

Abstract
Over the last several decades, our understanding of the genetic variation, pathophysiology, and complications of the hemolytic anemia associated with red cell pyruvate kinase deficiency (PKD) has expanded. Nonetheless, there remain significant gaps in our knowledge with regard to clinical care and monitoring. Treatment remains supportive with phototherapy and/or exchange transfusion in the newborn period, regular or intermittent red cell transfusions in children and adults, and splenectomy to decrease transfusion requirements and/or anemia related symptoms. In this article, we review the clinical diversity of PKD, the current standard of treatment and for supportive care, the complications observed, and future treatment directions.Am. J. Hematol. 90:825–830, 2015. © 2015 Wiley Periodicals, Inc.
Introduction
Red cell pyruvate kinase deficiency (PKD) is the most common glycolytic defect causing congenital non‐spherocytic hemolytic anemia. Pyruvate kinase converts phosphoenolpyruvate to pyruvate, creating 50% of the red cell total ATP. Red cell longevity is dependent on the ATP produced in glycolysis (Fig. 1). Thus, PKD leads to less ATP and a shortened red cell lifespan. The red cells in PKD are variably damaged with the youngest red cells, most dependent on glycolysis and increased ATP levels, at highest risk for destruction, while the older red cells are less severely affected 1, 2. Travel through the splenic capillaries causes damage to affected red cells, which are variably cleared by both the spleen and liver.
Figure 1.
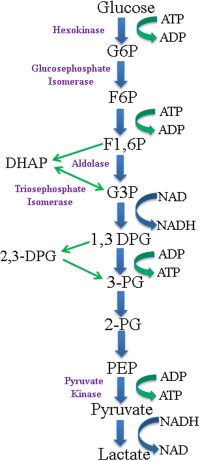
The Emden‐Meyerhof pathway. G6P, glucose‐6‐phosphate; F6P, fructose‐6‐phophate; F1,6P, fructose 1,6‐phosphate; DHAP, dihydroxyacetone phosphate; G3P, glucose‐3‐phosphate; 1,3‐DPG, 1,3‐diphosphoglycerate; 2,3‐DPG, 2,3‐diphosphoglycerate; 3‐PG, 3‐phosphoglycerate; 2‐PG, 2‐phosphoglycerate; PEP, phosphoenolpyruvate. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Human PK [E.C. nomenclature 2.7.1.40] is regulated through expression of four isoenzymes, with red cell PK restricted to the R‐type isoenzyme. Both erythrocyte and liver pyruvate kinase are expressed under the control of a 9.5 kilobase gene, the PK‐LR gene, located on chromosome 1q21. Over 200 mutations have been described in patients with PKD 3, 4. The most commonly reported are missense mutations, including 1529G→A in the United States and Europe, 1456C→T in Southern Europe, and 1468C→T in Asia 3, 4, 5, 6, 7, 8. The disease is transmitted as an autosomal recessive trait in which homozygotes and compound heterozygotes are common. The estimated prevalence of the disease based on the most common PK‐LR mutations associated in a Caucasian population is 51 per million population 9, 10. A particularly high frequency exists among the Pennsylvania Amish, associated with a homozygous 1436G→A mutation, and in the Romani communities, associated with a 1,149 base pair deletion and loss of exon 11 11. As with other autosomal recessive conditions, PKD is more common in specific populations due to the founder effect 12.
Reported cases are fewer than predicted, most likely related to a combination of undiagnosed PKD causing early/intra‐uterine death, under‐diagnosis, and misdiagnosis of mildly affected individuals. Also, there is under‐reporting of cases in general due to the small number of patients followed at individual centers and lack of a central reporting process. PKD has been shown to have a protective effect against replication of the malaria parasite in human red cells, although it is not clear whether PK‐LR mutant alleles are more prevalent in malaria endemic areas 13.
The clinical features of PKD are highly variable. Prenatal hydrops fetalis has been reported 14. After birth, infants with indirect hyperbilirubinemia often require phototherapy and/or exchange transfusion. PKD should always be considered in newborns with severe hyperbilirubinemia of unknown etiology. Infants and young children can be transfusion dependent for years until, or even after, splenectomy. Although the anemia tends to stabilize in adulthood, exacerbations occur with acute infections, stress, and pregnancy. Since 2,3 diphosphoglycerate is elevated in individuals with PKD, the anemia may be better tolerated than in other conditions, because the oxygen dissociation curve is shifted to favor unloading of oxygen in the tissues 15, 16. However, surveys of fatigue and quality of life in these patients have not been reported.
