injected subcutaneously with 100 µL recombinant mouse MOG or MOG35‐55 peptide (100 µg) emulsified with 100 μL complete Freunds adjuvant (200 μg Mycobacterium tuberculosis/mouse) followed by i.p injections of 200–300 ng pertussis toxin on days zero and two post‐immunization. EAE score was determined as previously described (Hesske et al., 2010; Zhang et al., 2014).
Preparation of Single Cell Suspensions
EAE mice were sacrificed by pentobarbital overdose or CO2 inhalation and transcardially perfused with ice‐cold PBS. Lymph node (LN) and spleen cell suspensions were obtained by mechanical dissociation of the organs and filtering through 40 μm cell strainers in ice‐cold PBS. Erythrocytes were lysed using ice‐cold ACK Lysis buffer (Sigma). Cells were counted using a Scepter Cell Counter (Millipore) and cell discrimination was 4.8–15 µm.
For CNS myeloid cell subset analysis, CNS tissue was minced and digested for thirty min at 37°C in Hank's balanced salt solution containing 50 µg/mL DNaseI (Roche) and 100 µg/mL collagenase/dispase (Roche). The digestion was quenched by cooling samples on ice, passed through a 100 μm mesh, pelleted, resuspended in 30% Percoll (GE Healthcare) and centrifuged at 15,000g for 30 min at 4°C. The myelin layer was removed and the mononuclear cells accumulated in the intermediate phase were collected and further processed.
For T cell cytokine analysis, CNS cells were prepared by homogenization of brains and spinal cords in 50% Percoll solution using a dounce homogenizer. This solution was underlain with 63% Percoll and overlain with 30% Percoll solution before centrifugation at 1000g for 30 min. Cells were then collected at the 30/50% interphase, filtered through a 40 μm cell strained and washed in ice‐cold PBS.
Bone Marrow Chimeras
LysMCreTgfbr2fl/fl on the C57BL6 (CD45.2) background were crossed with syngeneic CD45.1+ C57BL6 to obtain two syngeneic lines that allow the distinction of donor and recipient leukocytes based on the allelic difference of CD45.1 and CD45.2. Recipients were irradiated with a total of 10 gy in a split dose and subsequently reconstituted with 5 × 106 bone marrow cells by intravenous injection. The reconstituted animals were provided 2 g/L neomycin in the drinking water for two weeks and allowed to reconstitute for eight weeks before EAE was induced.
Flow Cytometry and Cell Sorting
Flow cytometry was performed using a CyFlow Space (Partec, Münster, Germany), Canto II or Fortessa (BD Bioscience), or Gallios (Beckman Coulter). Cell sorting was performed using a FACSAria (BD).
For extracellular staining single cell suspensions were incubated with antibody cocktails in PBS (Sigma) at 4°C. For intracellular staining cells were first incubated with PMA (50 ng/mL, Sigma), Ionomycin (1 μg/mL, Sigma Aldrich), and GolgiPlug (1 μL/mL, BD Biosciences) in complete DMEM (Sigma) for 4 h at 37°C before incubation with antibodies. Fixation/Permeablization and intracellular staining was conducted using the eBioscience Intracellular Staining Kit and the following antibodies: B220 (RA3‐6B2, Biolegend), CD3 (17A2, Biolegend), CD4 (GK1.5, Biolegend), CD8 (53‐6.7, eBioscience), CD11b (M1/70, Biolegend), CD11c (N418, Biolegend), CD44 (IM7, BD Biosciences), CD62L (MEL‐14, eBioscience), F4/80 (BM8, Biolegend) FoxP3 (FJK‐16s, eBioscience), IFN‐γ (XMG1.2, BD Biosciences), IL‐17 (TC11‐18H10, BD Biosciences), Ki67 (B56, BD Biosciences), Ly‐6C (HK1.4, Biolegend), Ly‐6G (1A8, BD Biosciences), and NK1.1 (PK136, BD Biosciences). Events were analyzed using Kaluza software (Beckman Coulter) and FlowJo (FlowJo LLC). Dead cells were excluded using the LIVE/DEAD Fixable Dead Cell Stain Kit (Invitrogen). Doublets were removed using FSC Area and FSC TOF or FSC Height. Gating strategies are provided in Supporting Information Figure S1.
BMDM/BMDC Culture and ELISA
BMDMs and BMDCs were prepared by flushing out the bone marrow cells from dissected femurs. BMDM single cell suspensions were prepared and cultured in DMEM (Sigma) supplemented with 20% FCS (Sigma) and M‐CSF (20 ng/mL) or in 50% DMEM/20% horse serum/30% L929 conditioned medium (Moransard et al., 2010) for seven days. BMDCs were generated in DMEM/20%FCS and 10 ng/mL GM‐CSF for seven days. Adherent cells were then replated in 48‐well plates and stimulated with LPS (50 ng/mL, Invitrogen), IFN‐γ or TGFβ (20 ng/mL, R&D) for 24 h. IL‐12p70 and IL‐23p19 levels in supernatants were analyzed using eBioscience Ready‐Set‐Go ELISA kits.
T Cell BMDC Coculture
C57BL6 mice were immunized with MOG35‐55 (as described above) and iLNs dissected 7 days later. CD4+ T cells were isolated using microbeads (Milteyni Biotec) according to manufacturer's instructions. 0.25 × 105 BMDCs (prepared as above) were plated in flat bottom 96‐well plates and stimulated with LPS (100 ng/mL) ± TGFβ1 (20 ng/mL) for 2 h and then washed three times and 0.4 × 106 T cells plated on top. BMDCs and T cells were cocultured for three days in the presence of 0, 20, or 60 μg MOG35‐55. Intracellular flow cytometry and cytometric bead array technology was used to analyse IL‐17 and IFNγ production directly in T cells or indirectly in the culture supernatant, respectively.
ROS Production Assay
BMDMs were stimulated with LPS (100 ng/mL) and IFN‐γ (20 ng/mL) ± TGFβ1 (20 ng/mL) for 24 h, and then pulsed with dihydrorhodamine‐123 (DHR‐123) for 10 min. Cells were washed, fixed, and DHR‐123 incorporation was determined by flow cytometry.
