

Summary
Body weight is determined via both metabolic and hedonic mechanisms. Metabolic regulation of body weight centres around the ‘body weight set point’, which is programmed by energy balance circuitry in the hypothalamus and other specific brain regions. The metabolic body weight set point has a genetic basis, but exposure to an obesogenic environment may elicit allostatic responses and upward drift of the set point, leading to a higher maintained body weight. However, an elevated steady‐state body weight may also be achieved without an alteration of the metabolic set point, via sustained hedonic over‐eating, which is governed by the reward system of the brain and can override homeostatic metabolic signals. While hedonic signals are potent influences in determining food intake, metabolic regulation involves the active control of both food intake and energy expenditure. When overweight is due to elevation of the metabolic set point (‘metabolic obesity’), energy expenditure theoretically falls onto the standard energy–mass regression line. In contrast, when a steady‐state weight is above the metabolic set point due to hedonic over‐eating (‘hedonic obesity’), a persistent compensatory increase in energy expenditure per unit metabolic mass may be demonstrable. Recognition of the two types of obesity may lead to more effective treatment and prevention of obesity.
Keywords: Body weight regulation, energy expenditure, food reward
Introduction
The mechanism by which body weight is regulated by metabolic signals has been studied for several decades. The initial proposal of a lipostatic mechanism for body weight regulation in 1950 1 triggered an intense search for lipostatic signals, including various parabiotic rodent models 2, 3, 4, which may act on the hypothalamic control centre 5, 6, 7. The discovery of leptin in 1994 by positional cloning of the obese gene 8 marked the beginning of an era in which our knowledge of the neuroendocrine regulation of body weight rapidly expanded. Overwhelming experimental evidence has now accumulated in support of a homeostatic regulatory system for body weight. Under such a homeostatic system, an individual's body weight in adulthood is usually maintained at a relatively constant level, fluctuating only slightly within a narrow range around the body weight ‘set point’. This is true not only in people with ‘normal’ weights but also in people who are overweight or obese. The exception is seen in people who are in a non‐steady state, with their body weights gradually increasing over time while they are developing obesity. We will deal with this exception in greater detail later. The controversy related to the concept of a body weight regulatory system is not so much about whether or not homeostatic mechanisms exist, or how they function, but about how this system would account for the current obesity epidemic. If body weight is determined by a genetically programmed set point, why are more and more people obese now than just a few short decades ago, when the population genome pool is considered identical to what we now have? Additionally, strictly with regard to body weight, why does it matter whether or not people eat unhealthy foods or consume energy dense junk foods and beverages since the set point model states that body weight will always return to an individual's set point range no matter how the weight may temporally deviate from it?
There have been alternative models to the set point concept to circumvent this problem that would seem to be irreconcilable with the assumptions of a strict homeostatic metabolic set point model. For example, the settling point model seems to fit the population data better by assuming no active metabolic regulation of body weight (as opposed to the set point model) (see 9). Explanations for the obesity epidemic are also provided by variants of the thrifty gene hypothesis, which focuses upon the adaptive value of enhanced energy storage in the presence of caloric abundance (see 10, 11, 12, 13, 14). Most of the models assume an asymmetrical defense of set point weight and recognize the fact that evolution has provided us with a set of genes that enable the body to defend itself more rigorously against weight loss than against weight gain (see 12, 15).
More recently, evidence has emerged to support the existence of a hedonic system controlling food intake and body weight. This system is operated by neuroendocrine pathways related to the reward characteristics of ingested foods 16, 17, 18 and is non‐homeostatic with regard to the body's metabolism and energetic balance. The dual central nervous system (CNS) mechanisms involved in homeostatic as well as hedonic regulation of body weight have been reviewed extensively in several previous reviews 19, 20, 21. Taken together, we believe that the obesity epidemic is a manifestation of a metabolic disorder that results in expression of an elevated body weight set point in some people (metabolic obesity), and also a hedonic disorder in other individuals that results in persistent and excessive net caloric intake which sustains body weight significantly above the individual's metabolic set point (hedonic obesity). In this article, we will review the evidence that supports this conclusion, as well as how the two types of obesity may be distinguished and treated differentially based upon their underlying aetiology.
Body weight regulation and underlying mechanisms of the common forms of obesity
Homeostatic mechanisms and body weight set point
Under relatively constant environmental conditions, an individual's body weight is ‘auto‐regulated’, i.e. the body senses and processes various metabolic signals regarding its energetic status and adjusts its metabolic responses without conscious control. The conscious mind can direct an individual's feeding behaviour and physical activities, but it is not a sustaining factor in the determination of body weight. The homeostatic regulation of body weight is similar to that of other physiological parameters, such as body temperature, blood pressure or blood glucose. The principle of all these homeostatic systems is analogous to a feedback thermostatic system, by which room temperature is regulated such that it is maintained within a narrow range around a pre‐set temperature. In general, body weight seems to oscillate in a similar manner 22 and is stable over a long period of time in most individuals; people may lose 5–10 lb or more in response to an acute illness 23, but regain it fairly rapidly after recovery from the illness without much conscious effort. Similarly, it is commonplace for many individuals to find that following a weight gain of 5–10 lb during a vacation or holiday season, their weight falls back to its previous level after they resume their ‘normal’ life.
Underlying such phenomena is a regulatory system consisting of an extremely complex neuroendocrine network termed the homeostatic energy balance circuitry. A full description of the components of this network and the supporting evidence thereof would require an extensive review of a large body of the literature. Thus, we refer the interested readers to several specific reviews on this topic 6, 24, 25, 26, 27, 28, 29, 30, 31. For the purpose of this article, it suffices to point out the existence, as supported by overwhelming evidence, of a CNS neuroendocrine energy balance circuitry, which is composed of specific nuclei in various brain regions, most prominently the hypothalamic arcuate nucleus (ARC), the paraventricular nucleus, the lateral hypothalamic area and the nucleus of solitary tract of the hindbrain 32. It is also important to point out that genetic, epigenetic, imprinting and early developmental factors may all influence how the circuitry functions in a given individual (see 21, 25). The most important feature of this CNS circuitry is the programmed body weight set point (see 6, 24), which demarcates a threshold level, above and below which, opposite sets of metabolic reactions will be activated in order to restore the set point body weight (see Fig. 1). More specifically, energy surplus or deficit leads to a change in body weight above or below the set point body weight, which is sensed by the energy balance circuitry of the brain through an array of feedback signals from the periphery. For example, leptin, an adipocyte‐derived hormone that is expressed in proportion to adipocyte size and fat mass 33, 34, 35, 36, 37, 38, 39, serves as one such feedback signal. The circulating leptin level (an indicator of the body's fatness) is sensed by specific neurons in the hypothalamus, particularly the POMC/CART and AgRP/NPY neurons, as well as other leptin‐responsive neurons in the homeostatic energy balance circuit (see 26, 27, 30, 40). Activation of these neurons then acts in concert to prompt anabolic or catabolic functions in response to changes in the circulating leptin level. Increased leptin signalling associated with weight gain elicits catabolic activities by increasing energy expenditure and suppressing food intake, promoting weight loss, whereas decreased leptin signalling associated with weight loss elicits anabolic activities by reducing energy expenditure and increasing food intake, promoting weight gain.
Figure 1.
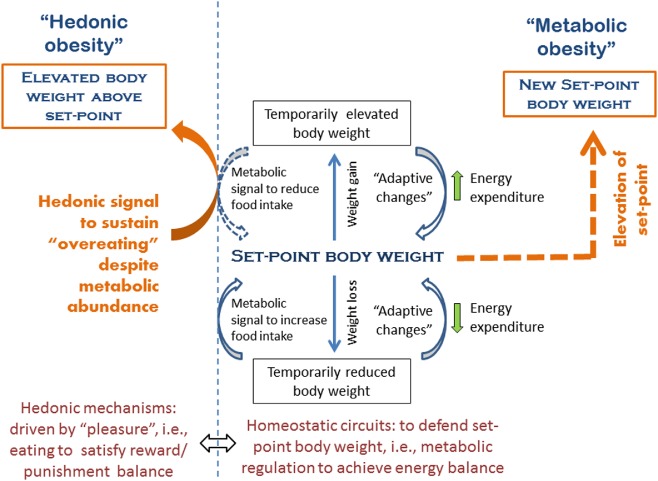
Metabolic and hedonic obesity as related to their respective mechanisms of weight gain. The homeostatic weight regulatory system located primarily in hypothalamus and brainstem accounts for weight regulation around a body weight set point. Deviation of body weight from this set point elicits a compensatory increase or decrease in food intake (cumulative over a long time period) and energy expenditure (both resting and non‐resting) in an opposite direction in order to restore the previous body weight set point. Obesity results from an elevation of the metabolic set point (see details in the text) that is characterized by an elevated body weight which is metabolically defended just as normal body weight is defended at its set point; we term an elevated body weight set point ‘metabolic obesity’. Hedonic eating is governed by the reward system to satisfy the need of pleasure and is non‐homeostatic with regard to energy balance. Dysfunction of the reward system may lead to hedonic over‐eating in susceptible individuals in the face of metabolic signals indicating an energy surplus, leading to sustained weight gain above the metabolic set point weight; we term this form of obesity ‘hedonic obesity’.