Since the first description of PKD in 1961 17, much has been learned about the genetic diversity of the disease, red cell clearance, clinical complications, and treatment with red cell transfusions and splenectomy. Nevertheless, the diagnosis of the condition remains challenging in many patients particularly those without a family history. The timing and modality of monitoring for complications remains unclear. Treatment remains supportive rather than curative.
Current State of Diagnosis
The possibility of PKD is considered in patients with active hemolysis with no findings suggestive of an acquired autoimmune process, red cell membrane defect, or hemoglobinopathy. The diagnosis in the absence of a known family history can be quite challenging. Patients can have a variable phenotype, normal or near‐normal red cell morphology, and enzyme testing can be falsely normal. Historically, enzyme testing has been the gold standard for diagnosis, and PK‐LR gene testing has been performed only on a research basis. More recently, PK‐LR gene testing has become clinically available and is now pursued to confirm equivocal cases.
Pyruvate kinase enzyme activity is the gold standard for initial testing for PKD. Although low enzyme activity is consistent with the diagnosis of PKD, falsely normal levels are sometimes encountered due to contamination with normal donor cells in transfused patients, incomplete leukocyte removal, persistence of the M2 (muscle) isoform in the mature red cell, or non‐physiologic substrate concentrations 18, 19. In the presence of hemolysis, comparing pyruvate kinase activity to the activity of other cell age dependent enzymes often provides evidence of PKD. For example, if PK activity is normal while the activity of other age dependent enzymes is increased, this is very suggestive of PKD 20. Notably, to the extent it has been evaluated, PK enzyme activity is not correlated with the degree of anemia or hemolysis but may correlate with reticulocytosis 21, 22.
Molecular diagnosis through sequencing the exons, flanking regions, and promotor of the PK‐LR gene is now clinically available. The interpretation of compound heterozygous mutations is limited without parent samples, as it may not be certain whether mutations are in cis or trans. Furthermore, large deletions and intron mutations at cryptic splice sites can be difficult to detect; thus leading to falsely negative results. Although the role of genetic testing is evolving, molecular analysis should be considered to confirm the diagnosis of PKD and also in cases when the diagnosis is not clear after initial enzyme testing (Table 1). Patients with clinical characteristics suggestive of PKD and challenging diagnostic testing results, should be referred to centers with a known interest and expertise in evaluating red cell enzymopathies. To date, no reported patients are predicted to be null on both alleles of the PK‐LR gene.
Table 1.
Framework for Diagnostic Testing for Pyruvate Kinase Deficiency
| PK enzyme activity testing: initial test, consider if any of the following are present: |
| 1. Congenital, chronic, non‐spherocytic, hemolytic anemia without evidence of immune mediated hemolysis, red cell membrane disorder, hemoglobin abnormality, or glucose‐6‐phosphate dehydrogenase deficiency |
| 2. Transfusion dependence since birth with no obvious etiology |
| 3. Unexplained severe neonatal hyperbilirubinemia |
| 4. Reticulocytosis that increases after splenectomy |
| 5. Family history of Pyruvate Kinase Deficiency |
| Molecular PK‐LR Analysis: Secondary/confirmatory test, consider after sending the enzyme activity testing if any of the following are present: |
| 1. Normal or low‐normal pyruvate kinase activity with elevated activity of other age dependent red cell enzymes |
| 2. Chronically transfused patient whose assayed enzyme activity could be falsely normal |
| 3. Low pyruvate kinase activity in the absence of a family history |
Patients with PKD have a variable laboratory phenotype ranging from mild anemia to transfusion dependent hemolysis. In a prior report of 61 patients, the median pre‐splenectomy hemoglobin was 9.8 g/dl with a range of 2.2‐14.4 g/dl 23. The reticulocyte count is increased but is not necessarily proportional to the severity of hemolysis. Moreover, since reticulocytes are preferentially sequestered in the spleen, splenectomy paradoxically causes a rise in both hemoglobin and reticulocytes 2, 24. Indirect bilirubin is typically increased and usually persists after splenectomy 23. Red cell morphology can be variable, including the presence of echinocytes, but may also be normal (Fig. 2). Zanella et al. have previously described a framework for categorizing patients by clinical severity (Table 2).
Figure 2.
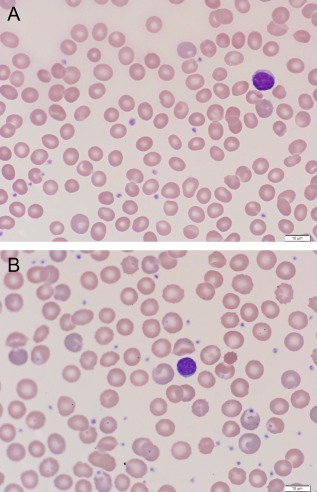
Blood smear findings in a transfusion dependent patient with pyruvate kinase deficiency before splenectomy (Figure 2A) and after splenectomy when no longer requiring red cell transfusions (Figure 2B). The red cell findings before splenectomy are mild, whereas, after splenectomy, polychromasia is more prominent and the characteristic echinocytes are more pronounced. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Table 2.
