

Abstract
Cilia arose early during eukaryotic evolution, and their structural components are highly conserved from the simplest protists to complex metazoan species. In recent years, the role of cilia in the ontogeny of vertebrate organs has received increasing attention due to a staggering correlation between human disease and dysfunctional cilia. In particular, the presence of cilia in both the developing and mature kidney has become a deep area of research due to ciliopathies common to the kidney, such as polycystic kidney disease (PKD). Interestingly, mutations in genes encoding proteins that localize to the cilia cause similar cystic phenotypes in kidneys of various vertebrates, suggesting an essential role for cilia in kidney organogenesis and homeostasis as well. Importantly, the genes so far identified in kidney disease have conserved functions across species, whose kidneys include both primary and motile cilia. Here, we aim to provide a comprehensive description of cilia and their role in kidney development, as well as highlight the usefulness of the zebrafish embryonic kidney as a model to further understand the function of cilia in kidney health.
Keywords: ciliopathy, development, kidney, mesonephros, metanephros, motile cilia, multiciliated cell, primary cilia, pronephros, zebrafish
1. Introduction
Cilia are present in various tissues throughout vertebrate development, and nearly ubiquitously present in the cells of mature organs (Davis, Brueckner, & Katsanis, 2006, Choksi et al., 2014). Based on their structural organization and function, cilia in vertebrates are divided into two classes: (1) motile cilia and (2) primary cilia (Figure 1A–C) (Vincensini, Blisnick, & Bastin, 2011). Motile cilia bear a radial “9 + 2” pattern, which is comprised of nine microtubule doublets and a pair of central microtubules (Figure 1B). Extensive studies have demonstrated the importance of fluid flow from cells with a single motile cilium to establish normal left‐right patterning of the early embryo (Bisgrove and Yost, 2006). Cells with multiple bundles of motile cilia, termed multiciliated cells (MCCs) are considered fully differentiated and nonmitotic (Figure 1C) (Brooks & Wallingford, 2014). Ciliary motility is achieved by dynein arms and radial spokes associated with microtubule structures (Vincensini et al., 2011). The major role of MCCs is to facilitate fluid propulsion by generating coordinated movements in multiple tissues of the body, including the respiratory system, the reproductive system, brain ventricles, and kidneys of lower vertebrates (Cowan, Gladwin, & Shelhamer, 2001; Jain et al., 2010; Kramer‐Zucker et al., 2005; Li, Cheng, Verdun, & Wingert, 2014; Liu, Narendra, Kramer‐Zucker, & Drummond, 2007; Ma & Jiang, 2007; Marra & Wingert, 2016; Wang et al., 2013; Worthington & Cathcart, 1963). For example, several studies in the zebrafish have illuminated roles for MCCs during kidney and brain formation (Kramer‐Zucker et al., 2005; Wang et al., 2013; Znosko et al., 2010). Thus, there is precedence for their involvement in vertebrate organ development, although additional roles for motile cilia and MCCs in higher vertebrate organogenesis have yet to be fully appreciated.
Figure 1.
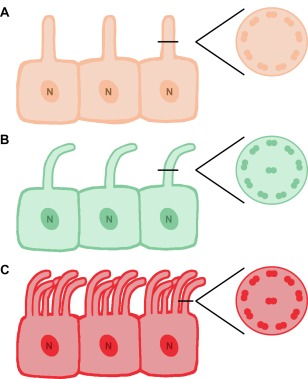
Types of ciliated cells and cilia. Schematic of epithelial cells with (A) primary cilia, (B) motile cilia, and (C) multiple motile cilia, also known as multiciliated cells. Respective cross sections depict the characteristic microtubule distribution of each cilia type. Abbreviations: N, nucleus
Different from motile cilia, primary cilia are typically present as a single organelle anchored on the cell surface (Vincensini et al., 2011) (Figure 1A). Primary cilia have a “9 + 0” pattern without the central pair of microtubules (Figure 1A). These organelles do not have dynein arms or their associated apparatus as energy sources, thus making them nonmotile. Rather, primary cilia play important sensory and signal transduction roles, as they house and interact with an elaborate machinery of membrane receptors, ion channels, transporter proteins, and various downstream effectors (Deane & Ricardo, 2012). In contrast to MCCs, cells with primary cilia are capable of undergoing mitosis (Deane & Ricardo, 2012). Primary cilia can be found in multiple tissues of the body throughout different developmental stages. Hence, primary cilia show potential for rolesin organ development, adult tissue homeostasis, and repair. The function of cilia, both primary and motile, in vertebrate kidney development has particularly begun to receive an increased amount of attention.