In addition to leptin, the CNS energy balance circuitry receives numerous other feedback signals that in various ways reflect the body's energy status (see 6, 26, 40, 41, 42, 43). Well‐recognized peripheral signals include insulin, which also serves to measure the body's adiposity (see 26, 40) and circulating levels of fatty acids, glucose and amino acids, which may reflect the fuel availability (see 6, 41). Ghrelin, cholecystokinin (CCK), glucagon‐like pepetide‐1 (GLP‐1) and various other gut hormones also provide feedback signals about current feeding status (see 42, 44). Direct nervous transmission of taste stimulation relayed by the cranial nerves and various visceral signals transmitted by the vagus nerve also participates (see 32, 43). Although many of these sensory systems are primarily involved in regulating short‐term energy balance, i.e. meal size and feeding frequency, some of them may also provide afferent signals to the homeostatic energy balance circuitry and work in concert with lipostatic signals, such as leptin and insulin, to regulate long‐term energy balance and, hence, body weight (see 26, 41, 43, 45). The direct participation by some of the meal‐related signals, such as ghrelin and peptide YY (PYY), in the regulation of the activities of ARC neurons (see 42, 44) may provide an explanation for the ability of short‐term feeding signals to cause changes in the long‐term energy balance.
Defining metabolic obesity: expression of an elevated body weight set point
There is little doubt that the body weight set point, like body height and other phenotypic characteristics, is influenced by genetic and environmental elements (see 25). It is estimated that 40–80% of the variance in body mass index is attributable to genetic elements 46, 47. There is also good evidence that the body weight set point is modifiable by environmental factors, and that persistent weight gain in response to environmental challenges can, in some cases, lead to an upward shift of body weight set point that will be metabolically defended 48, 49. This is well illustrated in rodent models. For example, outbred Sprague‐Dawley rats have a characteristic growth trajectory when they are fed ad libitum with a regular chow diet; any deviation from this trajectory due to caloric restriction or gavage overfeeding is temporary 50, 51. The rats will quickly return to its ‘set point’ weigh trajectory as soon as they are back to ad libitum chow diet. However, when the rats are fed a palatable high‐energy (HE) diet, about half of the rats become obese and they now follow a new growth trajectory, which is significantly above the original one 52, 53. Importantly, upon a prolonged period of HE diet feeding and weight gain, this new and elevated ‘growth trajectory’ is defended against overfeeding or caloric restriction just as in the lean rats when they are subjected to the same caloric manipulations 50. Obviously, the tendency for the expression of an elevated body weight set point in response to environmental changes is also encoded in individual rat's genetic constitution. In fact, about half of the outbred Sprague‐Dawley rats on HF diet do not become obese during 12–15 weeks of high‐fat (HF) feeding 50, 52, 53. Both obesity‐prone and obesity‐resistant traits can be selectively bred to produce two separate substrains that are then either 100% prone or 100% resistant to the weight‐altering effects of HE diets, respectively 53. The genetic elements in determining the outcomes of environmental influences demonstrated in rodents seem to be present in humans in a similar fashion as not everyone changes his/her set point weight when interacting with an ‘obesogenic’ environment. We term obesity caused by the expression of an elevated set point of body weight (and now defended metabolically) ‘metabolic obesity’ (Fig. 1).
The mechanisms by which dietary manipulations (e.g. the HE diet cited earlier) and weight gain lead to a higher defended set point weight are not fully understood. A large body of evidence suggests that diet‐induced obesity may lead to the development of secondary leptin resistance (i.e. acquired leptin resistance due to changes in environmental factors such as diet and/or weight gain) 12, 27, 54, 55, 56. Development of leptin resistance may be expected to ‘reset’ the body weight set point at a higher level by attenuating the catabolic effects of leptin. In addition to leptin resistance, functional impairment or structural modifications of any major component of the homeostatic energy balance circuitry may also be expected to alter the tonicity of this regulatory system, resulting in resetting the set point. For example, loss of synapses on hypothalamic POMC/CART neurons due to environment‐triggered reactive gliosis 48, 57 and diet‐induced inflammation and/or ageing of hypothalamic neurons 58, 59, 60 have been observed in obese mice fed a high‐fat diet (HFD), compared with their lean controls on normal chow diet. Indeed, permanent inflammation and neuronal damage were observed in the hypothalamus of long‐term HFD‐fed rats and mice, and similar damage was detected via magnetic resonance imaging (MRI) in the hypothalamus of adult obese humans 60. Mechanisms such as these, triggered by sustained intake of HF/sugar diets, likely participate in the eventual resetting of the body weight set point.
Hedonic control of food intake and evidence of altered hedonic pathways in obesity
The aforementioned set point weight regulation system is a homeostatic regulatory system located primarily in the hypothalamus and brainstem that processes internal metabolic signals, and it has been termed the ‘metabolic brain’ 21. Distinct from this ‘metabolic brain’ is the ‘cognitive and emotional brain’, which, in principle, guides food intake based upon the reward value of the food; the latter is governed by a different set of neuroendocrine signals and is non‐homeostatic with regard to energy balance. The brain regions responsible for this reward system are dispersed in the corticolimbic structures (see 61). Multiple neuronal pathways are involved in reward evaluation, which is also modulated by various other factors such as emotion, stress, arousal and metabolic status. This system integrates basic midbrain and hindbrain functions with more complex cortical functions involving arousal at the sight of palatable food items and the procurement of food. However, narrowly defined, the basic components of the reward system at the midbrain level mediate ‘liking’ (the level of pleasure or reward) and ‘wanting’ (the motivation or drive to consume food), which are subconscious processes 62. Implicit ‘liking’ is believed to be mediated by mu‐opioid receptor signalling as well as the CB1 cannabinoid receptor signalling networks (see 20) centred in the nucleus accumbens (NAc) of the ventral striatum and ventral pallidum, whereas implicit ‘wanting’ is chiefly encoded in the mesolimbic dopaminergic neurons that project from ventral tegmental area (VTA) to NAc. Importantly, structures involved in reward calculation (NAc, ventral pallidum and VTA) have connections to other brain areas, including the hippocampus, amygdala, gustatory and orbitofrontal cortex (via thalamus and substantia nigra), hypothalamus and brainstem (see 20). These connections are believed to play an important role in coordinating conscious and unconscious components related to hedonic eating.
Alterations in the food reward system leading to excessive caloric ingestion and obesity may be explained in part on the basis of two prevailing hypotheses related to the ‘liking’ and ‘wanting’ aspects of the reward system. The ‘gluttony hypothesis’ posits a positive correlation between the amount of dopaminergic signalling in an individual and the pleasure derived from the sensory experience of ingestion, whereas the ‘reward deficiency’ hypothesis suggests that a deficiency in dopaminergic signalling is the cause of overindulgence of the food in an attempt to achieve pleasure (see 21). Evidence obtained in animal and human studies supports both hypotheses 63, 64. For example, sustained exposure to sucrose or a palatable mixed diet in rats can lead to elevated dopamine release and transporter expression, as well as down‐regulation of dopamine D1 and D2 receptor expression in the NAc and dorsal striatum 63, 64, 65, 66. The changes seen in the dopamine reward pathway, particularly the down‐regulation of dopamine receptors, are similar to those seen in addictive states, and the concept of ‘food addiction’ has been proposed as an explanation for over‐eating and obesity 67. Advocates of the food addiction concept, in fact, compare the increasing consumption of rewarding food items potentially resulting from reward deficiency to the well‐known addictive process of drug tolerance 68.
Numerous animal studies demonstrate the relationship between defects in the mesolimbic reward system and the onset and/or maintenance of obesity. Geiger et al. demonstrated that laboratory rats fed a cafeteria diet for 15 weeks developed obesity, which was associated with reduced extracellular levels of dopamine in the NAc 69. While the dopaminergic response was not elicited in these obese rats by the regular chow diet, it was by the cafeteria diet, suggesting that a diet‐induced alteration in the food reward system stimulated the drive to indulge in the cafeteria diet and resulted in sustained overfeeding. Decreased excitability of the mu‐opioid receptor signalling system (encoding ‘liking’) has also been tested in animal models. Schwindinger et al. used a genetic mouse model deficient in G protein γ3‐subunit, which is believed to be essential in mediating mu‐opioid receptor signalling, and showed that these mice were resistant to diet‐induced obesity because of a lesser preference to the HF diet than their wild‐type littermates, which became obese on the same diet 70.
In human studies, functional MRI (fMRI) has shown correlations between obesity and overactivation of brain regions corresponding to many components of the reward system, including the gustatory cortex, somatosensory regions, and the limbic and paralimbic regions, in response to palatable food stimuli 71, 72, 73, 74, 75. A negative correlation was also seen between obesity and activation of frontal brain inhibitory regions, implicating a dysfunction related to increased impulsivity or decreased executive function 75, 76. Marked overactivation of many brain regions of the reward system is also consistently seen in obese patients with the rare congenital leptin deficiency 77, 78. Interestingly, an extraordinarily active rather than underactive frontopolar cortex, an area of the brain assumed to mediate response inhibition, was seen in an adolescent girl with congenital leptin deficiency 78. This neuronal phenomenon was correlated with strong cognitive control and a very strict behavioural pattern, which the subject appears to have acquired in compensation for the feeding stimulatory effects of leptin deficiency and resulting hyperphagia 78. Nevertheless, in general, obesity may be characterized by an overactivation of reward‐encoding brain regions and/or a deficiency in cortical inhibitory networks.
Not only is altered reward responding to food stimuli seen in the already obese, but Stice et al. demonstrated that many of these corticolimbic responses are predictive of future weight gain in adolescents 79, 80, 81. Thus, these data support the notion that hedonic eating can indeed lead to obesity.