Categorization of Pyruvate Kinase Deficiency Severity by Laboratory Findings and Transfusion Patterns
| Mild | Moderate | Severe | |
|---|---|---|---|
| Age at Diagnosis | Variable from childhood to adulthood | Birth/Infancy | |
| Neonatal Jaundice | 30–50% of patients, minority require exchange transfusion | 90% of patients, majority require exchange transfusions | |
| Transfusions | Rare, confined to exacerbations only | Common, confined to exacerbations only | Transfusion dependent before splenectomy |
| Median hemoglobin | |||
| Before Splenectomy | 11 g/dl | 9 g/dl | 6.8 g/dl |
| After Splenectomy | Rarely splenectomized | 10 g/dl | 8.4 g/dl |
| Complications | Iron overload | ||
| Common Molecular Findings | 1456T homozygotes or compound heterozygotes | 1529A homozygotes or compound heterozygotes | Stop codons, Frameshift, splicing, large deletions |
From Zanella et al. revised with minor modifications with permission 23.
Although multiple reports describe a genotype‐phenotype relationship, the clinical manifestations of PKD vary depending on many other factors including the patient's genetic background, presence of other red cell abnormalities, and differences in splenic function 23, 25. These findings are most readily noted in the variability among members of the same family with identical genotypes.
Current State of Therapies
Curative therapy for PKD is not currently available. Current treatment strategies, including maintaining an adequate hemoglobin through transfusion and, in many cases, splenectomy, have not changed since the disease was first described in 1961.
Care of the newborn infant with PKD is critical as severe hyperbilirubinemia is common, even in patients who later have mild phenotypes 23. Newborns often require phototherapy and/or exchange transfusions. Rare cases of fulminant hepatic failure related to PKD have been described, possibly related to expression of both liver and red cell pyruvate kinase by the PK‐LR gene 26. Chronic regular blood transfusions in infants are often necessary, although the anemia can improve with age 27. The optimal hemoglobin nadir and transfusion interval have not been reported. A hemoglobin nadir of 7‐9 g/dl is sufficient in most children; however, adults and infants may require a higher hemoglobin level. The anemia in adulthood is more constant with many adults only needing intermittent transfusions for acute hemolytic stressors. Elevated reticulocyte counts indicate rapid red cell turnover and thus a need for supplemental folic acid or a multivitamin containing folate but without iron.
Splenectomy as a treatment for PKD was first described in two patients in 1966 28. Since that time, total splenectomy has proven to be an effective therapy for eliminating or decreasing transfusion dependence, raising the baseline hemoglobin, and increasing the absolute reticulocyte count. As is true for many hematologic conditions, splenectomy as a therapeutic modality was adopted without controlled clinical trials. Data are available from observational experiences of large centers. Splenectomy usually results in a rise in hemoglobin of 1‐3 g/dl and is recommended in severely affected, transfusion dependent patients and in those who do not tolerate anemia 21, 29. Splenectomy may also improve the survival of transfused cells when splenomegaly is present. In most cases, splenectomy removes the need for regular transfusions. However, the optimal timing of splenectomy in transfusion dependent toddlers and children is unclear but involves weighing the risk of post‐splenectomy sepsis with the risk of allo‐sensitization and transfusional hemosiderosis. Following splenectomy, some patients may require intermittent transfusions associated with infections, and, in the minority of patients, there may be a continued requirement for transfusions. Splenectomy is associated with a life‐long risk of sepsis with encapsulated organisms. In addition, concern has been raised about the risk of thromboembolic disease after splenectomy for PKD, but the overall risk of post‐splenectomy thrombosis is not clear and does not appear to be as high as in other congenital hemolytic anemias 30.
With the emergence of antibiotic‐resistant pneumococci and concern for risk of post‐splenectomy thrombosis, partial splenectomy has been considered as an alternative to full splenectomy in multiple types of congenital hemolytic anemias. The goal of partial splenectomy, in which at least 80–90% of the spleen is removed, is to decrease hemolysis while leaving behind a functioning splenic remnant for phagocytic function. There are a few case reports documenting the results of partial splenectomy in PKD patients with two documented failures and one documented success with an increase in the baseline hemoglobin and reduction in transfusions 31, 32, 33. In practice, most clinicians do not recommend partial splenectomy for PKD.