During embryogenesis of higher vertebrates, kidney development is distinct from other organs in that it proceeds through a series of up to three successive phases, with each one becoming a more structurally advanced renal system. These three possible types of kidney systems are the pronephros, mesonephros, and metanephros (Saxen, 1987). In reptiles, birds and mammals, the pronephros and mesonephros are transient embryonic organs that subsequently degenerate. The final kidney form in these animals is the metanephros, which functions to filter the blood, excrete waste metabolites, and regulate electrolyte homeostasis. In mammals specifically, the pronephros has become a vestigial organ, although its genesis is nevertheless required for the proper subsequent stages of renal development (Bouchard, Souabni, Mandler, Neubuser, & Busslinger, 2002; James & Schultheiss, 2003; Mauch, Yang, Wright, Smith, & Schoenwolf, 2000; Shawlot & Behringer, 1995; Vize, Woolf, & Bard, 2003). Conversely, in lower vertebrates such as fish and amphibians, the pronephros serves as a functional excretory organ during development (Reimschuessel, 2001; Vize et al., 2003). Following degeneration of the pronephros, the mesonephros becomes the functioning kidney during the juvenile and adult stages of both fish and amphibians (Reimschuessel, 2001; Vize et al., 2003).
The basic functional and anatomical unit within all types of vertebrate kidneys is the nephron, a pipe‐like structure with highly segmented subdomains of epithelial cells that are distinct from one another in their expression profile of solute transporters and ion channels (McCampbell & Wingert, 2012; Romagnani, Lasagni, & Remuzzi, 2013). In each species, the number and anatomical arrangements of nephrons are a major aspect of the increasing complexity between the pronephros, mesonephros, and metanephros. At the beginning of each nephron is the glomerulus, which is responsible for filtering of the blood. The filtrate is then passed through a tubule comprised of a series of proximal and distal tubule segments that contain specialized epithelia which perform different modifications on the filtrate (namely reabsorption and secretion) followed by a collecting duct system. Within the nephron tubule, waste metabolites are excreted and urinary composition is further condensed by reabsorption of solutes, ions, proteins, and water before the final urine product is delivered to the collecting duct for disposal. These complex physiological tasks require an elegant composition of unique epithelial cell types and coordinating interplay during cellular fate specification (Costantini & Kopan, 2010; Desgrange & Cereghini, 2015; Little & McMahon, 2012). In the kidneys, nephrons alone contain more than a dozen epithelial cell types characterized by specific ultrastructural features and functions (Li & Wingert, 2013).
As such, the field of nephrology has been intrigued with how these distinct cell types are determined and the associated molecular mechanisms underlying vertebrate nephrogenesis, resulting in a precise architectural framework created to filter and regulate body fluid composition. Transformative research using the mouse model has been complemented over the past decades with the emergence of other advantageous animal models such as Xenopus laevis (frog) and Danio rerio (zebrafish) (Dressler, 2006). Using these experimental systems, there has been progress in discovering the mechanisms behind the genetic and cellular basis of renal development (Gerlach & Wingert, 2013). However, continuing endeavors by many labs are centered on furthering this still relatively incomplete understanding of how the cellular composition of the kidney is achieved through patterning and morphogenesis of renal progenitors, including the roles of cilia and the essential aspects of ciliated cells in nephron epithelial cell formation and function which we discuss in the following sections.
2. First indications to the role of cilia in kidney organogenesis
An increasing body of research has demonstrated the presence of primary cilia in developing and mature human kidneys (Figure 2), as well as their essential roles in the pathogenesis of polycystic kidney disease (PKD) (Calvet, 2002; Chapin & Caplan, 2010; Kolb, Woost, & Hopfer, 2004; Rodat‐Despoix & Delmas, 2009; Saraga‐Babić, Vukojević, Bočina, Drnašin, & Saraga, 2012; Satlin, Sheng, Woda, & Kleyman, 2001). Distinct from intercalated cells found in the collecting duct, the other mammalian nephron epithelia all possess a single nonmotile primary cilium (Figure 2D) (Saraga‐Babić et al., 2012). Under electron microscopy, these single primary cilia have been visualized as extending from the apical surface of the epithelial cells lining the nephron tubule and collecting duct, thus placing them under constant contact with urine flow in the nephron (Figure 2D) (Saraga‐Babić et al., 2012). Whereas the presence of primary cilia has been known for decades, it was not until recently that these tiny sensory antennae became appreciated for their suite of crucial physiological functions.