Hedonic obesity: elevated body weight sustained by hedonic over‐eating that overrides homeostatic metabolic signals
A primary characteristic of the hedonic system, with regard to the onset and maintenance of obesity, is its ability to override energy homeostatic signals in response to rewarding food items. In individuals susceptible to developing disorders of the hedonic system, the drive to eat can be strong enough to override inhibitory signals arising from the metabolic effects of food and potentially stimulate weight gain. We term obesity resulting from sustained hedonic over‐eating in the face of metabolic signals for energy surplus ‘hedonic obesity’ (Fig. 1). Evidence for hedonic signals overriding metabolic signals is strong in rodent models 82. For example, in the outbred diet‐induced obese rats mentioned earlier, despite the establishment of an elevated set point weight trajectory when fed an HE diet, these animals over‐eat when provided an even more palatable food (chocolate flavoured Ensure) and become even more obese, reaching weights significantly above their usual set point weight curve 83. As long as the chocolate Ensure is available, they maintain excess caloric intake and an elevated growth trajectory. Once the Ensure is removed, homeostatic regulation is re‐asserted and the body weight returns to its usual set point curve 83. In humans, it has been shown that obese children and adults display hyper‐responsiveness to food stimuli in the limbic and paralimbic regions in comparison to normal weight controls. Importantly, more obese individuals than normal weight controls display a failure to attenuate food‐elicited response after a meal 73, 74, suggesting that the satiety signal (metabolic) is being overridden by the reward signal (hedonic). Other supportive evidence for hedonic‐driven weight gain comes from population surveys. According to one recent study, the prevalence of ‘food addiction’, using the Yale Food Addiction Scale 84, was 5.4% in a general population, and these ‘food addicts’ were about 25 lb heavier than the non‐food addicts 85. Using the same scale, the percentage of ‘food addicts’ in an obese cohort was 7.7% 85. This number may appear to be low, but the true prevalence of obesity caused by hedonic over‐eating is likely greater than that represented by the percentage of obese individuals who qualify as food addicted. The phenomena of binge eating, stress‐induced eating, night eating and ‘grazing’ (snacking continuously throughout the day) may all contribute to obesity, and the prevalence of these conditions is certainly greater than that of food addiction. For example, only 57% of obese binge eaters meet the diagnostic criteria for food addiction by the Yale Food Addiction Scale according to one study 86. In summary, while we believe that the prevalence of hedonic obesity is greater than that of ‘food addiction’, its prevalence is unknown at this time.
Distinguishing hedonic obesity from metabolic obesity
Presence of the adaptive changes in energy expenditure in non‐set point body weights
Although not without controversy 87, 88, we believe that adaptive or compensatory changes in energy expenditure are present in individuals who have deviated from their respective set point weights 89, 90. The ‘adaptive changes’ have repeatedly been demonstrated, with particularly convincing data arising from well‐controlled human studies in metabolic wards 89 and other controlled environments 91.
In their well‐controlled human study at Rockefeller University 89, Leibel et al. recruited weight‐stable normal weight or obese study subjects. These participants were admitted to the clinical research centre, and the study was conducted essentially in‐house for the entire study period. The subjects were underfed or overfed, using a liquid formula diet, to lose or gain 10% of their usual body weights. After weights were stabilized at respective target levels (at least 14 d, and confirmed by respiratory quotient measurement), resting energy expenditure (REE) was determined by indirect calorimetry, and total energy expenditure was determined by differential excretion rates of 2 isotopes of water and by indirect calorimetry in a respiration chamber, in which physical activity was monitored. They found that a weight loss of 10% resulted in a significant reduction in resting and non‐REE (each was reduced by 3–4 kcal kg−1 fat‐free mass [FFM] d−1). Conversely, a 10% weight gain resulted in an increase in total energy expenditure of 8–9 kcal kg−1 FFM d−1, of which the change in non‐REE was the larger component. In their study, the magnitude of the adaptive changes in energy expenditure was not affected by sex or initial body weight. There is evidence that such adaptive changes in energy expenditure persist in otherwise weight‐stable individuals for as long as body weights remain deviated from the metabolic set point. This statement is supported by the demonstration of reduced energy expenditure in long‐term weight‐reduced individuals in several different studies 92, 93, 94.
Capture of the adaptive changes in energy expenditure exhibited in people with a non‐set point body weight
While an individual's energy expenditure in the waking hours may vary widely on a daily basis (in response to physical and mental activities and the thermal effects of digestion), REE is relatively constant under normal physiological conditions. Only a 2% variation (including measurement error) was seen in REE in the same individuals on different days 95). REE is proportional to the body's metabolic mass, another relatively constant parameter, which historically has been represented by various surrogate measurements, such as total body weight, FFM, total body surface area, individual organ mass or cellular mass 96.
REE correlates well with FFM 97, and scaling REE to FFM is widely used in various studies 89, 98, 99. Inter‐subject variance is largely accounted for by the differences in the individuals' FFM, so that while REE varies widely in different individuals, REE expressed per FFM is much closer among individuals. FFM can explain a major part of the inter‐subject variance (63%) 95. Fat mass (FM), age, and possibly race and sex may also explain a smaller but significant portion of the variance (FM 6.7%, age 1.7%) 95. Interestingly, in various regression modelling, there remains a large portion of the variance unexplained by FFM, FM, age, race, sex or other factors such as thyroid hormone levels 93, 95, 100, 101, 102. This unexplained ‘residual variance’ was estimated to be 26.6% of the total between‐individual variance 95.
What is this unexplained residual variance? We assume that in the general population, there exist people who exhibit a significant component in their REE that is mass‐independent. Such a component may be produced by some unusual and yet unrecognized physiological activity that, on the one hand, is present at rest but, on the other hand, is analogous to the non‐REE in that it is largely mass‐independent. Non‐REE produced by mental/physical work, stress, anxiety or any other physiological variations during the waking hours cannot be accounted for by metabolic mass as previously stated. Therefore, if a mass‐independent physiological activity is present in some individuals at rest, their REE would appear to be an outlier in the usual REE–mass relationship. The mass‐independent physiological variation we are interested in here is the ‘adaptive change’ in energy expenditure in individuals whose body weights are significantly deviated from their set point levels. We suggest that such adaptive changes are present in some of the weight‐gained and weight‐reduced people in the general population and may therefore constitute an inter‐individual variance that is at least part of the thus‐far‐unexplained ‘residual variance’. In other words, because of the presence of the mass‐independent energy expenditure at rest, it is likely that the ‘adaptive’ changes exhibited in people with a non‐set point weight may be captured simply by looking for the REE outliers on the usual REE‐FFM regression line 89, 103, 104, 105, 106, 107, 108, 109. Total energy‐FFM regression may also be used to capture the adaptive changes after weight loss or weight gain 89, but more sophisticated methodology in a controlled environment would have to be used.
Distinguishing ‘hedonic obesity’ from ‘metabolic obesity’
As stated previously, the measured energy expenditure of an obese individual can be compared with the predicted values based upon the REE–‘mass’ relationship of an appropriate reference group. While a ‘normal’ REE or total EE in an energy–‘mass’ relationship signifies a stable metabolic set point weight, a value that is significantly greater than what is predicted by the EE–‘mass’ regression line would signify an above‐the‐set point body weight, i.e. by definition ‘hedonic obesity’. Contrarily, the REE–‘mass’ relationship in people with ‘metabolic obesity’ is indistinguishable from that of normal weight individuals because the body weights are at their respective set points in both, except that the former is ‘obese’ by weight. Figure 2 illustrates the difference in REE/mass in a hypothetical person who develops metabolic obesity vs. hedonic obesity. We should emphasize that the above remains a testable hypothesis at this point, and validation would require future studies that directly address this issue.
Figure 2.
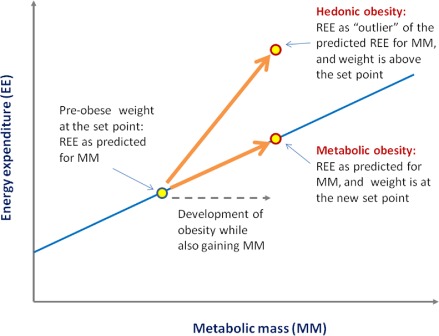
Distinct features in energy expenditure in ‘hedonic obesity’ vs. ‘metabolic obesity’. In this hypothetical illustration, we use resting energy expenditure (REE) regressed to ‘metabolic mass’ (MM). REE/MM falls onto the regression line in the case of normal weight and metabolic obesity, as both are at the respective set points, whereas REE/MM is significantly above the regression line (‘outlier’) in the case of hedonic obesity as the body weight is above the set point.
Although conceptually of critical importance, REE should not be the only clinical criterion used to predict whether an individual is at his/her set point weight. In making the distinction between ‘hedonic obesity’ and ‘metabolic obesity’, additional diagnostic tools should be used to corroborate the diagnostic evidence as a whole. These may include comparisons of daily caloric intake to calculated caloric needs per unit body mass (this method is perhaps more attainable in general clinical practice but likely less accurate) and potential use of the Yale Food Addiction Scale or addiction criteria in cases where compulsive over‐eating is suspected. Individuals with obesity and eating disorders or food addiction should practically be considered to have hedonic obesity until proven otherwise.
It is quite possible that in the real world, one may find few pure cases of ‘hedonic obesity’ or ‘metabolic obesity’ as the division is an artificial one. Both metabolic and hedonic forces are operating in any given individual. For example, it is quite conceivable that an individual who initially suffers from ‘hedonic obesity’ may eventually develop secondary leptin resistance if he/she remains obese long enough, leading to an elevation of the body weight set point, the hallmark of ‘metabolic obesity’. However, it is not inconceivable that, depending upon their individual genetic makeup, some may never develop ‘metabolic obesity’ in their lifetime, despite a persistent state of hedonic obesity. Of clinical importance is the identification of the predominant factor that underlies an individual's obesity phenotype at the time of clinical intervention.
