

Abstract
Recent reports suggest that the yeast Saccharomyces cerevisiae caspase‐related metacaspase, Mca1, is required for cell‐autonomous cytoprotective functions that slow cellular aging. Because the Mca1 protease has previously been suggested to be responsible for programmed cell death (PCD) upon stress and aging, these reports raise the question of how the opposing roles of Mca1 as a protector and executioner are regulated. One reconciling perspective could be that executioner activation may be restricted to situations where the death of part of the population would be beneficial, for example during colony growth or adaptation into specialized survival forms. Another possibility is that metacaspases primarily harbor beneficial functions and that the increased survival observed upon metacaspase removal is due to compensatory responses. Herein, we summarize data on the role of Mca1 in cell death and survival and approach the question of how a metacaspase involved in protein quality control may act as killer protein.
Keywords: aggregates, aging, apoptosis, caspase, lifespan, metacaspase, protein quality control
Abbreviations
- IPOD
insoluble protein deposit
- JUNQ
juxtanuclear quality control compartment
- NO
nitric oxide
- PCD
programmed cell death
- PQC
protein quality control
- ROS
reactive oxygen species
Introduction
Caspases are a family of proteases that have been the subject of extensive studies on account of their important role in activating programmed cell death (PCD) 1, 2. This program is under tight control, and initiator caspases are synthesized with a protective prodomain that needs to be removed in order to activate the caspase. Upon apoptotic stimuli, initiator caspases are processed and induce a cascade, by proteolytic activation of effector, or “executioner,” caspases. When activated, the effector caspases can then cleave a wide range of substrates, eliciting the typical cytological and morphological changes seen during apoptosis, a special form of PCD 3 (Fig.1). Apoptosis has been shown to be essential for proper development and tissue maintenance in multicellular organisms, where the controlled removal of damaged or undesired cells ensures the survival and fitness of the whole organism. Accordingly, perturbations of the apoptotic machinery have been linked to several pathologies such as tumor development, autoimmune, and neurodegenerative diseases 4. While apoptosis is an accepted and distinct mode of PCD, involving a genetically determined elimination of cells, it is important to note that other forms of programmed cell death have been described, which are elicited and executed without the involvement of caspases [5,6]5.
Figure 1.
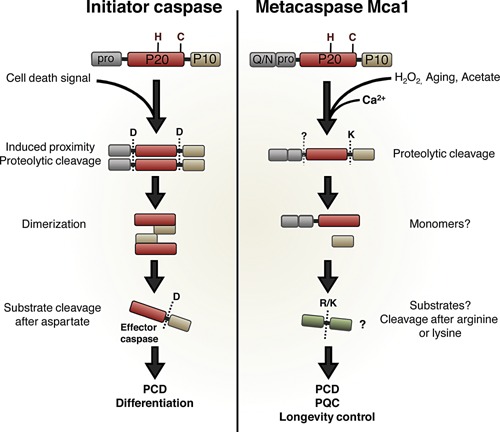
Schematic comparison of initiator caspases and the yeast metacaspase Mca1. Both proteases share the same overall structure: an N‐terminal prodomain, and a caspase domain consisting of two subunits, P20 and P10, with the conserved His‐Cys dyad located in the P20 domain. Both proteases also require processing to become enzymatically active. Activation of initiator caspases requires proteolytic cleavage after two different aspartate residues, hence separating the two subunits, and removing the prodomain. This cleavage event is followed by dimerization of two P20 subunits and two P10 subunits, forming the fully activated initiator caspase. Activation of metacaspases occurs by one lysine‐directed cleavage between P20 and P10 subunits. Although not essential for Mca1 activation, a possible second cleavage site has been identified, resulting in removal of the prodomain. Processing of metacaspases requires the presence of calcium. Once cleaved, metacaspases do not form dimers, and are thought to act as monomers. Initiator caspases activate effector caspases by aspartate directed cleavage, and effector caspases are then responsible for the proteolytic cascade leading to self‐degradation and cell death. Metacaspases cleave substrates after arginine or lysine, and downstream actions for metacaspases are less known, especially regarding the yeast metacaspase. PCD, programmed cell death; PQC, protein quality control.
Metacaspases differ from their caspase relatives in structure and substrate specificity
In studies on the evolution of cell death pathways, sequence analysis revealed that caspase‐related proteases termed paracaspases and metacaspases exists in several non‐metazoan organisms 6. Paracaspases are more similar to caspases and exist alongside caspases in several metazoans, where they have a role in the innate immune system 6, 7. Metacaspases, on the other hand, are the only caspase‐related proteins in plants, fungi, and protozoa. Metacaspases are further divided into two types: Type I, containing an N‐terminal prodomain, and type II, found only in plants, which lack an obvious prodomain 6. Recently a type III metacaspase, with a rearranged domain structure, was described in phytoplankton 8.