Histology/Immunohistochemistry
Mice were sacrificed with CO2. Histology was performed as described previously (Moransard et al., 2011). Spinal cords were removed and fixed in 4% buffered formalin and embedded in paraffin before staining with hematoxylin and eosin or Luxol fast blue to assess the degree of demyelination, MAC‐3 (BD Pharmingen) for DCs, macrophages and microglia, and CD3 for T cells (Serotec). An average area of 6.06 ± 1.23 mm2 spinal cord cross‐section was evaluated for the quantification of cell infiltrates and demyelination. Fluorescence immunostaining was performed using anti‐gp91phox (Nox2, BD Pharmingen) and anti‐MAC3 (BD Pharmingen) antibodies with DAPI (nuclei) counterstaining. Bound primary antibodies were visualized with the appropriate species‐specific Cy3‐ or Cy2‐conjugated secondary antibodies (all from Jackson ImmunoResearch Laboratories). Fluorescent myelin staining was performed by incubation of cryosections with 1:500 diluted anti‐PLP (plpc1, Thermo‐Fisher) and 1:500 diluted Oregon Green 488‐labeled secondary antibody (Molecular Probes/Thermo‐Fisher) plus DAPI counterstaining. For ROS quantification in the tissue DCFDA (1.25 µg in isotonic NaCl; Life Technologies) was injected 3 h before collecting tissues. Fresh cryosections were embedded in Mowiol and immediately photographed using a Leica DMI6000. The DCFDA fluorescence intensity of two‐four cross sections of three spinal cord levels was quantified using Image J and was normalized to the measured area.
qRT‐PCR
Total RNA was prepared from tissue and cell cultures using PeqGold (Peqlab) according to the manufacturer's protocols. Typically, 0.3 µg RNA was hybridized to random hexamers and then cDNA was synthesized using MuLV (Applied Biosystems). Quantitative real‐time PCR was performed on an AB7900 using 10 ng cDNA and Taqman primers and probes from Applied Biosystems. The results were normalized to the expression of 18S rRNA. To assess the extent of Tgfbr2 deletion we measured the presence of exon 4 using an amplicon on exon boundaries 3/4 relative to the undeleted part represented by exon boundary 6/7 of Tgfbr2.
MS Patient Cohorts
Ethics statement
All included patient and control materials and analyses in this study were approved by the Regional Ethical Review Board in Stockholm, Sweden (www.epn.se). Informed consent from all study participants or their parents was obtained. Investigations were carried out according to guidelines from the Declaration of Helsinki.
Patients included in the clinical association analyses were recruited from three Swedish nationwide epidemiological studies: EIMS (Hedström et al., 2009), GEMS (Hedström et al., 2014), and IMSE (Holmén et al., 2011). The EIMS study consists of incident and the GEMS study of prevalent cases of MS. The IMSE study is a phase IV study for Natalizumab and Fingolimod treatments for MS. In all cases, controls have been matched to cases with respect to age, gender, and area of residency. Patients with MS or other neurological disease included in expression analyses were recruited from the Neurological clinic at Karolinska University hospital (Khademi et al., 2011).
Genotyping
Genotyping was carried out using an Illumina custom array as part of a larger study replicating genetic association to MS within the international MS genetics consortium. In this custom array, 97 markers in the CYBB region on chromosome X were included. A total of 7701 MS patients and 6637 controls from Sweden were genotyped and passed quality control. Allele calling was carried out with an Illuminus caller. The quality control analyses for markers included minor allele frequency >0.02, success rate >0.98, Hardy Weinberg equilibrium among controls (P < 0.0001). For individuals the quality control included success rate >0.98, increased heterozygosity as determined as F (inbred coefficient) smaller than mean value minus three standard deviations. All these quality control steps were carried out using PLINK (Purcell et al., 2007). We identified population outliers using the SmartPCA program with standard settings (Price et al., 2006) and removed those that were outliers. Six PCA vectors, those with P < 0.05, were used for correcting for population stratification in the association analysis. We estimated relatedness between individuals using PLINK and removed one individual in reach pair with Pi_hat > 0.175.
Expression Analysis
Peripheral blood mononuclear cells from 28 clinically isolated syndrome (CIS), 121 MS patients and 36 patients with OND without an inflammatory state, such as neuralgia, paresthesia, sensory symptoms, vertigo, tension‐headache were isolated. RNA extraction was performed using the RNeasy MiniKit (Qiagen) including DNAse digestion, integrity of the RNA was analyzed with Bioanalyzer (Agilent Technologies), all samples had an RNA integrity above 8. cDNA libraries for sequencing were prepared using Illumina TruSeq kit (Illumina) and sequenced on an Illumina HiSeq 2000 machine. We generated data in fastq format using Illumina 1.8 quality scores. Paired‐end reads with a length of 100 bp with an average sequence depth of 36 million reads per sample. The reads were mapped to the H.Sapiens reference genome (NCBI v37, hg19) using STAR aligner. The Conditional Quantile Normalization (CQN) method, which accounts for the gene length bias and GC content bias, was used to generate CQN expression values at gene level from the RNA‐Seq count data. To correlate the gene expression to patient genotypes, the residual values were calculated from CQN expression values after correcting for batch‐effect (batch of RNA‐seq library preparation) and disease‐type (CIS grouped with MS and OND) using lmFit and eBayes from limma package.
Statistical Analysis
Association between genotypes and clinical variables were carried out with logistic regression in PLINK (17701901) including six PCA vectors to correct for population stratification. Twenty‐nine markers in or proximal to the CYBB gene were included after QC.
Comparison of expression levels between patients with MS and OND was carried out with Spearman's rank test on the residual values after correcting only for batch effect. The significance of difference in expression levels with genotype was tested with T‐test not assuming equal variance.
Results
Extensive Demyelination and Absence of Remission in Mice with Myeloid‐Specific Tgfbr2 Deficiency
To determine the role of TGFβ in myeloid cells during EAE we bred myeloid‐specific Cre‐mice (LysMCre) with Tgfbr2fl/fl mice to produce LysMCreTgfbr2fl/fl mice. We induced EAE in these and control mice (Tgfbr2fl/fl) by active immunization with the CNS‐specific antigen MOG and determined the clinical outcome. There were no differences in day of onset (day 11.6 ± 0.4 Tgfbr2fl/fl; day 11.5 ± 0.5 LysMCreTgfbr2fl/fl) or in disease score at peak of disease (score 2.49 ± 0.11 Tgfbr2fl/fl; score 2.63 ± 0.15 LysMCreTgfbr2fl/fl). However, while control mice entered the remission phase (day 17) with reduced disease severity, LysMCreTgfbr2fl/fl mice EAE scores remained at peak level throughout the period of observation (28 days; Fig. 1A).
Figure 1.