Clinical considerations in treating obesity
To the extent possible, successful obesity treatment must address the underlying aetiology of the condition. One obvious example in addressing the underlying cause is the use of recombinant leptin to treat the rare obesity due to leptin deficiency 110, 111. In treating the more common forms of obesity, we propose that practitioners make a distinction between hedonic causes and metabolic causes, as we have carried out in this article. This distinction recognizes the alterations in molecular mechanisms and CNS pathways that lead to the development of these two types of obesity in most individuals. We believe that conceptualizing metabolic obesity vs. hedonic obesity is the first step towards more rational and more tailored treatment. Behavioural and lifestyle modification, including traditional behaviour therapy and possibly pharmacotherapy, would be the primary treatment modality for hedonic obesity, whereas metabolic modification, pharmacological and/or other forms of intervention would be needed to treat metabolic obesity. Bariatric surgery, depending upon patient characteristics and the specific procedures used, appears to modify hedonic pathways and possibly alter the metabolic body weight set point and therefore may be suitable for treating both hedonic and metabolic obesity.
Current state of obesity treatment
Intensive behavioural therapy and lifestyle coaching delivered by trained healthcare professionals does appear to be effective for modest levels of weight loss. The efficacy of this treatment modality was demonstrated at the ‘population’ level in large‐scale randomized clinical trials, such as the Diabetes Prevention Program (DPP) 112 and the ‘Look AHEAD’ clinical trials 113. In these trials, behavioural and lifestyle modification consists of dietary guidance, exercise prescription and behavioural treatment aimed at enhancing adherence to dietary and exercise prescriptions 114. In the DPP trial, 38% of participants had achieved 7% or more weight loss at the average 2.8‐year follow‐up. The average weight loss in the lifestyle group was 5.6 kg, as compared to 0.1 and 2.1 kg, respectively, in the placebo and the metformin groups 112. In the even larger ‘Look AHEAD’ lifestyle intervention trial of over 5,000 people, an 8.6% weight loss was achieved at 1‐year follow‐up and 6% weight loss at the median 9.8‐year follow‐up 113, 115.
Despite this impressive achievement, more than a half of the participants in these trials were ‘non‐responders’ to lifestyle treatment, as defined by less than 7% weight loss. Moreover, in many weight management centres where treatment is actually more concentrated, results are even more disappointing. The percentage of the non‐responders to behavioural modification protocols appears to be much greater, perhaps in part because the patients seen in these centres represent the most difficult‐to‐treat. We submit that many of these individuals may have a formally diagnosed binge eating disorder, or meet food addiction criteria, and thus are not likely to respond to a generic lifestyle intervention protocol. Additionally, in people with intractable weight problems or repeated weight cycling, co‐existence of psychological disorders, such as depression, anxiety, impulse‐control disorder or other emotional problems, is frequently seen 116. The co‐existence of these psychological problems certainly makes treatment more difficult. Finally, we believe that there is a significant fraction of obese individuals who would not respond well to behavioural treatment alone as their obesity is metabolic in nature. Unless the body weight set point is lowered, any volitional weight loss would only be temporary.
Currently available medications approved for treatment of obesity are few. The mechanisms of action of most anti‐obesity medications are ‘unknown’ but may utilize aspects of the molecular and cellular pathways of both the hedonic and the metabolic systems. The recently Food and Drug Administration (FDA)‐approved anti‐obesity medication Qsymia (a combination of phentermine and topiramate) provides an excellent example. Phentermine is a sympathomimetic amine, which helps release noradrenaline (norepinephrine) and dopamine, and exerts anorexic effects through both the hedonic and the metabolic pathways 117, 118. Topiramate, the other active component of Qsymia, also produces anorexia, but exerts metabolic effects as well, possibly via increased substrate oxidation, mitochondrial metabolism and thermogenesis 119, 120. The appetite suppressive effects of lorcaserin, a selective serotonin receptor (5HT2CR) agonist recently approved by FDA for the treatment of obesity, may involve metabolic pathway modification as demonstrated by the finding that activation of 5HT2CR increases the POMC‐MC4R activity associated with hypophagia 121. However, lorcaserin's anorectic effects appear to be intertwined with a range of psychological effects of serotonin, which is known for its capability to modify anxiety, depression and aggression 122, 123. Contrave, which just attained FDA approval for treating obesity, also has dual effects on the hedonic and metabolic weight regulatory systems. In a recent study using fMRI to map activities in specific brain areas in response to food cues, this medication was shown to attenuate activities in hypothalamus but enhance activities in brain regions believed to be involved in hedonic control of food intake, i.e. the anterior cingulate, superior frontal, insular, superior parietal and hippocampal regions 124. Naltrexone, an active component of Contrave, is a mu‐opioid receptor antagonist and may be expected to attenuate 125 hedonic‐driven consumption of energy dense fats and sweets 126, 127, 128. Additionally, naltrexone is believed to block the beta‐endorphin‐mediated negative feedback inhibition of POMC‐neuron activation, thus potentially modulating the functionality of the metabolic energy balance circuitry encoded in the ARC of hypothalamus. Bupropion, the other active component of Contrave, by virtue of being a noradrenaline/dopamine reuptake inhibitor is believed to attenuate hedonic eating as well, but it also affects metabolic weight regulation by stimulating POMC/CART neurons 129. Quantitatively, the role of the metabolic and hedonic mechanisms in mediating the weight loss effects of each of the anti‐obesity medications remains to be determined.
Bariatric surgery, now also termed metabolic surgery 130, was initially developed to treat obesity by restrictive and/or malabsorptive means, but has turned out to be an effective treatment modality that produces profound changes in the regulation of body weight involving both the metabolic and the food reward systems 131, 132, 133, 134, 135. It has been shown that after Roux‐en‐Y gastric bypass (RYGB) surgery, for example, patients have enhanced taste sensitivity for sweet 136, consume less food and beverages high in sugar 137 and fat 137, 138, 139, and have decreased hunger ratings 139. Many of these changes may be due to altered hedonic responsivity in the patients 140. Modifications of the brain's reward system after surgery have been detected by fMRI 141 and positron emission tomography scanning using a selective radioligand for the dopamine receptors 142, 143. Increases in the postprandial levels of the gut hormones GLP‐1 and PYY seen in post‐RYGB patients 144, 145 and rat models 146 likely contribute to the observed weight loss outcomes. GLP‐1 is known to act on the caudal brainstem to regulate appetite, food intake and gastric emptying 147. The GLP‐1 receptor agonists exenatide 148 and liraglutide 149 both suppress appetite and cause weight loss. PYY acts on NPY/AgRP and POMC/CART neurons in the ARC of the hypothalamus 150, 151, 152 and inhibits appetite and food intake 150, 152, 153. Similar changes in GLP‐1 and PYY after RYGB were also seen after the sleeve gastrectomy 134, 154, 155. In concert with the post‐surgical changes of the hindgut hormones GLP‐1 and PYY, changes in foregut hormones, ghrelin and CCK, after sleeve gastrectomy and RYGB are believed to play an important role as well 154. However, weight loss and metabolic changes after these bariatric procedures may not be fully explained by the favourable changes in the gut hormones alone 156, 157. Post‐operative changes in the autonomic innervations and circulating bile acid levels are among those proposed to play a significant role as well 130, 134, 158, 159. Another area of intense research is the microbiota 160, 161, which may play a significant role in mediating some of the post‐surgical changes that alter the physiological responses to the food and their interactions with the brain. Increased energy expenditure was demonstrated after RYGB in some rodent models 162, 163 and in one human study 164, implying that the body weight set point may have been adjusted downwards after the surgery. However, most human studies show no increase in energy expenditure after RYGB 165, 166, 167, 168.
A strategy to consider in future clinical studies
We have observed that the non‐respondent rate for any single obesity treatment modality is usually high. This is particularly true for behavioural treatment and pharmacotherapy. We believe this is so because so far we have only measured the efficacy of each treatment in a mixed population of study subjects. We hypothesize that one or the other form of obesity, hedonic obesity or metabolic obesity, may favourably respond to a particular therapy depending upon which system the treatment targets. Note that the respondent rate is usually high with bariatric surgery. We suspect that this is because bariatric surgery has profound effects on both the hedonic and the metabolic systems, as just discussed. If this scenario holds true, we believe that in future clinical studies where a treatment modality is assessed for its efficacy, selecting respective study subjects with predominantly hedonic obesity or metabolic obesity for treatments targeting the food reward system or the metabolic system, respectively, should show better and more congruent treatment outcomes.
Conclusion
Body weight is regulated by two mechanistically discernible systems: the homeostatic metabolic system regulating both food intake and energy expenditure to keep the body weight at a relatively stable set point level and the non‐homeostatic food reward system regulating the hedonic control of food intake irrespective of the body's energetics. In recent decades, hedonic over‐eating has become an important factor in the obesity epidemic, in large part fuelled by the cheap and readily available processed foods. Sustained over‐eating of these ‘palatable’ energy dense foods can lead to hedonic obesity. These high‐calorie processed foods may also modify the homeostatic metabolic system in susceptible individuals, leading to upward shift of the body weight set point and causing metabolic obesity. Hedonic obesity is characterized by greater than normal energy expenditure per unit metabolic mass, whereas metabolic obesity with its body weight at the set point is characterized by the energy expenditure per unit metabolic mass within the expected normal range. Both hedonic and metabolic mechanisms are at work in any given individual, but derangement in either or both may lead to obesity. To the extent possible, consideration should be given in the management of obesity to the use of treatment modalities that target behavioural changes in the case of hedonic obesity and those that may modify the body weight set point in the case of metabolic obesity. Finally, while we are unable to change the genetics underlying either metabolic or hedonic obesity, we can change our obesogenic environment permissive for the development of these conditions. Therefore, in parallel to individual treatment of obesity, preventive measures should be taken at the population level, and that will require reforms in healthcare legislation and insurance policy 169, 170.