Cure through hematopoietic stem cell transplantation is theoretically possible. Bone marrow transplantation has been demonstrated to cure the PK deficiency and anemia in a mouse model of PKD 34. To date, one successful report in humans has been published of a 5 year old transfusion dependent, non‐splenectomized male who received an HLA‐matched sibling allogeneic transplant from his unaffected sister. At last report, 3 years after the transplant, the patient remained fully engrafted with normal PK enzyme activity 35. Since stratification for severity in PKD remains elusive, it is difficult to assess the risk to benefit ratio of transplant in individual patients and, in those patients in whom transplant is considered, consultation with centers with significant experience with PKD can be useful.
Current State of Supportive Care
Iron overload is common in PKD, in both chronically transfused and transfusion‐independent individuals. In transfusion‐independent individuals, additional factors for iron loading include splenectomy, a degree of ineffective erythropoiesis, and co‐inheritance of hereditary hemochromatosis mutations 36, 37, 38. Transfusional hemosiderosis is a predictable complication of chronic transfusion therapy and monitoring through measurements of plasma ferritin, liver iron burden by R2 (Ferriscan®), and/or liver, pancreas, and cardiac iron burden by T2* magnetic resonance imaging (MRI) methods is indicated. The age at which to begin these monitoring studies is not clear. Taking a guide from sickle cell anemia and thalassemia, evaluation of iron status after 10–20 red cell transfusions is a reasonable milepost, or, for non‐heavily transfused patients, when a patient can tolerate a non‐sedated MRI assessment. In addition, the type and frequency of monitoring for iron overload in transfusion‐independent or intermittently transfused patients (transfused for acute exacerbations only) is less clear. Plasma ferritin levels and transferrin saturation (iron/total iron binding capacity) measurements are simple screening tests, but they do not directly assess tissue iron burden, and the level at which more direct assessment, such as MRI, is indicated has not been determined for these measures.
Similarly to thalassemia major, data in individual patients with PKD suggests that ferritin may not correlate well with degree of iron overload by MRI 37. Given that hepatic iron overload has occurred in transfusion‐independent individuals with PKD, annual MRI monitoring may be indicated in all adult PKD patients 37, 39. Chelation may be necessary in many patients. Drawing from experience in treating thalassemia, chelation therapy may be indicated with ferritin >1,000 ng/ml, liver iron concentration >3 mg/g dry weight liver, and/or T2* cardiac iron <20 ms. Since ferritin may underestimate liver iron concentration, assessment by MRI should be considered when ferritin >500 ng/ml. Reports have described effective use of all three iron chelation agents available in the US, desferroxamine, deferasirox, and deferiprone 21, 37, 40.
Before splenectomy, all PKD patients should receive immunizations according to the Center for Disease Control guidelines for patients with asplenia, including the pneumococcal, meningococcal, and Haemophilus influenza vaccines. Post‐splenectomy, these vaccines should be boosted at regular intervals according to the same guidelines and any newly recommended vaccines should be administered in children and adults. Providers must be aware that these guidelines vary from time to time (http://www.cdc.gov/vaccines/schedules). Splenectomized patients should seek urgent medical care for all fevers ≥ 101.5 F due to the risk of sepsis with encapsulated organisms. These febrile, splenectomized individuals should have a blood culture drawn and then broad spectrum parenteral antibiotics should be administered. Penicillin prophylaxis generally is recommended for 1–2 years post‐splenectomy. Continued penicillin prophylaxis beyond this time period is more controversial. Some clinicians consider a lifetime course of prophylaxis, particularly for those patients who live a long distance from a medical center.
As in other congenital hemolytic anemias, gallstones occur at an earlier age than in the healthy population. Pancreatitis can occur as an acute complication of biliary tract disease. In contrast to hereditary spherocytosis, gallstones are also common in splenectomized PKD patients. Thus, the risk continues for gallstones to develop regardless of splenectomy status 21. For this reason, in order to avoid a second surgery and future complications, some advocate for a cholecystectomy at the time of splenectomy, even in the absence of gallstones. Concomitant Gilbert Syndrome should be suspected in patients with higher than expected indirect bilirubin measurements, which may predispose to a higher rate of gallstones. Genetic testing for the UGT1A1 promoter polymorphism of Gilbert syndrome may be indicated in such patients.
Successful pregnancies and deliveries have been reported in women with PKD 41, 42, 43, 44. Pregnancy is associated with increased hemolysis in some individuals; thus, regular transfusions may be necessary even in women who were transfusion‐independent before pregnancy. No guidelines exist regarding indications for blood transfusion during pregnancy, and it is not clear whether splenectomy status or other factors affect the degree of hemolysis in pregnancy. The increased hemolysis observed during pregnancy abates soon after delivery 43. Women should be counseled regarding the potential for increased hemolysis during pregnancy, and the requirement for close follow up with both an obstetrician and hematologist 41.