Similarly to their caspase relatives, metacaspases have been shown to be involved in regulated cell death during plant development and pathogen infection 9. Surprisingly, even single celled eukaryotes display responses and cytological alterations during stress that mimic those of PCD 10, 11, 12, 13, and in the yeast Saccharomyces cerevisiae, the single type I metacaspase Mca1 (Metacaspase‐1; standard name), also denoted Yca1 (Yeast caspase‐1), has been proposed to play a role – similar to an initiator caspase – in activating such a PCD pathway 14. This is intriguing because metacaspases show many similarities with their caspase relatives, including a caspase‐hemoglobinase fold and an active site containing a histidine and cysteine residue 15. However, there are also some significant differences 16: The sequence homology between caspases and metacaspases is rather low, and comparing the yeast metacaspase Mca1 to effector and initiator caspases (caspase‐3 and caspase‐9, respectively) reveals only 10–11% sequence homology 17. Consequently, there are several structural and functional differences between these protease relatives, as illustrated in Fig. 1. Both proteins are produced as inactive zymogens, with an N‐terminal prodomain and a caspase domain consisting of two subunits, P20 and P10 18. Residing in the P20 subunit of the caspase domain is the conserved His‐Cys dyad known to be required for the proteolytic activity of caspases and other members of the clan CD protease family 19, 20. Although both type I metacaspases and initiator caspases have a prodomain, metacaspases do not contain any of the well‐characterized motifs found in initiator caspases, such as DED and CARD domains 21. Instead, the prodomain of yeast Mca1 contains a proline‐rich stretch as seen for other type I metacaspases, as well as a QN rich domain, an aggregation‐prone motif present in many prion‐forming proteins 22.
Upon initiation of a cell death signal, the induced proximity of initiator caspases allows activational autoprocessing (Fig. 1) 23, 24. Such activation requires two aspartate‐directed cleavage events: separation of the P10 subunit, and removal of the prodomain. The processing is then followed by dimerization to form the final activated initiator caspase. Metacaspase activation is induced by various stressors and includes a similar separation of the P10 subunit but no removal of the prodomain. Another property of metacaspases that is not found among caspases is a dependence on calcium for activation 17, 25, 26. High levels of calcium induce further processing of Mca1 in vitro, occurring at either R72 or K86, thereby removing the prodomain (Fig. 1). However, this processing has so far not been shown to occur in vivo 17.
Structural analysis of Mca1 revealed that the yeast metacaspase is unable to undergo dimerization and, unlike canonical caspases, metacaspases presumably act as monomers 17. Metacaspases also lack the ability to cleave substrates after aspartate residues, which is the typical recognition site of caspases. Instead, metacaspases cleave proteins and peptides after arginine and lysine residues 26, 27, but little is known about substrates and downstream events following metacaspases activation in vivo. So far, only one protein has been identified as a possible substrate of the S. cerevisiae Mca1; the glycolytic enzyme GAPDH 28. The physiological relevance and effect of cleaving this substrate remain to be elucidated.
Taken together, data suggest that metacaspases, like the yeast Mca1, are distant relatives to caspases, with a different mode of activation and a different catalytic specificity (Fig. 1).
Does Mca1 act as a killer protein?
When yeast cells die as a result of age (in stationary phase) or particular stresses, they display numerous characteristics similar to those observed in cells undergoing PCD: these include nuclear fragmentation, decreased membrane integrity, and accumulation of intracellular ROS 5, 12, 29, 30, 31. The PCD‐like events in aged and deteriorated yeast cells have been proposed to be an altruistic response, where nutrients from the dead cells allow the remaining cells of the population to survive and replicate 32.
The cell death observed in yeast upon various stress treatments, including H2O2 exposure, was suggested to be initiated by Mca1, since deletion of this gene increased the survival of H2O2 stressed cells 5, 14, 33, 34, 35, 36, 37. Further, the cell death observed in wild type cells coincided with proteolytic processing of Mca1, similar to that which occurs in mammalian caspases upon their activation (Fig. 1) 14. Cells harboring a catalytically inactive version of Mca1 also survived better after H2O2 stress, strengthening the idea that the proteolytic activity of Mca1 causes cell death. PCD triggered by H2O2 has been suggested to be mediated by nitric oxide (NO) 38, which in plants have been shown to regulate metacaspase activity through S‐nitrosylation 38, 39.
Mca1‐dependent cell death has also been reported in early chronologically aged (stationary phase) cells, a phenomenon suggested to provide further evidence for an altruistic role of PCD in single cells because an Mca1‐deficient culture, after initially displaying better stationary phase survival (due to lack of killer function), showed a reduced ability to withstand subsequent, long‐term, stationary phase 32. These findings were reasoned to be the result of Mca acting as a true “killer‐protein” in an altruistic PCD pathway.
The theory of unicellular killer proteins and altruistic PCD is conceivable when considering microbial growth in colonies and clusters, where the population can exhibit “multicellular behavior.” In such clusters, PCD might serve to constrain cell number to ensure development and survival of the clone 40, 41. Classical bacterial examples include the congregation of starving myxobacteria followed by programmed death in part of a bacterial population allowing the remaining cells to develop fruiting bodies and spores 42. Similar to bacterial cases, the cell death observed in a yeast colony is localized to specific clusters of cells. In yeast, this cluster consists of the oldest cells located in the center 43. This is different from the results obtained in liquid cultures of yeast cells, which showed a spatially uniform death in the entire population 32. Moreover, the cell death observed in yeast colonies is independent of Mca1, which has been identified as the “initiator‐caspase” of cell death in liquid cultures 14. Thus, the programs of cell death elicited in liquid cultures and in colonies do not follow the same pattern, but rather rely on different factors. More data is clearly required concerning the nature of the different cell death programs triggered in low and high cell density cultures before making firm conclusions about their altruistic benefits, but it seems reasonable to assume that unicellular altruism can only be attained in crowded cultures. Also, if PCD is indeed operating in yeast cells under starvation conditions it will be interesting to elucidate how decisions are made between eliciting such a program in favor of sporulation, or whether these two processes run in parallel and co‐operate.