Conflict of interest statement
Dr. Yu reports no conflicts of interest; Dr. Vasselli reports grants and other support from Novo Nordsk, Inc., and grants from Pharma Science Nutrients, Inc., outside the submitted work; Dr. Zhang reports that she is an inventor in a patent on the leptin gene, licensed; Dr. Mechanick reports receiving honoraria for lectures and program development from Abbott Nutrition International, outside the submitted work; Dr. Korner reports grants from Covidien, outside the submitted work, and receiving honoraria for lectures from Takeda; Dr. Peterli reports grants from Ethicon Endosurgery Switzerland, outside the submitted work.
References
- 1. Kennedy GC. The hypothalamic control of food intake in rats. Proc R Soc Lond B Biol Sci 1950; 137: 535–549. [DOI] [PubMed] [Google Scholar]
- 2. Hervey GR. The effects of lesions in the hypothalamus in parabiotic rats. J Physiol 1959; 145: 336–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Coleman DL. Effects of parabiosis of obese with diabetes and normal mice. Diabetologia 1973; 9: 294–298. [DOI] [PubMed] [Google Scholar]
- 4. Harris RB, Hervey E, Hervey GR, Tobin G. Body composition of lean and obese Zucker rats in parabiosis. Int J Obes (Lond) 1987; 11: 275–283. [PubMed] [Google Scholar]
- 5. Bray GA, York DA. Hypothalamic and genetic obesity in experimental animals: an autonomic and endocrine hypothesis. Physiol Rev 1979; 59: 719–809. [DOI] [PubMed] [Google Scholar]
- 6. Harris RB. Role of set‐point theory in regulation of body weight. FASEB J 1990; 4: 3310–3318. [DOI] [PubMed] [Google Scholar]
- 7. Hirsch J, Leibel RL, Chua SC. A clinical perspective on peptides and food intake. Am J Clin Nutr 1992; 55: 296S–298S. [DOI] [PubMed] [Google Scholar]
- 8. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature 1994; 372: 425–432. [DOI] [PubMed] [Google Scholar]
- 9. Speakman JR, Levitsky DA, Allison DB et al Set points, settling points and some alternative models: theoretical options to understand how genes and environments combine to regulate body adiposity. Dis Model Mech 2011; 4: 733–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Neel JV. Diabetes mellitus: a ‘thrifty’ genotype rendered detrimental by ‘progress’? Am J Hum Genet 1962; 14: 353–362. [PMC free article] [PubMed] [Google Scholar]
- 11. Wendorf M, Goldfine ID. Archaeology of NIDDM. Excavation of the ‘thrifty’ genotype. Diabetes 1991; 40: 161–165. [DOI] [PubMed] [Google Scholar]
- 12. Flier JS. Clinical review 94: what's in a name? In search of leptin's physiologic role. J Clin Endocrinol Metab 1998; 83: 1407–1413. [DOI] [PubMed] [Google Scholar]
- 13. Prentice AM. Obesity and its potential mechanistic basis. Br Med Bull 2001; 60: 51–67. [DOI] [PubMed] [Google Scholar]
- 14. Wells JC. The evolution of human fatness and susceptibility to obesity: an ethological approach. Biol Rev Camb Philos Soc 2006; 81: 183–205. [DOI] [PubMed] [Google Scholar]
- 15. Leibel RL. Molecular physiology of weight regulation in mice and humans. Int J Obes (Lond) 2008; 32(Suppl. 7): S98–S108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kelley AE, Baldo BA, Pratt WE. A proposed hypothalamic‐thalamic‐striatal axis for the integration of energy balance, arousal, and food reward. J Comp Neurol 2005; 493: 72–85. [DOI] [PubMed] [Google Scholar]
- 17. Pierce RC, Kumaresan V. The mesolimbic dopamine system: the final common pathway for the reinforcing effect of drugs of abuse? Neurosci Biobehav Rev 2006; 30: 215–238. [DOI] [PubMed] [Google Scholar]
- 18. Berridge KC, Ho CY, Richard JM, DiFeliceantonio AG. The tempted brain eats: pleasure and desire circuits in obesity and eating disorders. Brain Res 2010; 1350: 43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saper CB, Chou TC, Elmquist JK. The need to feed: homeostatic and hedonic control of eating. Neuron 2002; 36: 199–211. [DOI] [PubMed] [Google Scholar]
- 20. Berthoud HR, Morrison C. The brain, appetite, and obesity. Annu Rev Psychol 2008; 59: 55–92. [DOI] [PubMed] [Google Scholar]
- 21. Berthoud HR. Metabolic and hedonic drives in the neural control of appetite: who is the boss? Curr Opin Neurobiol 2011; 21: 888–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khosla T, Billewicz WZ. Measurement of change in body‐weight. Br J Nutr 1964; 18: 227–239. [DOI] [PubMed] [Google Scholar]
- 23. Beisel WR. Effect of infection on human protein metabolism. Fed Proc 1966; 25: 1682–1687. [PubMed] [Google Scholar]
- 24. Leibel RL. Is obesity due to a heritable difference in ‘set point’ for adiposity? West J Med 1990; 153: 429–431. [PMC free article] [PubMed] [Google Scholar]
- 25. York D, Bouchard C. How obesity develops: insights from the new biology. Endocrine 2000; 13: 143–154. [DOI] [PubMed] [Google Scholar]
- 26. Schwartz MW, Woods SC, Porte D Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature 2000; 404: 661–671. [DOI] [PubMed] [Google Scholar]
- 27. Ahima RS, Saper CB, Flier JS, Elmquist JK. Leptin regulation of neuroendocrine systems. Front Neuroendocrinol 2000; 21: 263–307. [DOI] [PubMed] [Google Scholar]
- 28. Flier JS. Obesity wars: molecular progress confronts an expanding epidemic. Cell 2004; 116: 337–350. [DOI] [PubMed] [Google Scholar]
- 29. Elmquist JK, Coppari R, Balthasar N, Ichinose M, Lowell BB. Identifying hypothalamic pathways controlling food intake, body weight, and glucose homeostasis. J Comp Neurol 2005; 493: 63–71. [DOI] [PubMed] [Google Scholar]
- 30. Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature 2006; 443: 289–295. [DOI] [PubMed] [Google Scholar]
- 31. van de Wall E, Leshan R, Xu AW et al Collective and individual functions of leptin receptor modulated neurons controlling metabolism and ingestion. Endocrinology 2008; 149: 1773–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grill HJ, Hayes MR. Hindbrain neurons as an essential hub in the neuroanatomically distributed control of energy balance. Cell Metab 2012; 16: 296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Y, Guo KY, Diaz PA, Heo M, Leibel RL. Determinants of leptin gene expression in fat depots of lean mice. Am J Physiol Regul Integr Comp Physiol 2002; 282: R226–R234. [DOI] [PubMed] [Google Scholar]
- 34. Guo KY, Halo P, Leibel RL, Zhang Y. Effects of obesity on the relationship of leptin mRNA expression and adipocyte size in anatomically distinct fat depots in mice. Am J Physiol Regul Integr Comp Physiol 2004; 287: R112–R119. [DOI] [PubMed] [Google Scholar]
- 35. Maffei M, Halaas J, Ravussin E et al Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight‐reduced subjects. Nat Med 1995; 1: 1155–1161. [DOI] [PubMed] [Google Scholar]
- 36. Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet‐induced resistance to leptin action. Nat Med 1995; 1: 1311–1314. [DOI] [PubMed] [Google Scholar]
- 37. Considine RV, Sinha MK, Heiman ML et al Serum immunoreactive‐leptin concentrations in normal‐weight and obese humans. N Engl J Med 1996; 334: 292–295. [DOI] [PubMed] [Google Scholar]
- 38. Rosenbaum M, Nicolson M, Hirsch J et al Effects of gender, body composition, and menopause on plasma concentrations of leptin. J Clin Endocrinol Metab 1996; 81: 3424–3427. [DOI] [PubMed] [Google Scholar]
- 39. Schwartz MW, Peskind E, Raskind M, Boyko EJ, Porte D Jr. Cerebrospinal fluid leptin levels: relationship to plasma levels and to adiposity in humans. Nat Med 1996; 2: 589–593. [DOI] [PubMed] [Google Scholar]
- 40. York DA. Peripheral and central mechanisms regulating food intake and macronutrient selection. Obes Surg 1999; 9: 471–479. [DOI] [PubMed] [Google Scholar]
- 41. Seeley RJ, York DA. Fuel sensing and the central nervous system (CNS): implications for the regulation of energy balance and the treatment for obesity. Obes Rev 2005; 6: 259–265. [DOI] [PubMed] [Google Scholar]
- 42. Larder R, O'Rahilly S. Shedding pounds after going under the knife: guts over glory‐why diets fail. Nat Med 2012; 18: 666–667. [DOI] [PubMed] [Google Scholar]
- 43. Schwartz GJ, Zeltser LM. Functional organization of neuronal and humoral signals regulating feeding behavior. Annu Rev Nutr 2013; 33: 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Suzuki K, Jayasena CN, Bloom SR. The gut hormones in appetite regulation. J Obes 2011; 2011: 528401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ladenheim EE. Gastrointestinal regulatory peptides and central nervous system mechanisms of weight control. Curr Opin Endocrinol Diabetes Obes 2012; 19: 13–18. [DOI] [PubMed] [Google Scholar]
- 46. Bouchard C. Heredity and the path to overweight and obesity. Med Sci Sports Exerc 1991; 23: 285–291. [PubMed] [Google Scholar]
- 47. Bouchard C. Genetic determinants of regional fat distribution. Hum Reprod 1997; 12(Suppl. 1): 1–5. [DOI] [PubMed] [Google Scholar]