Folate requirement is likely higher during pregnancy related to increased hemolysis. Notably, women with PKD should not receive iron supplementation during pregnancy due to the high incidence and risk of iron overload in these individuals. Women with a prior transfusion history should be screened early in pregnancy for HIV and hepatitis viruses. Since iron overload is common in PKD, including in transfusion‐independent women, consideration must be given to maternal cardiac screening for myocardial hypertrophy related to anemia or dysfunction related to iron 44. Reported pregnancy complications include miscarriages, pre‐ecclampsia, and growth restricted infants; however, it is not known if the incidence of these types of complications is increased over healthy women 44.
Other Complications
Extramedullary hematopoiesis is a known complication of PKD, including lesions causing spinal cord compression 45, 46. Leg ulcers, similar to those reported in patients with sickle cell disease and hereditary spherocytosis, have been reported 47. Two reported PKD patients have developed bone marrow abnormalities at older ages, including a 70 year old who developed myelodysplasia with refractory anemia with ringed sideroblasts and 72 year old who developed chronic myelomonocytic leukemia 48, 49. Some have hypothesized that chronic ineffective erythropoiesis and hemolysis may predispose to clonal abnormalities, but this has not been verified for other types of anemias with similar hematologic findings 48.
Future Directions for Therapy
A gene therapy strategy has not been attempted in human PKD. Studies in mice have shown prolonged expression of PK in the blood and hematopoietic organs of mice after introduction of a viral vector expressing human liver‐type PK cDNA 50. In addition, other investigators have shown increased expression of PK in transgenic mice using a gene‐addition strategy, although this did not fully rescue the mice from their phenotype with continued evidence of ineffective erythropoiesis 51.
No specific drug therapies are available for treatment of PKD. Historic attempts to develop pharmacologic treatments, such as riboflavin and sulphydryl compounds, have been unsuccessful 52, 53, 54. A pharmacologic activator of red cell pyruvate kinase is presently in early phase clinical trials 55. This drug can both activate pyruvate kinase and enhance glycolytic flux in mice and increases ATP and decreases 2,3‐DPG in healthy individuals 55, 56. To date, there are no data from patients with PKD, but a clinical trial is under consideration.
Future Directions for Clinical Care
With a minimal number PKD cases reported in the literature, our understanding of the epidemiology, spectrum of clinical severity, and supportive care in this disease is limited. The Pyruvate Kinase Deficiency Natural History Study (clinicaltrials.gov, NCT02053480) is a longitudinal, multicenter, international patient registry which is collecting retrospective and current clinical information and patient reported outcome measures at enrollment and annually. All participants have PK‐LR mutation testing to confirm their diagnosis. With this information on a large number of phenotypically diverse PKD patients, it should be possible to establish clinical predictors of complications and develop clinical care guidelines. It is anticipated that such guidelines will be useful to generalists, hematologists, and obstetricians who follow a limited number of patients with PKD. Understanding the natural history of PKD will allow for an informed assessment of an affected child or adult's risk of future complications, the indications for specific monitoring, the need for splenectomy, as well as eligibility for future therapies.
Conclusions
Since PKD was first described 55 years ago, our understanding of the red cell defect, the clinical and genetic variability, supportive care, and potential complications has expanded. Despite this understanding, supportive care continues as the main clinical strategy, and evidence for monitoring for complications is lacking. In a rare disease with many potential complications, guidelines for supportive care and monitoring are critical. It is our hope that the international, Pyruvate Kinase Deficiency Natural History Study will improve our understanding of this rare anemia. As our understanding of PKD is enhanced, we anticipate a faster rate of progress in the discovery of new therapeutic interventions.
Conflict of interest:
R.G., A.Z., E.N., D.H.M., S.E., H.Y., and B.G. are scientific advisors for Agios Pharmaceuticals. R.G. is the P.I. of the Pyruvate Kinase Deficiency Natural History Study (NCT02053480), which is sponsored by Agios Pharmaceuticals. R.G. and B.G. assembled the co‐authors based on clinical expertise and prior publications in pyruvate kinase deficiency. The content of this manuscript was conceived, prepared, and written by the authors alone, independent of Agios Pharmaceuticals or any other commercial entity. R.G. and B.G. wrote the first draft of the manuscript, which was revised and commented by A.Z., E.N., D.H.M., S.E., and H.Y.