Mca1 is a cytoprotective protein that can retard aging
Reports of Mca1 acting as a pro‐death protein have been followed by studies suggesting that Mca1 might also possess cell‐autonomous, beneficial, functions for survival and homeostasis. For example, the yeast Mca1 appears to be required for proper cell cycle control, because cells lacking this metacaspase display an increased frequency of cell cycle arrest 44. Similar findings have been made for metacaspases in other organisms 45, 46. Mca1 has also been shown to be involved in protein quality control (PQC), as revealed by its direct physical interaction with protein aggregates 47, 48, 49, 50 and a defect observed in the removal of such aggregates in mca1Δ cells 48, 49. In C. albicans, the Mca1 metacaspase appears to cleave several chaperones suggesting that Mca1‐deficiency could boost PQC in this organism 51. However, genetic interaction analysis demonstrated that the Mca1 in S. cerevisiae is buffering against deficiencies in chaperone function, especially Hsp40, further strengthening the notion that Mca1 is a key component in cellular PQC. In fact, increased expression of MCA1 alone improves the ability of cells to remove heat‐induced aggregates, and counteracts the buildup of aggregated proteins seen during replicative aging of mother cells 48, hence suggesting that Mca1 is a limiting factor in the cellular defense against protein damage. In line with this notion, overproduction of Mca1 causes a robust extension of the replicative lifespan of cells 48. These results strongly advocate for Mca1 being involved also in pro‐survival functions.
But how, exactly, does Mca1 act in pro‐survival/longevity functions? The fact that lifespan extension achieved by Mca1 overproduction was dependent on the presence of the protein disaggregase Hsp104 suggests that management of damaged and misfolded proteins is key to Mca1‐dependent lifespan control. This control most likely involves the destruction of aberrant proteins since lifespan extension by Mca1 was also dependent on a functional proteasome: overexpression of Mca1 had no effect on lifespan in the absence of Rpn4, a transcriptional activator of proteasome genes 48. Based on these data, Mca1 may be involved in the removal of misfolded and aggregated proteins by facilitating their degradation by the proteasome (Fig. 2). Without Hsp104, Mca1 can still localize to the aggregates, but perhaps it needs the Hsp104 disaggregase activity to be able to access the misfolded proteins and assist in their destruction. There may be several ways in which Mca1 could facilitate the removal of such aberrant proteins, processes that may be linked to the possible function of Mca1 as a protease, co‐chaperone, or scaffold for other proteins (see below).
Figure 2.
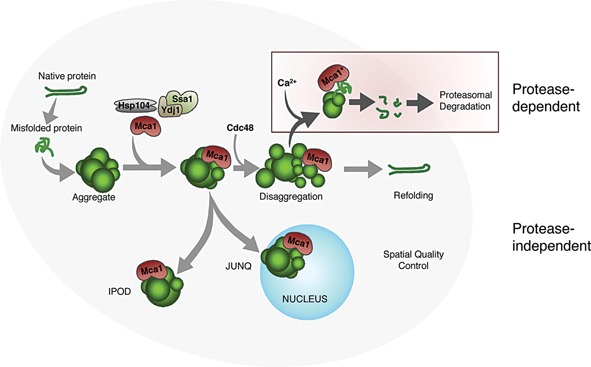
Two proposed modes of action, where Mca1 could act both in a protease‐dependent and in a protease‐independent way to manage protein damage. The protease‐dependent pathway is linked to Mca1 proteolytic activity (active Mca1 denoted as Mca1*) and proteasomal degradation. Recruitment of the segregase Cdc48, together with the disaggregase activity of Hsp104, provides access of Mca1* to substrates within aggregates. Mca1* could then cleave misfolded proteins into smaller fragments, facilitating their degradation by the proteasome. The protease‐independent role of Mca1 might be linked to a more chaperone‐like function, binding aggregates to stabilize structure and prevent further aggregation, thus providing easier access for chaperones to dissolve aggregates and refold misfolded proteins. Mca1 interactions could also facilitate the sequestration of aggregates into quality control compartments, as Mca1 localizes to aggregates in both the juxtanuclear JUNQ, as well as the peripheral IPOD compartment. The interaction between Mca1 and protein aggregates is likely mediated through the Q/N‐rich prodomain of Mca1.