- 48. Ravussin Y, Gutman R, Diano S et al Effects of chronic weight perturbation on energy homeostasis and brain structure in mice. Am J Physiol Regul Integr Comp Physiol 2011; 300: R1352–R1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bumaschny VF, Yamashita M, Casas‐Cordero R et al Obesity‐programmed mice are rescued by early genetic intervention. J Clin Invest 2012; 122: 4203–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Levin BE, Dunn‐Meynell AA. Defense of body weight against chronic caloric restriction in obesity‐prone and ‐resistant rats. Am J Physiol Regul Integr Comp Physiol 2000; 278: R231–R237. [DOI] [PubMed] [Google Scholar]
- 51. Drewry MM, Harris RB, Martin RJ. The effect of increased adiposity on food intake of juvenile rats. Physiol Behav 1989; 45: 381–386. [DOI] [PubMed] [Google Scholar]
- 52. Levin BE, Triscari J, Sullivan AC. Relationship between sympathetic activity and diet‐induced obesity in two rat strains. Am J Physiol 1983; 245: R364–R371. [DOI] [PubMed] [Google Scholar]
- 53. Levin BE, Dunn‐Meynell AA, Balkan B, Keesey RE. Selective breeding for diet‐induced obesity and resistance in Sprague‐Dawley rats. Am J Physiol 1997; 273: R725–R730. [DOI] [PubMed] [Google Scholar]
- 54. Arch JR. Central regulation of energy balance: inputs, outputs and leptin resistance. Proc Nutr Soc 2005; 64: 39–46. [DOI] [PubMed] [Google Scholar]
- 55. Scarpace PJ, Zhang Y. Leptin resistance: a prediposing factor for diet‐induced obesity. Am J Physiol Regul Integr Comp Physiol 2009; 296: R493–R500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vasselli JR, Scarpace PJ, Harris RB, Banks WA. Dietary components in the development of leptin resistance. Adv Nutr 2013; 4: 164–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Horvath TL, Sarman B, Garcia‐Caceres C et al Synaptic input organization of the melanocortin system predicts diet‐induced hypothalamic reactive gliosis and obesity. Proc Natl Acad Sci U S A 2010; 107: 14875–14880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McNay DE, Briancon N, Kokoeva MV, Maratos‐Flier E, Flier JS. Remodeling of the arcuate nucleus energy‐balance circuit is inhibited in obese mice. J Clin Invest 2012; 122: 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McNay DE, Speakman JR. High fat diet causes rebound weight gain. Mol Metab 2012; 2: 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Thaler JP, Yi CX, Schur EA et al Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest 2012; 122: 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sesack SR, Grace AA. Cortico‐basal ganglia reward network: microcircuitry. Neuropsychopharmacology 2010; 35: 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Berridge KC, Robinson TE, Aldridge JW. Dissecting components of reward: ‘liking’, ‘wanting’, and learning. Curr Opin Pharmacol 2009; 9: 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Avena NM, Rada P, Hoebel BG. Evidence for sugar addiction: behavioral and neurochemical effects of intermittent, excessive sugar intake. Neurosci Biobehav Rev 2008; 32: 20–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bello NT, Lucas LR, Hajnal A. Repeated sucrose access influences dopamine D2 receptor density in the striatum. Neuroreport 2002; 13: 1575–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bello NT, Sweigart KL, Lakoski JM, Norgren R, Hajnal A. Restricted feeding with scheduled sucrose access results in an upregulation of the rat dopamine transporter. Am J Physiol Regul Integr Comp Physiol 2003; 284: R1260–R1268. [DOI] [PubMed] [Google Scholar]
- 66. Johnson PM, Kenny PJ. Dopamine D2 receptors in addiction‐like reward dysfunction and compulsive eating in obese rats. Nat Neurosci 2010; 13: 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Volkow ND, Wang GJ, Tomasi D, Baler RD. Obesity and addiction: neurobiological overlaps. Obes Rev 2013; 14: 2–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ifland JR, Preuss HG, Marcus MT et al Refined food addiction: a classic substance use disorder. Med Hypotheses 2009; 72: 518–526. [DOI] [PubMed] [Google Scholar]
- 69. Geiger BM, Haburcak M, Avena NM, Moyer MC, Hoebel BG, Pothos EN. Deficits of mesolimbic dopamine neurotransmission in rat dietary obesity. Neuroscience 2009; 159: 1193–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Schwindinger WF, Borrell BM, Waldman LC, Robishaw JD. Mice lacking the G protein gamma3‐subunit show resistance to opioids and diet induced obesity. Am J Physiol Regul Integr Comp Physiol 2009; 297: R1494–R1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Stice E, Spoor S, Bohon C, Veldhuizen MG, Small DM. Relation of reward from food intake and anticipated food intake to obesity: a functional magnetic resonance imaging study. J Abnorm Psychol 2008; 117: 924–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Stoeckel LE, Kim J, Weller RE, Cox JE, Cook EW 3rd, Horwitz B. Effective connectivity of a reward network in obese women. Brain Res Bull 2009; 79: 388–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bruce AS, Holsen LM, Chambers RJ et al Obese children show hyperactivation to food pictures in brain networks linked to motivation, reward and cognitive control. Int J Obes (Lond) 2010; 34: 1494–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Martin LE, Holsen LM, Chambers RJ et al Neural mechanisms associated with food motivation in obese and healthy weight adults. Obesity (Silver Spring) 2010; 18: 254–260. [DOI] [PubMed] [Google Scholar]
- 75. Batterink L, Yokum S, Stice E. Body mass correlates inversely with inhibitory control in response to food among adolescent girls: an fMRI study. Neuroimage 2010; 52: 1696–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Volkow ND, Wang GJ, Telang F et al Inverse association between BMI and prefrontal metabolic activity in healthy adults. Obesity (Silver Spring) 2009; 17: 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Farooqi IS, Bullmore E, Keogh J, Gillard J, O'Rahilly S, Fletcher PC. Leptin regulates striatal regions and human eating behavior. Science 2007; 317: 1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Frank S, Heni M, Moss A et al Long‐term stabilization effects of leptin on brain functions in a leptin‐deficient patient. PLoS ONE 2013; 8: e65893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Stice E, Yokum S, Bohon C, Marti N, Smolen A. Reward circuitry responsivity to food predicts future increases in body mass: moderating effects of DRD2 and DRD4. Neuroimage 2010; 50: 1618–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yokum S, Ng J, Stice E. Attentional bias to food images associated with elevated weight and future weight gain: an fMRI study. Obesity (Silver Spring) 2011; 19: 1775–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yokum S, Gearhardt AN, Harris JL, Brownell KD, Stice E. Individual differences in striatum activity to food commercials predict weight gain in adolescents. Obesity (Silver Spring) 2014; 22: 2544–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Boggiano MM, Dorsey JR, Thomas JM, Murdaugh DL. The Pavlovian power of palatable food: lessons for weight‐loss adherence from a new rodent model of cue‐induced overeating. Int J Obes (Lond) 2009; 33: 693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Levin BE, Keesey RE. Defense of differing body weight set points in diet‐induced obese and resistant rats. Am J Physiol 1998; 274: R412–R419. [DOI] [PubMed] [Google Scholar]
- 84. Gearhardt AN, Corbin WR, Brownell KD. Preliminary validation of the Yale Food Addiction Scale. Appetite 2009; 52: 430–436. [DOI] [PubMed] [Google Scholar]
- 85. Pedram P, Wadden D, Amini P et al Food addiction: its prevalence and significant association with obesity in the general population. PLoS ONE 2013; 8: e74832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gearhardt AN, White MA, Masheb RM, Morgan PT, Crosby RD, Grilo CM. An examination of the food addiction construct in obese patients with binge eating disorder. Int J Eat Disord 2012; 45: 657–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Shetty PS. The Nestle Lecture. Chronic undernutrition and metabolic adaptation. Proc Nutr Soc 1993; 52: 267–284. [DOI] [PubMed] [Google Scholar]
- 88. Weinsier RL, Nagy TR, Hunter GR, Darnell BE, Hensrud DD, Weiss HL. Do adaptive changes in metabolic rate favor weight regain in weight‐reduced individuals? An examination of the set‐point theory. Am J Clin Nutr 2000; 72: 1088–1094. [DOI] [PubMed] [Google Scholar]
- 89. Leibel RL, Rosenbaum M, Hirsch J. Changes in energy expenditure resulting from altered body weight. N Engl J Med 1995; 332: 621–628. [DOI] [PubMed] [Google Scholar]
- 90. Rosenbaum M, Hirsch J, Gallagher DA, Leibel RL. Long‐term persistence of adaptive thermogenesis in subjects who have maintained a reduced body weight. Am J Clin Nutr 2008; 88: 906–912. [DOI] [PubMed] [Google Scholar]