References
- 1. Nathan DG, Oski FA, Miller DR, Gardner FH. Life‐span and organ sequestration of the red cells in pyruvate kinase deficiency. N Engl J Med 1968;278:73–81. [DOI] [PubMed] [Google Scholar]
- 2. Mentzer WC,J, Baehner, RL , Schmidt‐Schonbein H, et al. Selective reticulocyte destruction in erythrocyte pyruvate kinase deficiency. J Clin Invest 1971;50:688–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Warang P, Kedar P, Ghosh K, Colah R. Molecular and clinical heterogeneity in pyruvate kinase deficiency in India. Blood Cells Mol Dis 2013;51:133–137. [DOI] [PubMed] [Google Scholar]
- 4. Pissard S, Max‐Audit, I , Skopinski L, et al. Pyruvate kinase deficiency in France: A 3‐year study reveals 27 new mutations. Br J Haematol 2006;133:683–689. [DOI] [PubMed] [Google Scholar]
- 5. Zanella A, Bianchi P. Red cell pyruvate kinase deficiency: From genetics to clinical manifestations. Baillieres Best Pract Res Clin Haematol 2000;13:57–81. [DOI] [PubMed] [Google Scholar]
- 6. Baronciani L, Beutler E. Analysis of pyruvate kinase‐deficiency mutations that produce nonspherocytic hemolytic anemia. Proc Natl Acad Sci USA 1993;90:4324–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zarza R, Alvarez, R , Pujades A, et al. Molecular characterization of the PK‐LR gene in pyruvate kinase deficient Spanish patients. Red Cell Pathology Group of the Spanish Society of Haematology (AEHH). Br J Haematol 1998;103:377–382. [DOI] [PubMed] [Google Scholar]
- 8. Lenzner C, Nurnberg, P , Jacobasch G, et al. Molecular analysis of 29 pyruvate kinase‐deficient patients from central Europe with hereditary hemolytic anemia. Blood 1997;89:1793–1799. [PubMed] [Google Scholar]
- 9. Beutler E, Gelbart T. Estimating the prevalence of pyruvate kinase deficiency from the gene frequency in the general white population. Blood 2000;95:3585–3588. [PubMed] [Google Scholar]
- 10. Carey PJ, Chandler, J , Hendrick A, et al. Prevalence of pyruvate kinase deficiency in northern European population in the north of England. Northern Region Haematologists Group. Blood 2000;96:4005–4006. [PubMed] [Google Scholar]
- 11. Baronciani L, Beutler E. Molecular study of pyruvate kinase deficient patients with hereditary nonspherocytic hemolytic anemia. J Clin Invest 1995;95:1702–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Christensen RD, Yaish, HM , Johnson CB, et al. Six children with pyruvate kinase deficiency from one small town: Molecular characterization of the PK‐LR gene. J Pediatr 2011;159:695–697. [DOI] [PubMed] [Google Scholar]
- 13. Ayi K, Min‐Oo, G , Serghides L, et al. Pyruvate kinase deficiency and malaria. N Engl J Med 2008;358:1805–1810. [DOI] [PubMed] [Google Scholar]
- 14. Ferreira P, Morais, L , Costa R, et al. Hydrops fetalis associated with erythrocyte pyruvate kinase deficiency. Eur J Pediatr 2000;159:481–482. [DOI] [PubMed] [Google Scholar]
- 15. Delivoria‐Papadopoulos M, Oski FA, Gottlieb AJ. Oxygen‐hemoglobulin dissociation curves: Effect of inherited enzyme defects of the red cell. Science 1969;165:601–602. [DOI] [PubMed] [Google Scholar]
- 16. Oski FA, Marshall, BE , Cohen PJ, et al. The role of the left‐shifted or right‐shifted oxygen‐hemoglobin equilibrium curve. Ann Intern Med 1971;74:44–46. [DOI] [PubMed] [Google Scholar]
- 17. Tanaka KR, Valentine WN, Miwa S. Pyruvate kinase (PK) deficiency hereditary nonspherocytic hemolytic anemia. Blood 1962;19:267–295. [PubMed] [Google Scholar]
- 18. Lenzner C, Nurnberg, P , Thiele BJ, et al. Mutations in the pyruvate kinase L gene in patients with hereditary hemolytic anemia. Blood 1994;83:2817–2822. [PubMed] [Google Scholar]
- 19. Beutler E, Forman L, Rios‐Larrain E. Elevated pyruvate kinase activity in patients with hemolytic anemia due to red cell pyruvate kinase “deficiency”. Am J Med 1987;83:899–904. [DOI] [PubMed] [Google Scholar]