Since proteasome levels and the proteolytic capacity of cells are unaffected by an MCA1 deletion 33, 44, Mca1 may instead increase the in vivo capacity of the proteasome by making proteasome substrates more accessible for degradation (Fig. 2). Indeed, the degradation of a known misfolded substrate of the 26S proteasome was retarded by Mca1‐deficiency, whereas the opposite was true upon Mca1 overproduction 48. The proteolytic activity of Mca1 could be part of this mechanism, where Mca1 pre‐cleaves substrates, to facilitate their subsequent entry into and degradation by the proteasome 52. In this scenario, we hypothesize that the substrates of Mca1 could include a variety of protein species, such as proteins that are more prone to misfold and aggregate. Interestingly, the only reported substrate for the S. cerevisiae Mca1 so far, GAPDH, is a very abundant protein that has been reported to be part of aggregates in several organisms 28, 53, 54, 55, 56, 57.
However, the proteolytic activity of Mca1 does not seem to be the only function of Mca1 in lifespan control, because overexpression of the Mca1C276A mutant allele – which encodes a metacaspase that cannot undergo catalytic processing – resulted in a residual lifespan extension 48. One feature of the protease‐deficient Mca1 that may be of importance in PQC is the Q/N rich prodomain, suggested to be required for Mca1 localization to protein aggregates 47. Q/N rich proteins have been reported to bind and sequester aggregates of Huntingtin, thereby limiting its toxicity 58, and Mca1 is part of similar disease protein aggregates 59. It is possible that the aggregate‐interacting prodomain of Mca1 is stabilizing aggregate structures to prevent further aggregation, or it makes them more accessible for the chaperone machinery 52, 60.
Mca1 acting as a co‐chaperone could also aid in the spatial control of aberrant proteins (Fig. 2). Damaged proteins are sequestered to certain storage sites as a cytoprotective action and to facilitate degradation and refolding 61. This spatial quality control also enables the retention of damaged proteins in the mother cell during cell division 62, 63. Although these quality control compartments seem not to be dependent on Mca1 for their formation (our unpublished data), Mca1 localizes both to sites of the juxtanuclear quality control compartment (JUNQ) and to the peripheral insoluble protein deposit (IPOD) 48. Located at these aggregation sites, Mca1 could be acting to keep aggregates together, and/or recruiting certain protective factors (see section below). In line with this suggested function, Mca1 suppression of the cytotoxic effect of Wwm1 overproduction might be a result of sequestering this protein to cytosolic inclusions, thereby preventing its localization to the nucleus 64.
Another role of Mca1 in PQC may be related to the metacaspase serving as a scaffold required for recruiting other proteins, such as Cdc48, to quality control compartments. Cdc48 is an AAA ATPase involved in many cellular processes, including proteasomal degradation, and extraction of ubiquitinylated proteins from large complexes through its segregase function 65. CDC48/p97 has also been shown to be involved in the formation and clearance of insoluble aggregates in mammalian cells, where it may aid resolution through a function similar to Hsp104 in yeast 66. Interestingly, the fitness of conditional yeast mutants displaying reduced Cdc48 activity is markedly reduced by deleting MCA1, and Cdc48 and Mca1 interact physically 48, 49. That this interaction is physiologically relevant is supported by data demonstrating that localization of Cdc48 to sites of protein aggregation is, in part, dependent on Mca1 49. Thus, it is possible that Mca1 recruits Cdc48 to protein aggregates were this segregase could co‐operate with Hsp104 in extracting proteins from aggregates to aid their refolding and/or degradation.
Can Mca1 act in both pro‐death and pro‐survival pathways?
Is it possible to reconcile the seemingly opposing functions of Mca1 as killer and a protector? One reconciling perspective could be that the killer function is tightly controlled such that programmed death is only set into action when there is a potential benefit for the population, such as during growth limitations in congregated cultures or colonies. An example of such control could be that the catalytic cleavage of Mca1 by removal of the prodomain – as reported in vitro upon excess calcium addition – could function as a molecular switch to regulate Mca1 killer function in vivo, and that a governor restricts switching such that it only occurs if cues indicate high cell densities (similar to quorum sensing in bacteria). Since calcium is essential for the initial proteolytic cleavage of metacaspases and for their further proteolytic activity 17, such a regulatory pathway could also be under strict spatial control and restricted to sites experiencing temporal increases in calcium concentrations 17. This theory is in line with PCD upon H2O2 treatment being linked to activational processing of Mca1 (Fig. 3A), which was previously suggested to elicit a caspase‐like activity of Mca1. However, this notion remains controversial due to uncertainties in the protocols and reagents used, including the caspase inhibitor FITC‐VAD‐FMK, which appears unspecific, and the use of non‐metacaspase substrates 26, 67, 68.
Figure 3.
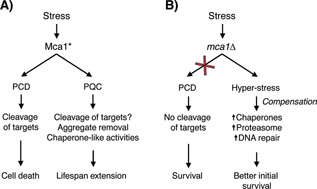
The dual role of Mca1 in cell death and cell survival. The figure schematically describes two different scenarios that could explain the increased survival of Mca1‐deficient cells observed upon H2O2 treatment and aging. A: Reports have shown that Mca1 is processed, and presumably activated (active Mca1 denoted as Mca1*), upon H2O2 exposure, and that this in turn causes downstream effects leading to cellular death. The same processing and activation of Mca1 is seen in replicatively aged cells, where Mca1 has a beneficial role in protein quality control and lifespan extension. B: Deletion of Mca1 results in a better survival after H2O2 exposure and in stationary phase. This finding can be explained by Mca1 acting as a killer protein and by the observation that the removal of such a protein enhances survival. Another explanation could be that the removal of Mca1, acting as an important player in cytoprotection, causes upregulation of compensatory stress resistance factors, including several chaperones, hence indirectly enhancing survival (compensation).