- 91. Johannsen DL, Knuth ND, Huizenga R, Rood JC, Ravussin E, Hall KD. Metabolic slowing with massive weight loss despite preservation of fat‐free mass. J Clin Endocrinol Metab 2012; 97: 2489–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Leibel RL, Hirsch J. Diminished energy requirements in reduced‐obese patients. Metabolism 1984; 33: 164–170. [DOI] [PubMed] [Google Scholar]
- 93. Weyer C, Walford RL, Harper IT et al Energy metabolism after 2 y of energy restriction: the biosphere 2 experiment. Am J Clin Nutr 2000; 72: 946–953. [DOI] [PubMed] [Google Scholar]
- 94. van Gemert WG, Westerterp KR, van Acker BA et al Energy, substrate and protein metabolism in morbid obesity before, during and after massive weight loss. Int J Obes Relat Metab Disord 2000; 24: 711–718. [DOI] [PubMed] [Google Scholar]
- 95. Johnstone AM, Murison SD, Duncan JS, Rance KA, Speakman JR. Factors influencing variation in basal metabolic rate include fat‐free mass, fat mass, age, and circulating thyroxine but not sex, circulating leptin, or triiodothyronine. Am J Clin Nutr 2005; 82: 941–948. [DOI] [PubMed] [Google Scholar]
- 96. Heymsfield SB, Thomas D, Bosy‐Westphal A, Shen W, Peterson CM, Muller MJ. Evolving concepts on adjusting human resting energy expenditure measurements for body size. Obes Rev 2012; 13: 1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ravussin E, Lillioja S, Anderson TE, Christin L, Bogardus C. Determinants of 24‐hour energy expenditure in man. Methods and results using a respiratory chamber. J Clin Invest 1986; 78: 1568–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ravussin E, Bogardus C. Relationship of genetics, age, and physical fitness to daily energy expenditure and fuel utilization. Am J Clin Nutr 1989; 49: 968–975. [DOI] [PubMed] [Google Scholar]
- 99. Bogardus C, Lillioja S, Ravussin E et al Familial dependence of the resting metabolic rate. N Engl J Med 1986; 315: 96–100. [DOI] [PubMed] [Google Scholar]
- 100. Booyens J, McCance RA. Individual variations in expenditure of energy. Lancet 1957; 272: 225–229. [DOI] [PubMed] [Google Scholar]
- 101. Heymsfield SB, Gallagher D, Kotler DP, Wang Z, Allison DB, Heshka S. Body‐size dependence of resting energy expenditure can be attributed to nonenergetic homogeneity of fat‐free mass. Am J Physiol Endocrinol Metab 2002; 282: E132–E138. [DOI] [PubMed] [Google Scholar]
- 102. Gallagher D, Belmonte D, Deurenberg P et al Organ‐tissue mass measurement allows modeling of REE and metabolically active tissue mass. Am J Physiol 1998; 275: E249–E258. [DOI] [PubMed] [Google Scholar]
- 103. Sims EA, Danforth E Jr, Horton ES, Bray GA, Glennon JA, Salans LB. Endocrine and metabolic effects of experimental obesity in man. Recent Prog Horm Res 1973; 29: 457–496. [DOI] [PubMed] [Google Scholar]
- 104. Forbes GB, Welle SL. Lean body mass in obesity. Int J Obes (Lond) 1983; 7: 99–107. [PubMed] [Google Scholar]
- 105. de Boer JO, van Es AJ, Roovers LC, van Raaij JM, Hautvast JG. Adaptation of energy metabolism of overweight women to low‐energy intake, studied with whole‐body calorimeters. Am J Clin Nutr 1986; 44: 585–595. [DOI] [PubMed] [Google Scholar]
- 106. Weigle DS, Sande KJ, Iverius PH, Monsen ER, Brunzell JD. Weight loss leads to a marked decrease in nonresting energy expenditure in ambulatory human subjects. Metabolism 1988; 37: 930–936. [DOI] [PubMed] [Google Scholar]
- 107. Valtuena S, Blanch S, Barenys M, Sola R, Salas‐Salvado J. Changes in body composition and resting energy expenditure after rapid weight loss: is there an energy‐metabolism adaptation in obese patients? Int J Obes Relat Metab Disord 1995; 19: 119–125. [PubMed] [Google Scholar]
- 108. Bessard T, Schutz Y, Jequier E. Energy expenditure and postprandial thermogenesis in obese women before and after weight loss. Am J Clin Nutr 1983; 38: 680–693. [DOI] [PubMed] [Google Scholar]
- 109. Geissler CA, Miller DS, Shah M. The daily metabolic rate of the post‐obese and the lean. Am J Clin Nutr 1987; 45: 914–920. [DOI] [PubMed] [Google Scholar]
- 110. O'Rahilly S, Farooqi IS, Yeo GS, Challis BG. Minireview: human obesity‐lessons from monogenic disorders. Endocrinology 2003; 144: 3757–3764. [DOI] [PubMed] [Google Scholar]
- 111. Licinio J, Caglayan S, Ozata M et al Phenotypic effects of leptin replacement on morbid obesity, diabetes mellitus, hypogonadism, and behavior in leptin‐deficient adults. Proc Natl Acad Sci U S A 2004; 101: 4531–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Knowler WC, Barrett‐Connor E, Fowler SE et al Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346: 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wing RR. Long‐term effects of a lifestyle intervention on weight and cardiovascular risk factors in individuals with type 2 diabetes mellitus: four‐year results of the Look AHEAD trial. Arch Intern Med 2010; 170: 1566–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wadden TA, McGuckin BG, Rothman RA, Sargent SL. Lifestyle modification in the management of obesity. J Gastrointest Surg 2003; 7: 452–463. [DOI] [PubMed] [Google Scholar]
- 115. Wing RR, Bolin P, Brancati FL et al Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N Engl J Med 2013; 369: 145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ohsiek S, Williams M. Psychological factors influencing weight loss maintenance: an integrative literature review. J Am Acad Nurse Pract 2011; 23: 592–601. [DOI] [PubMed] [Google Scholar]
- 117. Igel LI, Powell AG, Apovian CM, Aronne LJ. Advances in medical therapy for weight loss and the weight‐centric management of type 2 diabetes mellitus. Curr Atheroscler Rep 2012; 14: 60–69. [DOI] [PubMed] [Google Scholar]
- 118. Colon‐Gonzalez F, Kim GW, Lin JE, Valentino MA, Waldman SA. Obesity pharmacotherapy: what is next? Mol Aspects Med 2013; 34: 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Tremblay A, Chaput JP, Berube‐Parent S et al The effect of topiramate on energy balance in obese men: a 6‐month double‐blind randomized placebo‐controlled study with a 6‐month open‐label extension. Eur J Clin Pharmacol 2007; 63: 123–134. [DOI] [PubMed] [Google Scholar]
- 120. McElroy SL, Hudson JI, Capece JA, Beyers K, Fisher AC, Rosenthal NR. Topiramate for the treatment of binge eating disorder associated with obesity: a placebo‐controlled study. Biol Psychiatry 2007; 61: 1039–1048. [DOI] [PubMed] [Google Scholar]
- 121. Lam DD, Przydzial MJ, Ridley SH et al Serotonin 5‐HT2C receptor agonist promotes hypophagia via downstream activation of melanocortin 4 receptors. Endocrinology 2008; 149: 1323–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Lucki I. The spectrum of behaviors influenced by serotonin. Biol Psychiatry 1998; 44: 151–162. [DOI] [PubMed] [Google Scholar]
- 123. Drevets WC, Thase ME, Moses‐Kolko EL et al Serotonin‐1A receptor imaging in recurrent depression: replication and literature review. Nucl Med Biol 2007; 34: 865–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Wang GJ, Tomasi D, Volkow ND et al Effect of combined naltrexone and bupropion therapy on the brain's reactivity to food cues. Int J Obes (Lond) 2014; 38: 682–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Klein G, Rossi GC, Waxman AR et al The contribution of MOR‐1 exons 1–4 to morphine and heroin analgesia and dependence. Neurosci Lett 2009; 457: 115–119. [DOI] [PubMed] [Google Scholar]
- 126. Grissom NM, Lyde R, Christ L et al Obesity at conception programs the opioid system in the offspring brain. Neuropsychopharmacology 2014; 39: 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Nathan PJ, Bullmore ET. From taste hedonics to motivational drive: central mu‐opioid receptors and binge‐eating behaviour. Int J Neuropsychopharmacol 2009; 12: 995–1008. [DOI] [PubMed] [Google Scholar]
- 128. Shin AC, Pistell PJ, Phifer CB, Berthoud HR. Reversible suppression of food reward behavior by chronic mu‐opioid receptor antagonism in the nucleus accumbens. Neuroscience 2010; 170: 580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Mercer SL. ACS chemical neuroscience molecule spotlight on contrave. ACS Chem Neurosci 2011; 2: 484–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Gass M, Beglinger C, Peterli R. Metabolic surgery‐principles and current concepts. Langenbecks Arch Surg 2011; 396: 949–972. [DOI] [PubMed] [Google Scholar]
- 131. van de Sande‐Lee S, Pereira FR, Cintra DE et al Partial reversibility of hypothalamic dysfunction and changes in brain activity after body mass reduction in obese subjects. Diabetes 2011; 60: 1699–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Tam CS, Berthoud HR, Bueter M et al Could the mechanisms of bariatric surgery hold the key for novel therapies? Report from a Pennington Scientific Symposium. Obes Rev 2011; 12: 984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Bruce JM, Hancock L, Bruce A et al Changes in brain activation to food pictures after adjustable gastric banding. Surg Obes Relat Dis 2012; 8: 602–608. [DOI] [PubMed] [Google Scholar]