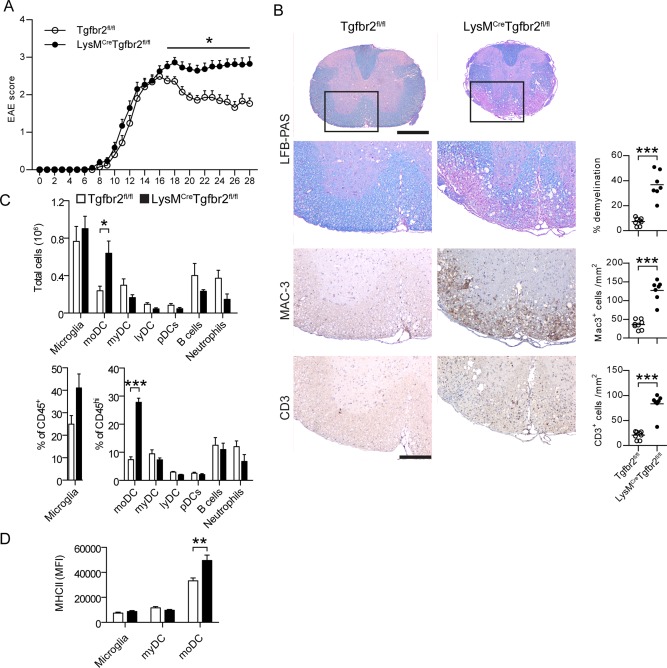
Loss of TGFβ‐signaling in myeloid cells results in chronic EAE without remission. EAE was induced by immunization with MOG emulsified in CFA. Clinical score was assessed on a daily basis (A). Data are pooled from three experiments. n = 25 (Tgfbr2fl/fl), 38 (LysMCreTgfbr2fl/fl). B: Spinal cords taken at day 28 post‐immunization stained with LFB‐PAS, anti‐Mac‐3, and anti‐CD3. n = 7‐8/group. C: Analysis of CNS cell subsets by flow cytometry at day 28 postimmunization n = 3–5/group. moDC monocyte‐derived DC, myDC conventional myeloid DC, lyDC lymphoid DC pDC plasmacytoid DC. D: Surface MHC II expression n = 9/group. Data in A, C, and D are presented as means ± SEM. * P < 0.05 *** P < 0.001 (A, Mann‐Whitney test; B–D, Student's unpaired t‐test). Scale bar = 500 μm (spinal cords) and 200 μm (magnifications). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Histological analysis of the spinal cord during the chronic stage (day 26) demonstrated pronounced demyelination and extensive cell infiltration in LysMCreTgfbr2fl/fl mice. Quantification of Luxol Fast Blue/Periodic Acid Schiff staining demonstrated a five‐fold increased demyelination in LysMCreTgfbr2fl/fl (36.7 ± 4.1%) compared with in control (7.2 ± 1.1%) mice (Fig. 1B). Assessment of cell infiltrates identified Mac3+ and CD3+ T cells as the most prominent populations and revealed a 3–4‐fold increase in these cells in LysMCreTgfbr2fl/fl infiltrates compared to in control mice. No differences in cell infiltration were evident during the peak of disease at day 16 (Supp. Info. Fig. S2).
To determine which myeloid (Mac3+) cell subsets were increased in the CNS of LysMCreTgfbr2fl/fl mice during chronic EAE we analyzed CNS mononuclear cells ex vivo by flow cytometry. We recorded no significant differences in frequency or absolute numbers of CNS‐resident microglia, B cells or neutrophils, but instead a 2–3‐fold increase in the frequency and absolute number of moDCs was apparent (Fig. 1C). No differences were observed in myeloid, lymphoid or plasmacytoid DCs between the two strains. Furthermore, we observed increased MHC class II expression on moDCs but not on microglia or myeloid DCs in LysMCreTgfbr2fl/fl mice (Fig. 1D).
Similar analyses conducted at peak of disease (day 16) revealed no significant differences in any CNS infiltrating cell subset, consistent with our histological findings (Supp. Info. Fig. S3). LysMCreTgfbr2fl/fl spleens and iLNs were also analyzed in the steady state, this revealing normal representation of the main immunological subsets (Supp. Info. Fig. S4).
These results demonstrate that TGFβ responsiveness in the myeloid compartment is important for remission from EAE. Loss thereof leads to retention of T cells and activated moDCs in the spinal cord that is concomitant with severe demyelination and the absence of clinical remission during the chronic stage of EAE.
Loss of TGFβ Responsiveness in Blood‐Derived Myeloid Cells but Not Resident Microglia Causes Nonremitting EAE
To determine whether the moDCs retained within the CNS during nonremitting EAE were deficient in Tgfbr2 we sorted moDCs and microglia from mice with chronic EAE and performed qRT‐PCR analysis. LysM expression was 30‐fold higher in moDCs than in microglia (Fig. 2A) and we detected a high degree of recombination of the Tgfbr2 locus in moDCs. High expression of LysM and loss of Tgfbr2 suggest an activated state of the moDCs, which was indeed evident based on increased MHC class II positivity in the spinal cords of LysMCreTgfbr2fl/fl mice. In contrast, we could not detect any recombination in microglia (Fig. 2B).
Figure 2.
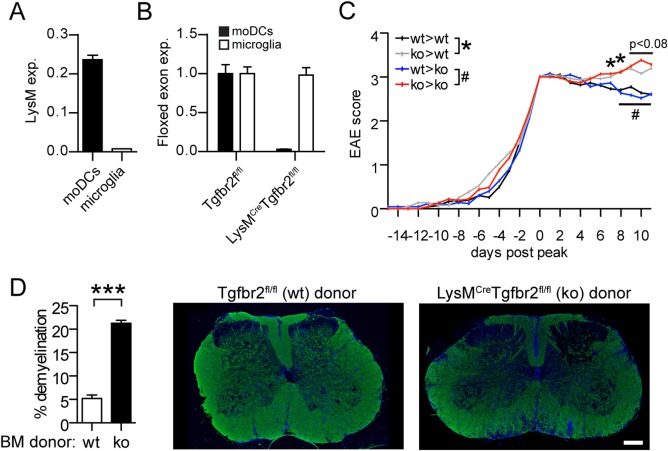
Tgfbr2 deficiency in moDCs and not in microglia causes severe chronic EAE. moDCs (CD45hi CD11b+ F4/80+ CD11c+) and microglia (CD45intCD11b+) were sorted from the CNS of EAE animals and quantitative RT‐PCR analysis performed of mRNA encoding (A) LysM (relative to 18S RNA) and (B) of the floxed exon 3/4 boundary of Tgfbr2 in relation to the unfloxed exon 6/7 boundary to assess recombination efficiency. Data in A and B are presented as means ± SEM. n = 3 mice/group. C: EAE scores from Tgfbr2fl/fl (wt) and LysMCreTgfbr2fl/fl (ko) chimeras. Data are pooled from three separate experiments and analyzed from peak of disease (first time score ≥ 3) and presented as means. n = 18–21/group. D: Demyelination in chimeras determined by fluorescent anti‐PLP staining. n = 5/group. Scale bar 250 µm. * and # P < 0.05 *** P < 0.001 (C, Mann‐Whitney test; D, Student's unpaired t‐test). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
To address the importance of moDCs versus microglia more specifically, LysMCreTgfbr2fl/fl or control (Tgfbr2fl/fl) mice were lethally irradiated and reconstituted with either LysMCreTgfbr2fl/fl or control bone marrow. Eight weeks later, these chimeric mice were immunized with MOG to induce EAE. Both LysMCreTgfbr2fl/fl and control mice that were reconstituted with LysMCreTgfbr2fl/fl bone marrow developed significantly more severe EAE symptoms in the chronic phase than did mice reconstituted with control bone marrow (Fig. 2C). Histological analysis of spinal cords in the chimeras revealed a significantly higher degree of demyelination in mice reconstituted with LysMCreTgfbr2fl/fl bone marrow compared to control bone marrow (Fig. 2D).
Taken together these results demonstrate that remission in EAE requires TGFβ responsiveness in moDCs but not in microglia.