Whether linked to proteolytic activation or not, processing of Mca1 occurs in replicatively old cells, where the consequences of Mca1 overexpression – in part dependent on Mca1 processing – result in an improved PQC and an extended lifespan 48. Thus, it appears that the proteolytic activity of Mca1 can be both cytotoxic and cytoprotective, depending on cellular contexts. There are examples of such a dichotomy in the case of the 26S proteasome, where elevated activity is detrimental during heat shock of cells with limited Hsp70 chaperone activity 69, 70, but beneficial in aging cells 70. The detrimental activity of the 26S proteasome under specific conditions highlights that an unrestrained protein quality control (e.g. degradation of partially aberrant proteins that could display residual activity) may be harmful and cause cell death. It is possible that Mca1 may be part of such an unrestrained quality control.
The discussion above is based on the premise that an MCA1 deletion improves survival during stress and stationary phase due the fact that Mca1 is, in this context, acting in a direct fashion as a pro‐death protein 14, 32, 48. An alternative explanation is that an MCA1 deletion results in indirect effects and triggers responses compensating for the loss of Mca1, which could greatly affect survival. It is known that mutations in stress defense genes can trigger hyper‐induction if global stress responses, such as the super‐induction if the heat shock response in DnaK or DnaJ chaperone mutants of E. coli 71, and the constitutive stress gene activation and thermotolerance of yeast Act3 mutants 72. Likewise, Mca1 deficiency has been shown to cause upregulation of many stress‐responsive genes, including chaperones of the Hsp70 family (Ssa1, Ssa2, and Ssa4) and the disaggregase Hsp104 as well as genes involved in protein degradation and DNA repair 44, 47. Thus, mca1Δ cells display a boosted defense system that could explain improved initial survival during stationary phase and H2O2 and acetic acid exposure (Fig. 3B).
It should be noted also that the studies of chronological aging demonstrated that the initial increased survival (at the population level) displayed by Mca1‐deficient cells (up to 23 days old) was reversed as cells grew older. Similarly, for H2O2 treatment, the benefits of an MCA1 deletion were lost during aging, and old mca1Δ cells were more sensitive to H2O2 32. Thus, the older the cells are, the more they rely on functional Mca1 to survive. This dependence on a suspected killer protein for long‐term survival was rationalized to be due to benefits of an early, altruistic, cell death program. However, it could also be the result of a beneficial, non‐death role of Mca1.
Conclusions and outlook
Following the identification of the yeast metacaspase Mca1 as a protein that initiates PCD, Mca1 has also been shown to display several non‐death functions linked to PQC 33, 44, 47. The role of Mca1 in PQC could be related to the protein acting as: (i) a typical metacaspase protease, perhaps in junction with the proteasome; (ii) a co‐chaperone dependent on its Q/N‐rich domain, and/or; (iii) a protein scaffold for the recruitment of PQC factors, such as the segregase Cdc48, to sites of protein aggregation (Fig. 2). These suggested functions of Mca1 are not mutually exclusive, because binding to aggregates could be succeeded by activational cleavage and induction of proteolytic functions.
Mca1‐mediated cell death could be a result of over activation of Mca1 activity upon prolonged or severe stress, and the type of death that ensues might be linked to Mca1’s role in PQC. Because an over committed proteasome system can cause cell death under specific stress conditions that elicit protein misfolding, so could over activation of Mca1. Possibly, such improper activation of Mca1 upon stress might be linked to a breakdown in cellular calcium regulation since Ca2+ is required for autocatalytic activation of metacaspases. An alternative explanation for the improved survival of Mca1‐deficient cells during stress could be an indirect effect of the induction of a compensatory response.
There are clearly many interesting facts still to be learned about metacaspases in both multi‐and unicellular organisms, and the yeast Mca1 offers a unique opportunity to acquire some knowledge about the evolutionary significance of this intriguing protein family.
Acknowledgments
Work in the Nystrom laboratory was supported by grants from the Swedish Natural Research Council (VR), the Knut and Alice Wallenberg Foundation (Wallenberg Scholar), and ERC (Advanced Grant; QualiAge).
The authors have declared no conflict of interest.