- 134. Stefater MA, Wilson‐Perez HE, Chambers AP, Sandoval DA, Seeley RJ. All bariatric surgeries are not created equal: insights from mechanistic comparisons. Endocr Rev 2012; 33: 595–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Frank S, Wilms B, Veit R et al Altered brain activity in severely obese women may recover after Roux‐en Y gastric bypass surgery. Int J Obes (Lond) 2014; 38: 341–348. [DOI] [PubMed] [Google Scholar]
- 136. Burge JC, Schaumburg JZ, Choban PS, DiSilvestro RA, Flancbaum L. Changes in patients' taste acuity after Roux‐en‐Y gastric bypass for clinically severe obesity. J Am Diet Assoc 1995; 95: 666–670. [DOI] [PubMed] [Google Scholar]
- 137. Kenler HA, Brolin RE, Cody RP. Changes in eating behavior after horizontal gastroplasty and Roux‐en‐Y gastric bypass. Am J Clin Nutr 1990; 52: 87–92. [DOI] [PubMed] [Google Scholar]
- 138. Olbers T, Bjorkman S, Lindroos A et al Body composition, dietary intake, and energy expenditure after laparoscopic Roux‐en‐Y gastric bypass and laparoscopic vertical banded gastroplasty: a randomized clinical trial. Ann Surg 2006; 244: 715–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Thirlby RC, Bahiraei F, Randall J, Drewnoski A. Effect of Roux‐en‐Y gastric bypass on satiety and food likes: the role of genetics. J Gastrointest Surg 2006; 10: 270–277. [DOI] [PubMed] [Google Scholar]
- 140. Schultes B, Ernst B, Wilms B, Thurnheer M, Hallschmid M. Hedonic hunger is increased in severely obese patients and is reduced after gastric bypass surgery. Am J Clin Nutr 2010; 92: 277–283. [DOI] [PubMed] [Google Scholar]
- 141. Ochner CN, Kwok Y, Conceicao E et al Selective reduction in neural responses to high calorie foods following gastric bypass surgery. Ann Surg 2011; 253: 502–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Dunn JP, Cowan RL, Volkow ND et al Decreased dopamine type 2 receptor availability after bariatric surgery: preliminary findings. Brain Res 2010; 1350: 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Steele KE, Prokopowicz GP, Schweitzer MA et al Alterations of central dopamine receptors before and after gastric bypass surgery. Obes Surg 2010; 20: 369–374. [DOI] [PubMed] [Google Scholar]
- 144. Korner J, Bessler M, Cirilo LJ et al Effects of Roux‐en‐Y gastric bypass surgery on fasting and postprandial concentrations of plasma ghrelin, peptide YY, and insulin. J Clin Endocrinol Metab 2005; 90: 359–365. [DOI] [PubMed] [Google Scholar]
- 145. Korner J, Bessler M, Inabnet W, Taveras C, Holst JJ. Exaggerated glucagon‐like peptide‐1 and blunted glucose‐dependent insulinotropic peptide secretion are associated with Roux‐en‐Y gastric bypass but not adjustable gastric banding. Surg Obes Relat Dis 2007; 3: 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Shin AC, Zheng H, Townsend RL, Sigalet DL, Berthoud HR. Meal‐induced hormone responses in a rat model of Roux‐en‐Y gastric bypass surgery. Endocrinology 2010; 151: 1588–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Hayes MR, Skibicka KP, Grill HJ. Caudal brainstem processing is sufficient for behavioral, sympathetic, and parasympathetic responses driven by peripheral and hindbrain glucagon‐like‐peptide‐1 receptor stimulation. Endocrinology 2008; 149: 4059–4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin‐4) on glycemic control and weight over 30 weeks in metformin‐treated patients with type 2 diabetes. Diabetes Care 2005; 28: 1092–1100. [DOI] [PubMed] [Google Scholar]
- 149. Astrup A, Rossner S, Van Gaal L et al Effects of liraglutide in the treatment of obesity: a randomised, double‐blind, placebo‐controlled study. Lancet 2009; 374: 1606–1616. [DOI] [PubMed] [Google Scholar]
- 150. Batterham RL, Cowley MA, Small CJ et al Gut hormone PYY(3–36) physiologically inhibits food intake. Nature 2002; 418: 650–654. [DOI] [PubMed] [Google Scholar]
- 151. Acuna‐Goycolea C, van den Pol AN. Peptide YY(3–36) inhibits both anorexigenic proopiomelanocortin and orexigenic neuropeptide Y neurons: implications for hypothalamic regulation of energy homeostasis. J Neurosci 2005; 25: 10510–10519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Riediger T, Bothe C, Becskei C, Lutz TA. Peptide YY directly inhibits ghrelin‐activated neurons of the arcuate nucleus and reverses fasting‐induced c‐Fos expression. Neuroendocrinology 2004; 79: 317–326. [DOI] [PubMed] [Google Scholar]
- 153. Sloth B, Holst JJ, Flint A, Gregersen NT, Astrup A. Effects of PYY1‐36 and PYY3‐36 on appetite, energy intake, energy expenditure, glucose and fat metabolism in obese and lean subjects. Am J Physiol Endocrinol Metab 2007; 292: E1062–E1068. [DOI] [PubMed] [Google Scholar]
- 154. Peterli R, Steinert RE, Woelnerhanssen B et al Metabolic and hormonal changes after laparoscopic Roux‐en‐Y gastric bypass and sleeve gastrectomy: a randomized, prospective trial. Obes Surg 2012; 22: 740–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Peterli R, Borbely Y, Kern B et al Early results of the Swiss Multicentre Bypass or Sleeve Study (SM‐BOSS): a prospective randomized trial comparing laparoscopic sleeve gastrectomy and Roux‐en‐Y gastric bypass. Ann Surg 2013; 258: 690–694, discussion 695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Chambers AP, Kirchner H, Wilson‐Perez HE et al The effects of vertical sleeve gastrectomy in rodents are ghrelin independent. Gastroenterology 2013; 144: 50–52, e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Wilson‐Perez HE, Chambers AP, Ryan KK et al Vertical sleeve gastrectomy is effective in two genetic mouse models of glucagon‐like Peptide 1 receptor deficiency. Diabetes 2013; 62: 2380–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Tadross JA, le Roux CW. The mechanisms of weight loss after bariatric surgery. Int J Obes (Lond) 2009; 33(Suppl. 1): S28–S32. [DOI] [PubMed] [Google Scholar]
- 159. Shin AC, Berthoud HR. Obesity surgery: happy with less or eternally hungry? Trends Endocrinol Metab 2013; 24: 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Cotillard A, Kennedy SP, Kong LC et al Dietary intervention impact on gut microbial gene richness. Nature 2013; 500: 585–588. [DOI] [PubMed] [Google Scholar]
- 161. Zhao L. The gut microbiota and obesity: from correlation to causality. Nat Rev Microbiol 2013; 11: 639–647. [DOI] [PubMed] [Google Scholar]
- 162. Stylopoulos N, Hoppin AG, Kaplan LM. Roux‐en‐Y gastric bypass enhances energy expenditure and extends lifespan in diet‐induced obese rats. Obesity (Silver Spring) 2009; 17: 1839–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Hatoum IJ, Stylopoulos N, Vanhoose AM et al Melanocortin‐4 receptor signaling is required for weight loss after gastric bypass surgery. J Clin Endocrinol Metab 2012; 97: E1023–E1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Faria SL, Faria OP, Cardeal Mde A, Ito MK, Buffington C. Diet‐induced thermogenesis and respiratory quotient after Roux‐en‐Y gastric bypass surgery: a prospective study. Surg Obes Relat Dis 2014; 10: 138–143. [DOI] [PubMed] [Google Scholar]
- 165. Benedetti G, Mingrone G, Marcoccia S et al Body composition and energy expenditure after weight loss following bariatric surgery. J Am Coll Nutr 2000; 19: 270–274. [DOI] [PubMed] [Google Scholar]
- 166. Das SK, Roberts SB, McCrory MA et al Long‐term changes in energy expenditure and body composition after massive weight loss induced by gastric bypass surgery. Am J Clin Nutr 2003; 78: 22–30. [DOI] [PubMed] [Google Scholar]
- 167. Carrasco F, Papapietro K, Csendes A et al Changes in resting energy expenditure and body composition after weight loss following Roux‐en‐Y gastric bypass. Obes Surg 2007; 17: 608–616. [DOI] [PubMed] [Google Scholar]
- 168. Iannelli A, Martini F, Rodolphe A et al Body composition, anthropometrics, energy expenditure, systemic inflammation, in premenopausal women 1 year after laparoscopic Roux‐en‐Y gastric bypass. Surg Endosc 2014; 28: 500–507. [DOI] [PubMed] [Google Scholar]
- 169. Allen PJ, Batra P, Geiger BM, Wommack T, Gilhooly C, Pothos EN. Rationale and consequences of reclassifying obesity as an addictive disorder: neurobiology, food environment and social policy perspectives. Physiol Behav 2012; 107: 126–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Garvey W, Mechanick JI, Einhorn D, The American Association of Clinical Endocrinologists , the American College of Endocrinology . 2014 Advanced Framework for a New Diagnosis of Obesity as a Chronic Disease. 2014. [DOI] [PMC free article] [PubMed]