- 20. Titapiwatanakun R, Hoyer JD, Crain K, Arndt CA. Relative red blood cell enzyme levels as a clue to the diagnosis of pyruvate kinase deficiency. Pediatr Blood Cancer 2008;51:819–821. [DOI] [PubMed] [Google Scholar]
- 21. Zanella A, Fermo E, Bianchi P, Valentini G. Red cell pyruvate kinase deficiency: Molecular and clinical aspects. Br J Haematol 2005;130:11–25. [DOI] [PubMed] [Google Scholar]
- 22. Pekrun A, Lakomek, M , Eber S, et al. [Diagnosis of pyruvate kinase deficiency in the presence of an elevated reticulocyte count]. Dtsch Med Wochenschr 1995;120:1620–1624. [DOI] [PubMed] [Google Scholar]
- 23. Zanella A, Fermo, E , Bianchi P, et al. Pyruvate kinase deficiency: The genotype‐phenotype association. Blood Rev 2007;21:217–231. [DOI] [PubMed] [Google Scholar]
- 24. Matsumoto N, Ishihara, T , Nakashima K, et al. Sequestration and destruction of reticulocyte in the spleen in pyruvate kinase deficiency hereditary nonspherocytic hemolytic anemia. Nihon Ketsueki Gakkai Zasshi 1972;35:525–537. [PubMed] [Google Scholar]
- 25. Rider NL, Strauss, KA , Brown K, et al. Erythrocyte pyruvate kinase deficiency in an old‐order Amish cohort: Longitudinal risk and disease management. Am J Hematol 2011;86:827–834. [DOI] [PubMed] [Google Scholar]
- 26. Raphael MF, Van Wijk, R , Schweizer JJ, et al. Pyruvate kinase deficiency associated with severe liver dysfunction in the newborn. Am J Hematol 2007;82:1025–1028. [DOI] [PubMed] [Google Scholar]
- 27. Boivin P, Ottenwaelter T. [Hereditary haemolytic anaemia due to pyruvate kinase deficiency. Prognosis of neonatal forms (author's transl)]. Nouv Presse Med 1982;11:917–919. [PubMed] [Google Scholar]
- 28. Necheles TF, Finkel HE, Sheehan RG, Allen DM. Red cell pyruvate kinase deficiency. The effect of splenectomy. Arch Intern Med 1966;118:75–78. [PubMed] [Google Scholar]
- 29. Zanella A, Bianchi P, Fermo E. Pyruvate kinase deficiency. Haematologica 2007;92:721–723. [DOI] [PubMed] [Google Scholar]
- 30. Chou R, DeLoughery TG. Recurrent thromboembolic disease following splenectomy for pyruvate kinase deficiency. Am J Hematol 2001;67:197–199. [DOI] [PubMed] [Google Scholar]
- 31. Sandoval C, Stringel G, Weisberger J, Jayabose S. Failure of partial splenectomy to ameliorate the anemia of pyruvate kinase deficiency. J Pediatr Surg 1997;32:641–642. [DOI] [PubMed] [Google Scholar]
- 32. Diesen DL, Zimmerman, SA , Thornburg CD, et al. Partial splenectomy for children with congenital hemolytic anemia and massive splenomegaly. J Pediatr Surg 2008;43:466–472. [DOI] [PubMed] [Google Scholar]
- 33. Rice HE, Oldham, KT , Hillery CA, et al. Clinical and hematologic benefits of partial splenectomy for congenital hemolytic anemias in children. Ann Surg 2003;237:281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morimoto M, Kanno, H , Asai H, et al. Pyruvate kinase deficiency of mice associated with nonspherocytic hemolytic anemia and cure of the anemia by marrow transplantation without host irradiation. Blood 1995;86:4323–4330. [PubMed] [Google Scholar]
- 35. Tanphaichitr VS, Suvatte, V , Issaragrisil S, et al. Successful bone marrow transplantation in a child with red blood cell pyruvate kinase deficiency. Bone Marrow Transplant 2000;26:689–690. [DOI] [PubMed] [Google Scholar]
- 36. Zanella A, Berzuini A, Colombo MB, et al. Iron status in red cell pyruvate kinase deficiency: Study of Italian cases. Br J Haematol 1993;83:485–490. [DOI] [PubMed] [Google Scholar]
- 37. Marshall SR, Saunders PW, Hamilton PJ, Taylor PR. The dangers of iron overload in pyruvate kinase deficiency. Br J Haematol 2003;120:1090–1091. [DOI] [PubMed] [Google Scholar]