Loss of TGFβ Responsiveness in moDCs Induces Th1 Polarization
To determine if Tgfbr2 deficiency in moDCs influenced T cell differentiation during EAE we isolated CNS cells and analyzed T cell cytokine production. No significant differences were observed in the frequencies of IFN‐γ or IL‐17‐producing CD4+ T cells or FoxP3+ Tregs at the peak of disease (Fig. 3A–C; day 16). During the chronic phase, however, the frequencies of IFN‐γ‐producing CD4+ T cells were significantly increased, whereas IL‐17‐producing CD4+ T cells were significantly decreased in the CNS of LysMCreTgfbr2fl/fl compared with in control mice (Fig. 3A–C; day 24). No significant differences in the percentages of IFN‐γ+IL‐17+ double‐positive T cells or of FoxP3+ Tregs were observed at all time points analyzed (Fig. 3A–C). The differences evident in the chronic phase were independent of events during the priming phase, since analysis of day 7 iLN revealed no differences in the frequencies of proliferating (Ki67+), activated (CD62L‐CD44+), IFN‐γ+, IL‐17+, or FoxP3+ T cells (Supp. Info. Fig. S4).
Figure 3.
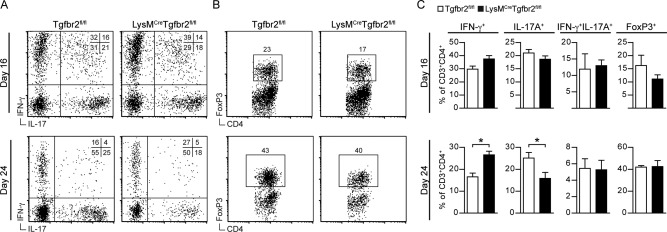
Severe chronic EAE is characterized by IFN‐γ‐producing T cells in the CNS. CNS mononuclear cells were isolated at peak of EAE (day 16) or during the chronic stage (day 24) and stained for surface CD3, CD4, and intracellular IFN‐γ, IL‐17A, and FoxP3. Representative dot plots of CNS cells gated on CD3±CD4± (A, B) and corresponding frequencies of IFN‐γ+, IL‐17±, IFN‐γ+IL‐17±, and FoxP3± T‐cells at day 16 and day 24 (C). Data are presented as means ± SEM. n = 4‐5 mice/group. Data are representative of two (day 16) or three (day 24) independent experiments. * P < 0.05 (Student's unpaired t‐test).
To address whether loss of TGFβ responsiveness in CNS‐infiltrating moDCs could skew T cells toward IFN‐γ production we measured IL‐12 production in cultured BMDCs and BMDMs as a factor that could polarize T cells to a Th1 phenotype. LysMCreTgfbr2fl/fl and control BMDCs produced similar levels of IL‐12 following LPS stimulation and TGFβ was able to attenuate LPS‐induced IL‐12 production in control BMDCs. In contrast, no significant reduction in IL‐12 production was detected in LysMCreTgfbr2fl/fl BMDCs upon TGFβ costimulation (Fig. 4A). The same results were obtained with BMDMs, although lower levels of IL‐12 were recorded in these cultures. No differences in IL‐23 levels in either BMDCs or BMDMs were detected in the presence of TGFβ in any of the strains (Fig. 4A).
Figure 4.
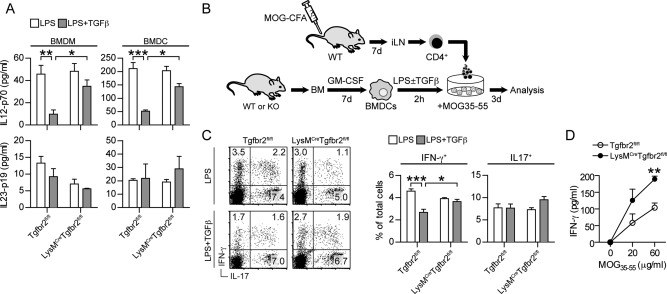
Loss of Tgfbr2 in dendritic cells polarizes T cells to produce IFN‐γ. Levels of IL‐12p70 and IL‐23p19 in culture supernatants from (A) BMDMs and BMDCs stimulated for 24 h with the indicated cytokines. B: Experimental setup of BMDC and T cell coculture. C: Intracellular cytokine staining of CD4+ T cells from day 7 MOG‐CFA immunized mice after coculture with BMDCs and 20 µg MOG35‐55. BMDCs were pre‐stimulated for 2 h with the indicated cytokines. D: Soluble IFN‐γ in culture supernatants from LPS + TGFβ condition. Data are presented as means ± SEM. n = 3 mice/group. * P < 0.05 ** P < 0.01 *** P < 0.001 (A,C, one way ANOVA with Bonferroni post test; D, Student's unpaired t‐test).
This indicated that APC‐T cell interactions in the CNS in the absence of TGFβ signaling could further skew MOG‐specific T cells towards a Th1 phenotype. To model this interaction more specifically we co‐cultured CD4+ T cells from day 7 iLN of MOG35‐55 immunized wildtype mice with BMDCs from LysMCreTgfbr2fl/fl or control mice. BMDCs were prestimulated with LPS in the presence or absence of TGFβ for 2 h and then cocultured with MOG‐specific T‐cells and MOG35‐55 peptide (Fig. 4B). Analysis of T cell cytokine production revealed frequencies of IFN‐γ+ and IL‐17+ T cells following coculture with LPS‐stimulated BMDCs to be similar in both strains. However, IFN‐γ+ T cell frequencies were significantly reduced when T cells were cocultured with LPS/TGFβ‐stimulated BMDCs from control mice whereas when cocultured with LysMCreTgfbr2fl/fl BMDCs, TGFβ did not affect the numbers of IFN‐γ producing T cells. Furthermore, extracellular levels of IFN‐γ were increased in culture supernatants as assessed by cytometric bead array measurement (Fig. 4D). However, TGFβ‐stimulated BMDCs did not affect frequencies of IL‐17+ T cells in either strain (Fig. 4C).
Collectively, these results demonstrate that lack of TGFβ responsiveness in BMDCs can cause T cells to produce more IFN‐γ, likely via an inability to down regulate IL‐12 production.
IFNγ Regulates Nox‐2 and ROS Production in moDCs during Chronic EAE
Tissue damage in EAE and MS is at least partly mediated by the production of toxic molecules from infiltrating myeloid cells (Schuh et al., 2014). Since we had observed increased numbers of inflammatory moDCs (Fig. 1C) and severe tissue destruction (Fig. 1B) in LysMCreTgfbr2fl/fl mice with chronic EAE, we tested the possible involvement of ROS or NOS pathways by measuring the expression of the NADPH oxidase (Nox) subunits and of inducible nitric oxide synthase (iNOS) in total spinal cords or sorted moDCs during chronic EAE (day 24). Only the Nox2‐subunit was upregulated in LysMCreTgfbr2fl/fl mice (Fig. 5A) and no differences were observed in other subunits. iNOS levels were not altered in LysMCreTgfbr2fl/fl total spinal cords or moDCs, excluding the importance of nitric oxide in this model (Fig. 5A and data not included). Sorting of CNS myeloid populations revealed that Nox2 levels were only upregulated during EAE in CNS‐isolated moDCs, but not in resident microglia or infiltrating neutrophils from LysMCreTgfbr2fl/fl mice (Fig. 5B).
Figure 5.