References
- 1. Aravind L, Dixit VM, Koonin EV. 2001. Apoptotic molecular machinery: vastly increased complexity in vertebrates revealed by genome comparisons. Science 291: 1279–84. [DOI] [PubMed] [Google Scholar]
- 2. Yuan J, Horvitz HR. 2004. A first insight into the molecular mechanisms of apoptosis. Cell 116: S53–6, p following : S9. [DOI] [PubMed] [Google Scholar]
- 3. Kumar S. 2007. Caspase function in programmed cell death. Cell Death Differ 14: 32–43. [DOI] [PubMed] [Google Scholar]
- 4. Favaloro B, Allocati N, Graziano V, Di Ilio C, et al. 2012. Role of apoptosis in disease. Aging 4: 330–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Madeo F, Carmona‐Gutierrez D, Ring J, Buttner S, et al. 2009. Caspase‐dependent and caspase‐independent cell death pathways in yeast. Biochem Biophys Res Commun 382: 227–31. [DOI] [PubMed] [Google Scholar]
- 6. Uren AG, O'Rourke K, Aravind LA, Pisabarro MT, et al. 2000. Identification of paracaspases and metacaspases: two ancient families of caspase‐like proteins, one of which plays a key role in MALT lymphoma. Mol Cell 6: 961–7. [DOI] [PubMed] [Google Scholar]
- 7. Thome M, Charton JE, Pelzer C, Hailfinger S. 2010. Antigen receptor signaling to NF‐kappaB via CARMA1, BCL10, and MALT1. Cold Spring Harb Perspect Biol 2: 0a003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Choi CJ, Berges JA. 2013. New types of metacaspases in phytoplankton reveal diverse origins of cell death proteases. Cell Death Dis 4: e490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tsiatsiani L, Van Breusegem F, Gallois P, Zavialov A, et al. 2011. Metacaspases. Cell Death Differ 18: 1279–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ameisen JC, Idziorek T, Billaut‐Mulot O, Loyens M, et al. 1995. Apoptosis in a unicellular eukaryote (Trypanosoma cruzi): implications for the evolutionary origin and role of programmed cell death in the control of cell proliferation, differentiation and survival. Cell Death Differ 2: 285–300. [PubMed] [Google Scholar]
- 11. Lewis K. 2000. Programmed death in bacteria. Microbiol Mol Biol Rev 64: 503–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Madeo F, Frohlich E, Frohlich KU. 1997. A yeast mutant showing diagnostic markers of early and late apoptosis. J Cell Biol 139: 729–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Segovia M, Haramaty L, Berges JA, Falkowski PG. 2003. Cell death in the unicellular chlorophyte Dunaliella tertiolecta. A hypothesis on the evolution of apoptosis in higher plants and metazoans. Plant Physiol 132: 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Madeo F, Herker E, Maldener C, Wissing S, et al. 2002. A caspase‐related protease regulates apoptosis in yeast. Mol Cell 9: 911–7. [DOI] [PubMed] [Google Scholar]
- 15. Aravind L, Koonin EV. 2002. Classification of the caspase‐hemoglobinase fold: detection of new families and implications for the origin of the eukaryotic separins. Proteins 46: 355–67. [DOI] [PubMed] [Google Scholar]
- 16. Koonin EV, Aravind L. 2002. Origin and evolution of eukaryotic apoptosis: the bacterial connection. Cell Death Differ 9: 394–404. [DOI] [PubMed] [Google Scholar]
- 17. Wong AH, Yan C, Shi Y. 2012. Crystal structure of the yeast metacaspase Yca1. J Biol Chem 287: 29251–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thornberry NA, Lazebnik Y. 1998. Caspases: enemies within. Science 281: 1312–6. [DOI] [PubMed] [Google Scholar]
- 19. Mottram JC, Helms MJ, Coombs GH, Sajid M. 2003. Clan CD cysteine peptidases of parasitic protozoa. Trends Parasitol 19: 182–7. [DOI] [PubMed] [Google Scholar]
- 20. Chen JM, Rawlings ND, Stevens RA, Barrett AJ. 1998. Identification of the active site of legumain links it to caspases, clostripain and gingipains in a new clan of cysteine endopeptidases. FEBS Lett 441: 361–5. [DOI] [PubMed] [Google Scholar]
- 21. Chang HY, Yang X. 2000. Proteases for cell suicide: functions and regulation of caspases. Microbiol Mol Biol Rev 64: 821–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alberti S, Halfmann R, King O, Kapila A, et al. 2009. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 137: 146–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Parrish AB, Freel CD, Kornbluth S. 2013. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb Perspect Biol 5: a008672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bao Q, Shi Y. 2007. Apoptosome: a platform for the activation of initiator caspases. Cell Death Differ 14: 56–65. [DOI] [PubMed] [Google Scholar]
- 25. McLuskey K, Rudolf J, Proto WR, Isaacs NW, et al. 2012. Crystal structure of a Trypanosoma brucei metacaspase. Proc Natl Acad Sci USA 109: 7469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Watanabe N, Lam E. 2005. Two Arabidopsis metacaspases AtMCP1b and AtMCP2b are arginine/lysine‐specific cysteine proteases and activate apoptosis‐like cell death in yeast. J Biol Chem 280: 14691–9. [DOI] [PubMed] [Google Scholar]