- 38. Zanella A, Bianchi P, Iurlo A, et al. Iron status and HFE genotype in erythrocyte pyruvate kinase deficiency: Study of Italian cases. Blood Cells Mol Dis 2001;27:653–661. [DOI] [PubMed] [Google Scholar]
- 39. Finkenstedt A, Bianchi, P , Theurl I, et al. Regulation of iron metabolism through GDF15 and hepcidin in pyruvate kinase deficiency. Br J Haematol 2009;144:789–793. [DOI] [PubMed] [Google Scholar]
- 40. Deeren D. Deferasirox in pyruvate kinase deficiency. Ann Hematol 2009;88:397. [DOI] [PubMed] [Google Scholar]
- 41. Dolan LM, Ryan M, Moohan J. Pyruvate kinase deficiency in pregnancy complicated by iron overload. Bjog 2002;109:844–846. [DOI] [PubMed] [Google Scholar]
- 42. Fanning J, Hinkle RS. Pyruvate kinase deficiency hemolytic anemia: Two successful pregnancy outcomes. Am J Obstet Gynecol 1985;153:313–314. [DOI] [PubMed] [Google Scholar]
- 43. Amankwah KS, Dick BW, Dodge S. Hemolytic anemia and pyruvate kinase deficiency in pregnancy. Obstet Gynecol 1980;55:42S–44S. [DOI] [PubMed] [Google Scholar]
- 44. Wax JR, Pinette MG, Cartin A, Blackstone J. Pyruvate kinase deficiency complicating pregnancy. Obstet Gynecol 2007;109:553–555. [DOI] [PubMed] [Google Scholar]
- 45. Rutgers MJ, van der Lugt PJ, van Turnhout JM. Spinal cord compression by extramedullary hemopoietic tissue in pyruvate‐kinase‐deficiency‐caused hemolytic anemia. Neurology 1979;29:510–513. [DOI] [PubMed] [Google Scholar]
- 46. Hipkins R, Thompson, J , Naidoo P, et al. Images in haematology. Paravertebral extramedullary haemopoiesis associated with pyruvate kinase deficiency. Br J Haematol 2009;147:275. [DOI] [PubMed] [Google Scholar]
- 47. Muller‐Soyano A, Tovar de Roura, E , Duke PR, et al. Pyruvate kinase deficiency and leg ulcers. Blood 1976;47:807–813. [PubMed] [Google Scholar]
- 48. Ryan C, Percy M, O'Brien D, et al. Myelodysplastic syndrome in a patient with hereditary pyruvate kinase deficiency. Hematol J 2004;5:91–92. [DOI] [PubMed] [Google Scholar]
- 49. Vives‐Corrons JL, Florensa L, Muncunill J, et al. Chronic myelomonocytic leukemia associated with hereditary pyruvate kinase deficiency and multiple acquired erythrocyte abnormalities. Acta Haematol 1979;61:168–174. [DOI] [PubMed] [Google Scholar]
- 50. Tani K, Yoshikubo T, Ikebuchi K, et al. Retrovirus‐mediated gene transfer of human pyruvate kinase (PK) cDNA into murine hematopoietic cells: Implications for gene therapy of human PK deficiency. Blood 1994;83:2305–2310. [PubMed] [Google Scholar]
- 51. Kanno H, Utsugisawa, T , Aizawa S, et al. Transgenic rescue of hemolytic anemia due to red blood cell pyruvate kinase deficiency. Haematologica 2007;92:731–737. [DOI] [PubMed] [Google Scholar]
- 52. Zanella A, Rebulla P, Giovanetti AM, et al. Effects of sulphydryl compounds on abnormal red cell pyruvate kinase. Br J Haematol 1976;32:373–385. [DOI] [PubMed] [Google Scholar]
- 53. Blume KG, Arnold H, Hasslinger K, Lohr GW. Effect of riboflavin treatment on human red cell pyruvate kinase deficiency. Clin Chim Acta 1976;71:331–334. [DOI] [PubMed] [Google Scholar]
- 54. Staal GE, van Berkel TJ, Nijessen JG, Koster JF. Normalisation of red blood cell pyruvate kinase in pyruvate kinase deficiency by riboflavin treatment. Clin Chim Acta 1975;60:323–327. [DOI] [PubMed] [Google Scholar]
- 55. Yang H, Merica E, Chen Y, et al. Phase 1 single (SAD) and multiple (MAD) ascending dose studies of the safety, tolerability, and pharmacokinetics/pharmacodynamics (PK/PD) of AG‐348, a first‐in‐ ‐class allosteric activator of pyruvate kinase‐R, in healthy subjects. Am Soc Hematol 2014;124:4007. [DOI] [PubMed] [Google Scholar]
- 56. Kung C, Hill, C , Chen Y, et al. AG‐348 activation of pyruvate kinase in vivo enhances red cell glycolysis in mice. Am Soc Hematol, 2014;124:4010. [Google Scholar]