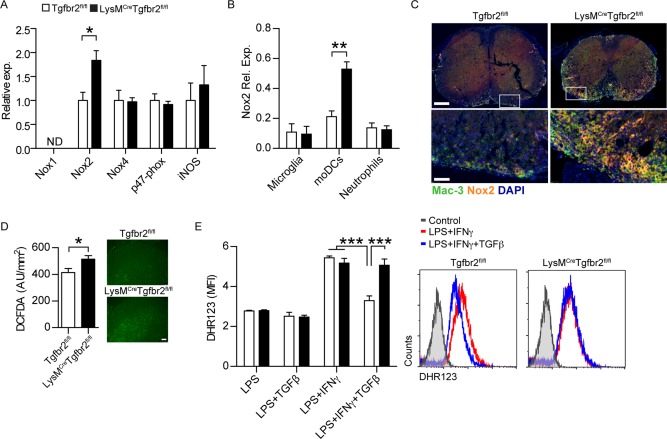
IFN‐γ regulates ROS‐production in moDCs during EAE. qRT‐PCR analysis of mRNA encoding Nox1, Nox2, Nox4, p47‐phox, and iNOS in (A) spinal cord homogenate or Nox2 in (B) sorted microglia (CD45int CD11b+), moDCs (CD45hi CD11b+ F4/80+ CD11c+), and neutrophils (CD45hi CD11b+ Ly6G+) from chronic stage EAE animals. n = 3 experiments with 8–12 pooled animals/group and experiment. Expression is shown relative to 18S RNA. C: Immunofluorescent staining of chronic stage EAE spinal cords for Nox2 and Mac3. Scale bar 50 and 250 µm. D: ROS‐levels in chronic stage EAE spinal cord sections as determined by DCFDA staining. n = 5 mice/group. Scale bar 100 µm. E: ROS production as measured by DHR123‐incorporation in BMDMs stimulated 24 h with the indicated cytokines. n = 3 mice/group. Data are presented as means ± SEM. *P < 0.05 *** P < 0.001 (A–D: Student's unpaired t‐test, E: one‐way ANOVA with Bonferroni post‐test). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
These findings were further substantiated by immunofluorescent staining of spinal cords using a Nox2 isoform‐specific antibody, demonstrating an elevation in Nox2 staining in spinal cord‐infiltrating Mac3+ cells in LysMCreTgfbr2fl/fl mice during the chronic disease state (Fig. 5C). We could also demonstrate increased levels of ROS in LysMCreTgfbr2fl/fl spinal cords compared with control mice by employing DCFDA staining (Fig. 5D).
These results suggested that Nox2‐mediated ROS production in moDCs is regulated by TGFβ. To confirm this we assessed ROS production through measuring fluorescent dihydrorohadmine‐123 (DHR123) incorporation into BMDMs. LPS stimulation alone did not increase ROS levels compared with in unstimulated BMDMs. However, when IFN‐γ was included as a costimulus ROS production was elevated in both LysMCreTgfbr2fl/fl and control BMDMs. TGFβ was able to attenuate ROS production to baseline levels in control mice whereas ROS levels in LysMCreTgfbr2fl/fl BMDMs were not affected by the presence of TGFβ (Fig. 5E).
Expression of CYBB Delineates Human MS and Its Severity
We analyzed 29 SNP markers in the human NOX2/CYBB (cytochrome b‐245, beta polypeptide) gene. Twenty of the SNPs in high linkage disequilibrium (LD) were nominally associated with MS severity. We measured severity of disease in three different ways: (a) Comparing patients who had switched to a second‐line treatment (Nataliumab, Rituximab, or Fingolimod) compared to those receiving standard treatments. These second‐line treatments were until recently only used for patients with highly active disease in Sweden; (b) Patients with high MSSS compared with those with low MSSS. The definition of high was set to the highest quartile and low as the lowest quartile of MSSS in the patient cohort; (c) A combination of second‐line treatment and MSSS where those patients with second‐line treatment and MSSS above median were compared with those with standard treatment and MSSS in the lowest quartile. Since the identified SNPs were in very high LD with close to identical P‐values and Odds ratios, we chose the first one (rs72619425) as a representative for the following figures. The odds ratio for the A allele of rs72619425 increased when considering only second‐line treatment to MSSS, and when combining both measures (1.06, 1.16, and 1.36, respectively; Fig. 6A), presumably identifying increasingly more severe MS cases. The expression of CYBB was increased in PBMCs from these patients compared to patients with OND (P < 0.05 Fig. 6B). The expression was higher among individuals carrying the A allele compared with G homozygotes in PBMC (P < 0.04, Fig. 6C).
Figure 6.
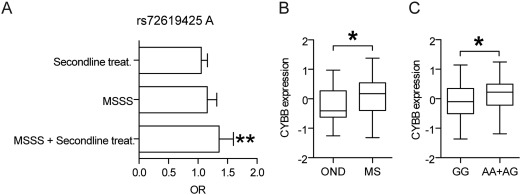
Effect of CYBB alleles on MS. A: Odds ratio for rs72619425 A for different measures of severity of MS. Second‐line treatment patients (Natalizumab, Rituximab, or Fingolimod n = 2189) were compared with standard treatments (n = 5500, ns). Patients with high MSSS value (highest quartile n = 1497, MSSS > 6.464) had a tendency for higher frequency of the A allele compared with those with low MSSS (lowest quartile n = 1711, MSSS < 1.282, P = 0.06). Patients who received second‐line treatment and had high MSSS (above median 4.022 n = 739) had a higher frequency of the A allele than patients receiving standard treatment and low MSSS (lowest quartile MSSS < 1.282; n = 1160; P = 0.003). B: Comparison of expression levels of CYBB in PBMC from MS patients (n = 120) compared with patients with other neurological disease (OND, n = 36). Expression was measured with RNAseq and quantified as residual values after correction for batch effects using a linear model fit. * P < 0.05 (Student's unpaired t‐test). C: Expression of CYBB in PBMC is increased among carriers of the rs72619425 A alleles. Expression was analyzed in patients with MS (n = 87), CIS (n = 19), and OND (n = 31) according to genotype; AA + AG (n = 41) and GG (n = 96). Expression was measured with RNA‐seq and quantified as residual values after correction for batch effect (RNA‐seq library preparation) and disease status (MS or OND) using a linear model fit. * P < 0.05 (Student's unpaired t‐test).
Discussion
The present study demonstrates that TGFβ responsiveness in the myeloid compartment is critical in controlling autoimmune encephalomyelitis, and that its specific deletion in moDCs causes severe EAE without remission. This effect can be attributed to the control of three major pathways: IL‐12 production by moDCs, IFN‐γ production by Th1 cells, and subsequent ROS production by moDCs.
In previous studies in which a dominant‐negative Tgfbr2 was expressed in CD11c+ DCs the major findings were that TGFβ‐DC interplay is a key event in the control of autoimmune encephalomyelitis (Laouar et al., 2008) and especially in controlling Th17 differentiation (Speck et al., 2014). This T cell subset was critical in disease initiation, whereas NK cells and microglia played no role in that model. Furthermore, TGFβ was demonstrated to be a potent suppressor of DC derivation by GM‐CSF, but not of DC activation by LPS (Speck et al., 2014).