- 27. Vercammen D, van de Cotte B, De Jaeger G, Eeckhout D, et al. 2004. Type II metacaspases Atmc4 and Atmc9 of Arabidopsis thaliana cleave substrates after arginine and lysine. J Biol Chem 279: 45329–36. [DOI] [PubMed] [Google Scholar]
- 28. Silva A, Almeida B, Sampaio‐Marques B, Reis MI, et al. 2011. Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) is a specific substrate of yeast metacaspase. Biochim Biophys Acta 1813: 2044–9. [DOI] [PubMed] [Google Scholar]
- 29. Laun P, Pichova A, Madeo F, Fuchs J, et al. 2001. Aged mother cells of Saccharomyces cerevisiae show markers of oxidative stress and apoptosis. Mol Microbiol 39: 1166–73. [PubMed] [Google Scholar]
- 30. Ludovico P, Sousa MJ, Silva MT, Leao C, et al. 2001. Saccharomyces cerevisiae commits to a programmed cell death process in response to acetic acid. Microbiology 147: 2409–15. [DOI] [PubMed] [Google Scholar]
- 31. Ribeiro GF, Corte‐Real M, Johansson B. 2006. Characterization of DNA damage in yeast apoptosis induced by hydrogen peroxide, acetic acid, and hyperosmotic shock. Mol Biol Cell 17: 4584–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Herker E, Jungwirth H, Lehmann KA, Maldener C, et al. 2004. Chronological aging leads to apoptosis in yeast. J Cell Biol 164: 501–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Khan MA, Chock PB, Stadtman ER. 2005. Knockout of caspase‐like gene, YCA1, abrogates apoptosis and elevates oxidized proteins in Saccharomyces cerevisiae . Proc Natl Acad Sci USA 102: 17326–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guaragnella N, Pereira C, Sousa MJ, Antonacci L, et al. 2006. YCA1 participates in the acetic acid induced yeast programmed cell death also in a manner unrelated to its caspase‐like activity. FEBS Lett 580: 6880–4. [DOI] [PubMed] [Google Scholar]
- 35. Bettiga M, Calzari L, Orlandi I, Alberghina L, et al. 2004. Involvement of the yeast metacaspase Yca1 in ubp10Delta‐programmed cell death. FEMS Yeast Res 5: 141–7. [DOI] [PubMed] [Google Scholar]
- 36. Lefevre S, Sliwa D, Auchere F, Brossas C, et al. 2012. The yeast metacaspase is implicated in oxidative stress response in frataxin‐deficient cells. FEBS Lett 586: 143–8. [DOI] [PubMed] [Google Scholar]
- 37. Reiter J, Herker E, Madeo F, Schmitt MJ. 2005. Viral killer toxins induce caspase‐mediated apoptosis in yeast. J Cell Biol 168: 353–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Almeida B, Buttner S, Ohlmeier S, Silva A, et al. 2007. NO‐mediated apoptosis in yeast. J Cell Sci 120: 3279–88. [DOI] [PubMed] [Google Scholar]
- 39. Belenghi B, Romero‐Puertas MC, Vercammen D, Brackenier A, et al. 2007. Metacaspase activity of Arabidopsis thaliana is regulated by S‐nitrosylation of a critical cysteine residue. J Biol Chem 282: 1352–8. [DOI] [PubMed] [Google Scholar]
- 40. Vachova L, Cap M, Palkova Z. 2012. Yeast colonies: a model for studies of aging, environmental adaptation, and longevity. Oxid Med Cell Longev 2012: 601836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Palkova Z, Wilkinson D, Vachova L. 2014. Aging and differentiation in yeast populations: elders with different properties and functions. FEMS Yeast Res 14: 96–108. [DOI] [PubMed] [Google Scholar]
- 42. Claessen D, Rozen DE, Kuipers OP, Sogaard‐Andersen L, et al. 2014. Bacterial solutions to multicellularity: a tale of biofilms, filaments and fruiting bodies. Nat Rev Microbiol 12: 115–24. [DOI] [PubMed] [Google Scholar]
- 43. Vachova L, Palkova Z. 2005. Physiological regulation of yeast cell death in multicellular colonies is triggered by ammonia. J Cell Biol 169: 711–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee RE, Puente LG, Kaern M, Megeney LA. 2008. A non‐death role of the yeast metacaspase: Yca1p alters cell cycle dynamics. PLoS One 3: e2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ambit A, Fasel N, Coombs GH, Mottram JC. 2008. An essential role for the Leishmania major metacaspase in cell cycle progression. Cell Death Differ 15: 113–22. [DOI] [PubMed] [Google Scholar]
- 46. Helms MJ, Ambit A, Appleton P, Tetley L, et al. 2006. Bloodstream form Trypanosoma brucei depend upon multiple metacaspases associated with RAB11‐positive endosomes. J Cell Sci 119: 1105–17. [DOI] [PubMed] [Google Scholar]
- 47. Lee RE, Brunette S, Puente LG, Megeney LA. 2010. Metacaspase Yca1 is required for clearance of insoluble protein aggregates. Proc Natl Acad Sci USA 107: 13348–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hill SM, Hao X, Liu B, Nystrom T. 2014. Life‐span extension by a metacaspase in the yeast Saccharomyces cerevisiae . Science 344: 1389–92. [DOI] [PubMed] [Google Scholar]
- 49. Shrestha A, Puente LG, Brunette S, Megeney LA. 2013. The role of Yca1 in proteostasis. Yca1 regulates the composition of the insoluble proteome. J Proteomics 81: 24–30. [DOI] [PubMed] [Google Scholar]