The differences in these studies and the present one are most likely due to the different targeting systems employed. In support of this, a recent report has demonstrated that the use of CD11c‐targeting systems predominantly affects conventional DCs (cDCs) such as myeloid and lymphoid DCs (Croxford et al., 2015). Importantly, as also highlighted in that study, LysM‐Cre efficiently targets moDCs, which are the pathogenic myeloid cells during EAE (Fife et al., 2000; Croxford et al., 2015). Indeed, LysM‐Cre has been used in several studies to target the myeloid lineage, including neutrophils, monocytes and microglia (Malipiero et al., 2006; Goldmann et al., 2013; Wieghofer et al., 2015). In this study, we determined that our approach was efficiently targeting moDCs and not microglia, and we confirmed the importance of peripheral cells compared to CNS microglia by using bone marrow chimeras.
We further demonstrate that loss of TGFβ responsiveness causes a specific retention of activated MHC class II+ moDCs but not of other myeloid cell populations such as neutrophils or conventional DCs in the CNS, this being paralleled by an increased frequency of IFN‐γ‐producing T cells. The accumulation of Th1 cells is likely to occur as a result of APC and T cell interactions at the site of inflammation because Th1 numbers are not increased in the periphery or the CNS at any timepoint before the onset of chronic EAE. We modeled the interaction between moDCs and T cells in vitro and determined that Tgfbr2 deficiency in myeloid cells alone could account for IFN‐γ production in T cells via increased IL‐12 production. This is of particular interest because it implicates Th1 cells in the perpetuation of severe disease rather than in the initiation phase. Interestingly, while Th17 or IFN‐γ/IL‐17 double positive cells are early infiltrators into the CNS prior to EAE disease debut and prior to Th1 infiltration, Th1 cells were previously reported to dominate at later timepoints (Murphy et al., 2010). Our findings indicate that lack of TGFβ responsiveness leads to accumulation of Th1 cells during the chronic disease state. These Th1 cells secrete IFN‐γ that induces the enhanced oxidative response in moDCs.
The brain is especially vulnerable to oxidative damage due to its high oxygen consumption (20% of total body basal oxygen consumption), the local abundance of iron and accumulation of metal ions, coupled with the relatively lower expression levels of antioxidants (Harris and Amor, 2011). In pathological scenarios such as EAE/MS there is sustained oxidative/carbonyl stress that overwhelms these antioxidant mechanisms so that a pro‐inflammatory, dysregulated oxidative environment prevails, particularly during the secondary progressive phase of MS (Gonsette, 2008). ROS production by peripheral‐derived myeloid cells and by resident microglia thus contributes to demyelination and axonal damage (Lassmann, 2003) and oxidative DNA damage is evident in MS plaques (Vladimirova et al., 1998). Moreover, in one study it was reported that mononuclear cells from patients produce higher ROS levels upon activation (Vladimirova et al., 1998), while other studies describe TGFβ dysregulation in blood mononuclear cells (Hollifield et al., 2003; Meoli et al., 2011). It is thus very likely that in MS patients a similar vicious cycle is at work as in our mouse model and that antioxidative and/or TGFβ treatments could lead to substantial amelioration of the chronic progressive disease phase.
While MOG‐EAE is a useful model of human disease, we further investigated our findings by analyzing gene expression data in well‐characterized human MS cohorts. Not only was the CYBB gene significantly associated with MS compared with OND, but it also correlated with disease severity in an allele‐specific manner. As we focused on a restricted set of markers based on our original hypothesis, this association should be regarded as being exploratory, since the P value does not reach genome‐wide significance in models in which the whole genome is screened.
In conclusion, we identify a novel myeloid cell‐T cell activation loop active in the CNS that is critical for the chronic disease stage, and that could be therapeutically targeted by increasing TGFβ responsiveness.
Supporting information
Supporting Information 1
Supporting Information 2
Supporting Information 3
Supporting Information 4
Supporting Information 5
Acknowledgment
The authors appreciate the technical expertise of Sean Carlson and Friederike Ackermann and acknowledge the genotyping performed via the International Multiple Sclerosis Genetics Consortium as well as histological support by Matthias Heikenwälder. The computations were performed on resources provided by SNIC through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) under Project b2011139. Adriano Fontana is a Hertie Senior Research Professor for Neuroscience of the Gemeinnützige Hertie Stiftung. Doron Merkler holds stipendiary professorships of the Swiss National Science Foundation. Tobias Suter was supported by the CRPP Multiple Sclerosis.
References
- Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FMV. 2011. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci 14:1142–1149. [DOI] [PubMed] [Google Scholar]
- Becher B. 2001. The clinical course of experimental autoimmune encephalomyelitis and inflammation is controlled by the expression of CD40 within the central nervous system. J Exp Med 193:967–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biollaz G, Bernasconi L, Cretton C, Püntener U, Frei K, Fontana A, Suter T. 2009. Site‐specific anti‐tumor immunity: Differences in DC function, TGF‐β production and numbers of intratumoral Foxp3+ Treg. Eur J Immunol 39:1323–1333. [DOI] [PubMed] [Google Scholar]
- Codarri L, Greter M, Becher B. 2013. Communication between pathogenic T cells and myeloid cells in neuroinflammatory disease. Trends Immunol 34:114–119. [DOI] [PubMed] [Google Scholar]
- Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B. 2011. RORγt drives production of the cytokine GM‐CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol 12:560–567. [DOI] [PubMed] [Google Scholar]
- Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, Clausen BE, Jung S, Greter M, Becher B. 2015. The Cytokine GM‐CSF Drives the Inflammatory Signature of CCR2+ Monocytes and Licenses Autoimmunity. Immunity 43:502–514. [DOI] [PubMed] [Google Scholar]
- Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ. 2000. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J Exp Med 192:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford AL. 1996. Microglia induce CD4 T lymphocyte final effector function and death. J Exp Med 184:1737–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Zhang N, Yopp AC, Chen D, Mao M, Chen D, Zhang H, Ding Y, Bromberg JS. 2004. TGF‐beta Induces Foxp3 + T‐Regulatory Cells from CD4 + CD25 ‐ Precursors. Am J Transplant 4:1614–1627. [DOI] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Müller PF, Wolf Y, Varol D, Yona S, Brendecke SM, Kierdorf K, Staszewski O, Datta M, Luedde T, Heikenwalder M, Jung S, Prinz M. 2013. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci 16:1618–1626. [DOI] [PubMed] [Google Scholar]
- Gonsette RE. 2008. Neurodegeneration in multiple sclerosis: The role of oxidative stress and excitotoxicity. J Neurol Sci 274:48–53. [DOI] [PubMed] [Google Scholar]
- Greter M, Helft J, Chow A, Hashimoto D, Mortha A, Agudo‐Cantero J, Bogunovic M, Gautier EL, Miller J, Leboeuf M, Lu G, Aloman C, Brown BD, Pollard JW, Xiong H, Randolph GJ, Chipuk JE, Frenette PS, Merad M. 2012. GM‐CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity 36:1031–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, Noelle RJ, Becher B. 2005. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med 11:328–334. [DOI] [PubMed] [Google Scholar]
- Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, Yona S. 2014. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol 14:571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RA, Amor S. 2011. Sweet and sour–oxidative and carbonyl stress in neurological disorders. CNS Neurol Disord Drug Targets 10:82–107. [DOI] [PubMed] [Google Scholar]
- Harris RA. 2014. Spatial, Temporal, and Functional Aspects of Macrophages during “The Good, the Bad, and the Ugly” Phases of Inflammation. Front Immunol 5:612‐ [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Zhang H, Yung A, Villeda SA, Jaeger PA, Olayiwola O, Fainberg N, Wyss‐Coray T. 2014. ALK5‐dependent TGF‐β signaling is a major determinant of late‐stage adult neurogenesis. Nat Neurosci 17:943–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedström AK, Bäärnhielm M, Olsson T, Alfredsson L. 2009. Tobacco smoking, but not Swedish snuff use, increases the risk of multiple sclerosis. Neurology 73:696–701. [DOI] [PubMed] [Google Scholar]
- Hedström AK, Hillert J, Olsson T, Alfredsson L. 2014. Alcohol as a Modifiable Lifestyle Factor Affecting Multiple Sclerosis Risk. JAMA Neurol 71:300‐ [DOI] [PubMed] [Google Scholar]
- Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hövelmeyer N, Waisman A, Rülicke T, Prinz M, Priller J, Becher B, Aguzzi A. 2005. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med 11:146–152. [DOI] [PubMed] [Google Scholar]
- Hesske L, Vincenzetti C, Heikenwalder M, Prinz M, Reith W, Fontana A, Suter T. 2010. Induction of inhibitory central nervous system‐derived and stimulatory blood‐derived dendritic cells suggests a dual role for granulocyte‐macrophage colony‐stimulating factor in central nervous system inflammation. Brain 133:1637–1654. [DOI] [PubMed] [Google Scholar]
- Hollifield RD, Harbige LS, Pham‐Dinh D, Sharief MK. 2003. Evidence for Cytokine Dysregulation in Multiple Sclerosis: Peripheral Blood Mononuclear Cell Production of Pro‐inflammatory and Anti‐inflammatory Cytokines During Relapse and Remission. Autoimmunity 36:133–141. [DOI] [PubMed] [Google Scholar]
- Holmén C, Piehl F, Hillert J, Fogdell‐Hahn A, Lundkvist M, Karlberg E, Nilsson P, Dahle C, Feltelius N, Svenningsson A, Lycke J, Olsson T. 2011. A Swedish national post‐marketing surveillance study of natalizumab treatment in multiple sclerosis. Mult Scler 17:708–719. [DOI] [PubMed] [Google Scholar]
- Johns LD, Sriram S. 1993. Experimental allergic encephalomyelitis: Neutralizing antibody to TGFβ1 enhances the clinical severity of the disease. J Neuroimmunol 47:1–7. [DOI] [PubMed] [Google Scholar]
- Khademi M, Kockum I, Andersson ML, Iacobaeus E, Brundin L, Sellebjerg F, Hillert J, Piehl F, Olsson T. 2011. Cerebrospinal fluid CXCL13 in multiple sclerosis: a suggestive prognostic marker for the disease course. Mult Scler 17:335–343. [DOI] [PubMed] [Google Scholar]
- King IL, Dickendesher TL, Segal BM. 2009. Circulating Ly‐6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood 113:3190–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalive PH, Paglinawan R, Biollaz G, Kappos EA, Leone DP, Malipiero U, Relvas JB, Moransard M, Suter T, Fontana A. 2005. TGF‐beta‐treated microglia induce oligodendrocyte precursor cell chemotaxis through the HGF‐c‐Met pathway. Eur J Immunol 35:727–737. [DOI] [PubMed] [Google Scholar]
- Lanz TV, Ding Z, Ho PP, Luo J, Agrawal AN, Srinagesh H, Axtell R, Zhang H, Platten M, Wyss‐Coray T, Steinman L. 2010. Angiotensin II sustains brain inflammation in mice via TGF‐β. J Clin Invest 120:2782–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laouar Y, Town T, Jeng D, Tran E, Wan Y, Kuchroo VK, Flavell RA. 2008. TGF‐beta signaling in dendritic cells is a prerequisite for the control of autoimmune encephalomyelitis. Proc Natl Acad Sci USA 105:10865–10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann H. 2003. Axonal injury in multiple sclerosis. J Neurol Neurosurg Psychiatr 74:695–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Amarnath S, Chen W. 2006. Requirement of CD28 signaling in homeostasis/survival of TGF‐beta converted CD4+CD25+ Tregs from thymic CD4+CD25‐ single positive T cells. Transplantation 82:953–964. [DOI] [PubMed] [Google Scholar]
- Luo J, Ho PP, Buckwalter MS, Hsu T, Lee LY, Zhang H, Kim D‐K, Kim S‐J, Gambhir SS, Steinman L, Wyss‐Coray T. 2007. Glia‐dependent TGF‐β signaling, acting independently of the TH17 pathway, is critical for initiation of murine autoimmune encephalomyelitis. J Clin Invest 117:3306–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malipiero U, Koedel U, Pfister H‐W, Levéen P, Bürki K, Reith W, Fontana A. 2006. TGFbeta receptor II gene deletion in leucocytes prevents cerebral vasculitis in bacterial meningitis. Brain 129:2404–2415. [DOI] [PubMed] [Google Scholar]
- Meoli EM, Oh U, Grant CW, Jacobson S. 2011. TGF‐β signaling is altered in the peripheral blood of subjects with multiple sclerosis. J Neuroimmunol 230:164–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Mack M, Schmidt H, Brück W, Djukic M, Zabel MD, Hille A, Priller J, Prinz M. 2009. CCR2+Ly‐6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain 132:2487–2500. [DOI] [PubMed] [Google Scholar]
- Moransard M, Dann A, Staszewski O, Fontana A, Prinz M, Suter T. 2011. NG2 expressed by macrophages and oligodendrocyte precursor cells is dispensable in experimental autoimmune encephalomyelitis. Brain 134:1315–1330. [DOI] [PubMed] [Google Scholar]
- Moransard M, Sawitzky M, Fontana A, Suter T. 2010. Expression of the HGF receptor c‐met by macrophages in experimental autoimmune encephalomyelitis. Glia 58:559–571. [DOI] [PubMed] [Google Scholar]