- 50. Dick SA, Megeney LA. 2013. Cell death proteins: an evolutionary role in cellular adaptation before the advent of apoptosis. BioEssays 35: 974–83. [DOI] [PubMed] [Google Scholar]
- 51. Leger T, Garcia C, Ounissi M, Lelandais G, et al. 2015. The metacaspase (Mca1p) has a dual role in Farnesol‐induced apoptosis in Candida albicans . Mol Cell Proteomics 14: 93–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kampinga HH. 2014. Cell biology. A cell death avenue evolved from a life‐saving path. Science 344: 1341–2. [DOI] [PubMed] [Google Scholar]
- 53. Burke JR, Enghild JJ, Martin ME, Jou YS, et al. 1996. Huntingtin and DRPLA proteins selectively interact with the enzyme GAPDH. Nat Med 2: 347–50. [DOI] [PubMed] [Google Scholar]
- 54. Koshy B, Matilla T, Burright EN, Merry DE, et al. 1996. Spinocerebellar ataxia type‐1 and spinobulbar muscular atrophy gene products interact with glyceraldehyde‐3‐phosphate dehydrogenase. Hum Mol Genet 5: 1311–8. [DOI] [PubMed] [Google Scholar]
- 55. Wang Y, Meriin AB, Costello CE, Sherman MY. 2007. Characterization of proteins associated with polyglutamine aggregates: a novel approach towards isolation of aggregates from protein conformation disorders. Prion 1: 128–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Guzhova IV, Lazarev VF, Kaznacheeva AV, Ippolitova MV, et al. 2011. Novel mechanism of Hsp70 chaperone‐mediated prevention of polyglutamine aggregates in a cellular model of huntington disease. Hum Mol Genet 20: 3953–63. [DOI] [PubMed] [Google Scholar]
- 57. Lazarev VF, Sverchinskyi DV, Ippolitova MV, Stepanova AV, et al. 2013. FActors affecting aggregate formation in cell models of huntington's disease and amyotrophic lateral sclerosis. Acta Naturae 5: 81–9. [PMC free article] [PubMed] [Google Scholar]
- 58. Kayatekin C, Matlack KE, Hesse WR, Guan Y, et al. 2014. Prion‐like proteins sequester and suppress the toxicity of huntingtin exon 1. Proc Natl Acad Sci USA 111: 12085–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Park SH, Kukushkin Y, Gupta R, Chen T, et al. 2013. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell 154: 134–45. [DOI] [PubMed] [Google Scholar]
- 60. Bozhkov PV, Smertenko AP, Zhivotovsky B. 2010. Aspasing out metacaspases and caspases: proteases of many trades. Sci Signal 3: pe48. [DOI] [PubMed] [Google Scholar]
- 61. Kaganovich D, Kopito R, Frydman J. 2008. Misfolded proteins partition between two distinct quality control compartments. Nature 454: 1088–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Spokoini R, Moldavski O, Nahmias Y, England JL, et al. 2012. Confinement to organelle‐associated inclusion structures mediates asymmetric inheritance of aggregated protein in budding yeast. Cell Rep 2: 738–47. [DOI] [PubMed] [Google Scholar]
- 63. Song J, Yang Q, Yang J, Larsson L, et al. 2014. Essential genetic interactors of SIR2 required for spatial sequestration and asymmetrical inheritance of protein aggregates. PLoS Genet 10: e1004539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Szallies A, Kubata BK, Duszenko M. 2002. A metacaspase of Trypanosoma brucei causes loss of respiration competence and clonal death in the yeast Saccharomyces cerevisiae . FEBS Lett 517: 144–50. [DOI] [PubMed] [Google Scholar]
- 65. Baek GH, Cheng H, Choe V, Bao X, et al. 2013. Cdc48: a swiss army knife of cell biology. J Amino Acids 2013: 183421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kobayashi T, Manno A, Kakizuka A. 2007. Involvement of valosin‐containing protein (VCP)/p97 in the formation and clearance of abnormal protein aggregates. Genes Cells 12: 889–901. [DOI] [PubMed] [Google Scholar]
- 67. Vachova L, Palkova Z. 2007. Caspases in yeast apoptosis‐like death: facts and artefacts. FEMS Yeast Res 7: 12–21. [DOI] [PubMed] [Google Scholar]
- 68. Hardwick JM, Cheng WC. 2004. Mitochondrial programmed cell death pathways in yeast. Dev Cell 7: 630–2. [DOI] [PubMed] [Google Scholar]
- 69. Baxter BK, Craig EA. 1998. Isolation of UBP3, encoding a de‐ubiquitinating enzyme, as a multicopy suppressor of a heat‐shock mutant strain of S. cerevisiae . Curr Genet 33: 412–9. [DOI] [PubMed] [Google Scholar]
- 70. Oling D, Eisele F, Kvint K, Nystrom T. 2014. Opposing roles of Ubp3‐dependent deubiquitination regulate replicative life span and heat resistance. EMBO J 33: 747–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Liberek K, Georgopoulos C. 1993. Autoregulation of the Escherichia coli heat shock response by the DnaK and DnaJ heat shock proteins. Proc Natl Acad Sci USA 90: 11019–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gorzer I, Schuller C, Heidenreich E, Krupanska L, et al. 2003. The nuclear actin‐related protein Act3p/Arp4p of Saccharomyces cerevisiae is involved in transcription regulation of stress genes. Mol Microbiol 50: 1155–71. [DOI] [PubMed] [Google Scholar]