- Murphy AC, Lalor SJ, Lynch MA, Mills KHG. 2010. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav Immun 24:641–651. [DOI] [PubMed] [Google Scholar]
- Ostroukhova M. 2006. Treg‐mediated immunosuppression involves activation of the Notch‐HES1 axis by membrane‐bound TGF. J Clin Invest 116:996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsa R, Andresen P, Gillett A, Mia S, Zhang XM, Mayans S, Holmberg D, Harris RA. 2012. Adoptive Transfer of Immunomodulatory M2 Macrophages Prevents Type 1 Diabetes in NOD Mice. Diabetes 61:2881–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. 2006. Principal components analysis corrects for stratification in genome‐wide association studies. Nat Genet 38:904–909. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd‐Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC. 2007. PLINK: A tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet 81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racke MK, Dhib‐Jalbut S, Cannella B, Albert PS, Raine CS, McFarlin DE. 1991. Prevention and treatment of chronic relapsing experimental allergic encephalomyelitis by transforming growth factor‐beta 1. J Immunol 146:3012–3017. [PubMed] [Google Scholar]
- Racke MK, Siram S, Carlino J, Cannella B, Raine CS, McFarlin DE. 1993. Long‐term treatment of chronic relapsing experimental allergic encephalomyelitis by transforming growth factor‐β2. J Neuroimmunol 46:175–183. [DOI] [PubMed] [Google Scholar]
- Sawcer S, Hellenthal G, Pirinen M, Spencer CCA, Patsopoulos NA, Moutsianas L, Dilthey A, Su Z, Freeman C, Hunt SE, Edkins S, Gray E, Booth DR, Potter SC, Goris A, Band G, Oturai AB, Strange A, Saarela J, Bellenguez C, Fontaine B, Gillman M, Hemmer B, Gwilliam R, Zipp F, Jayakumar A, Martin R, Leslie S, Hawkins S, Giannoulatou E, D'alfonso S, Blackburn H, Martinelli Boneschi F, Liddle J, Harbo HF, Perez ML, Spurkland A, Waller MJ, Mycko MP, Ricketts M, Comabella M, Hammond N, Kockum I, McCann OT, Ban M, Whittaker P, Kemppinen A, Weston P, Hawkins C, Widaa S, Zajicek J, Dronov S, Robertson N, Bumpstead SJ, Barcellos LF, Ravindrarajah R, Abraham R, Alfredsson L, Ardlie K, Aubin C, Baker A, Baker K, Baranzini SE, Bergamaschi L, Bergamaschi R, Bernstein A, Berthele A, Boggild M, Bradfield JP, Brassat D, Broadley SA, Buck D, Butzkueven H, Capra R, Carroll WM, Cavalla P, Celius EG, Cepok S, Chiavacci R, Clerget‐Darpoux F, Clysters K, Comi G, Cossburn M, Cournu‐Rebeix I, Cox MB, Cozen W, Cree BAC, Cross AH, Cusi D, Daly MJ, Davis E, de Bakker PIW, Debouverie M, D'hooghe MB, Dixon K, Dobosi R, Dubois B, Ellinghaus D, Elovaara I, Esposito F, Fontenille C, Foote S, Franke A, Galimberti D, Ghezzi A, Glessner J, Gomez R, Gout O, Graham C, Grant SFA, Guerini FR, Hakonarson H, Hall P, Hamsten A, Hartung H‐P, Heard RN, Heath S, Hobart J, Hoshi M, Infante‐Duarte C, Ingram G, Ingram W, Islam T, Jagodic M, Kabesch M, Kermode AG, Kilpatrick TJ, Kim C, Klopp N, Koivisto K, Larsson M, Lathrop M, Lechner‐Scott JS, Leone MA, Leppä V, Liljedahl U, Bomfim IL, Lincoln RR, Link J, Liu J, Lorentzen ÅR, Lupoli S, Macciardi F, Mack M, Marriott M, Martinelli V, Mason D, McCauley JL, Mentch F, Mero IL, Mihalova T, Montalban X, Mottershead J, Myhr K‐M, Naldi P, Ollier W, Page A, Palotie A, Pelletier J, Piccio L, Pickersgill T, Piehl F, Pobywajlo S, Quach HL, Ramsay PP, Reunanen M, Reynolds R, Rioux JD, Rodegher M, Roesner S, Rubio JP, Rückert IM, Salvetti M, Salvi E, Santaniello A, Schaefer CA, Schreiber S, Schulze C, Scott RJ, Sellebjerg F, Selmaj KW, Sexton D, Shen L, Simms‐Acuna B, Skidmore S, Sleiman PMA, Smestad C, Sørensen PS, Søndergaard HB, Stankovich J, Strange RC, Sulonen A‐M, Sundqvist E, Syvänen A‐C, Taddeo F, Taylor B, Blackwell JM, Tienari P, Bramon E, Tourbah A, Brown MA, Tronczynska E, Casas JP, Tubridy N, Corvin A, Vickery J, Jankowski J, Villoslada P, Markus HS, Wang K, Mathew CG, Wason J, Palmer CNA, Wichmann HE, Plomin R, Willoughby E, Rautanen A, Winkelmann J, Wittig M, Trembath RC, Yaouanq J, Viswanathan AC, Zhang H, Wood NW, Zuvich R, Deloukas P, Langford C, Duncanson A, Oksenberg JR, Pericak‐Vance MA, Haines JL, Olsson T, Hillert J, Ivinson AJ, De Jager PL, Peltonen L, Stewart GJ, Hafler DA, Hauser SL, McVean G, Donnelly P, Compston A. 2011. Genetic risk and a primary role for cell‐mediated immune mechanisms in multiple sclerosis. Nature 476:214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh C, Wimmer I, Hametner S, Haider L, Van Dam A‐M, Liblau RS, Smith KJ, Probert L, Binder CJ, Bauer J, Bradl M, Mahad D, Lassmann H. 2014. Oxidative tissue injury in multiple sclerosis is only partly reflected in experimental disease models. Acta Neuropathol 128:247–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sie C, Korn T, Mitsdoerffer M. 2014. Th17 cells in central nervous system autoimmunity. Exp Neurol 262:18–27. [DOI] [PubMed] [Google Scholar]
- Speck S, Lim J, Shelake S, Matka M, Stoddard J, Farr A, Kuchroo V, Laouar Y. 2014. TGF‐β signaling initiated in dendritic cells instructs suppressive effects on Th17 differentiation at the site of neuroinflammation. PLoS One 9:e102390‐ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter T, Biollaz G, Gatto D, Bernasconi L, Herren T, Reith W, Fontana A. 2003. The brain as an immune privileged site: Dendritic cells of the central nervous system inhibit T cell activation. Eur J Immunol 33:2998–3006. [DOI] [PubMed] [Google Scholar]
- Vladimirova O, O'Connor J, Cahill A, Alder H, Butunoi C, Kalman B. 1998. Oxidative damage to DNA in plaques of MS brains. Mult Scler 4:413–418. [DOI] [PubMed] [Google Scholar]
- Wieghofer P, Knobeloch K‐P, Prinz M. 2015. Genetic targeting of microglia. Glia 63:1–22. [DOI] [PubMed] [Google Scholar]
- Wyss‐Coray T, Borrow P, Brooker MJ, Mucke L. 1997. Astroglial overproduction of TGF‐β1 enhances inflammatory central nervous system disease in transgenic mice. J Neuroimmunol 77:45–50. [DOI] [PubMed] [Google Scholar]
- Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, Wu PM, Doykan CE, Lin J, Cotleur AC, Kidd G, Zorlu MM, Sun N, Hu W, Liu L, Lee JC, Taylor SE, Uehlein L, Dixon D, Gu J, Floruta CM, Zhu M, Charo IF, Weiner HL, Ransohoff RM. 2014. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med 211:1533–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X‐M, Lund H, Mia S, Parsa R, Harris RA. 2014. Adoptive transfer of cytokine‐induced immunomodulatory adult microglia attenuates experimental autoimmune encephalomyelitis in DBA/1 mice. Glia 62:804–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information 1
Supporting Information 2
Supporting Information 3
Supporting Information 4
Supporting Information 5