Figure 2.
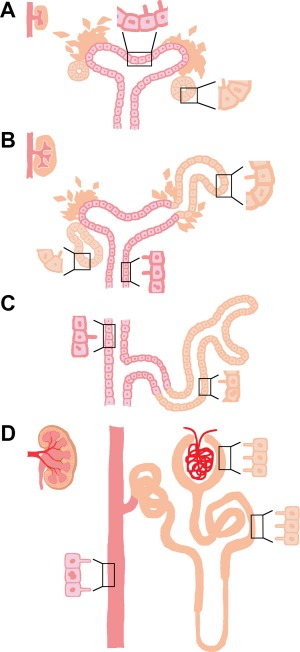
Cilia in mammalian kidney development. (A) Primary cilia are first seen on nonproliferating epithelial cells of the renal vesicle (RV) and ureteric bud (UB) during kidney development. (B) As the RV grows into a comma‐shaped early nephron, primary cilia remain on nonproliferating cells of the nascent nephron and growing UB as it expands the collecting duct system. (C) Nonproliferating cells of the collecting duct and S‐shaped nephron have primary cilia. (D) Primary cilia continue to be present on epithelial cells of the mature nephron tubule and collecting duct. Proliferating cells are drawn with a dividing nucleus.
In adult mammalian kidneys, primary cilia (distinguished by their “9 + 0” structure) in the nephron are typically 2–4 μm in length, and have been reported in both Bowman's capsule and the tubule (Figure 2D) (Webber & Lee, 1975). However, as a dynamic organelle, the presence, length, and composition of primary cilia are under constant regulation in order to fulfill essential functions such as signaling transduction (Marshall & Rosenbaum, 2001). In most eukaryotes, the assembly and maintenance of cilia relies on the phenomenon of intraflagellar transport (IFT) (Kozminski, Johnson, Forscher, & Rosenbaum, 1993; Kozminski, Beech, & Rosenbaum, 1995). IFT represents the bidirectional transport of particles along the microtubular doublets of the cilium and serves as a shuttle system centered upon the basal body, which is a nucleation site for the growth of cilia microtubules (Carvalho‐Santos, Azimzadeh, Pereira‐Leal, & Bettencourt‐Dias, 2011).
As such, IFT is important for the delivery of components from the cytoplasm to the cilium and recycling of turnover products. By doing so, IFT regulates the dynamic balance between cilia assembly and disassembly, and hence determines the length of the organelle (Carvalho‐Santos et al., 2011). This active dynamism of cilia constituents relies on the trafficking of membrane proteins to and from the cilium, which is accomplished by a specialized protein complex known as the BBSome. Localized to the cilia, malfunctions of BBsome components are related to a severe genetic disorder called Bardet–Biedl syndrome, which is characterized by complex ciliopathic conditions affecting multiple body systems in the patients (Jin & Nachury, 2009). Whereas numerous studies have described primary cilia as cellular sensory organs that play essential roles for conducting signaling pathways in adult tissues (Marshall & Nonaka, 2006; Singla & Reiter, 2006; Berbari, O'Connor, Haycraft, & Yoder, 2009), more recent work has begun to explore if and how primary cilia mediate organogenesis during vertebrate development, especially for the kidney.
A direct piece of evidence that primary cilia may play essential roles in proper kidney development came from studying the inherited kidney developmental disorders such as autosomal recessive polycystic kidney disease (ARPKD) (Deane & Ricardo, 2007). ARPKD can be diagnosed by the appearance of renal and hepatic cysts, with an incidence of 1 in 20,000 live births (Jang et al., 2011). Progressive expansion of fluid‐filled cysts results in massive enlargement of the kidneys and often leads to renal failure (Yoder, 2007). Genome analysis of human ARPKD and a related autosomal dominant disease,ARPKD, has identified several mutations leading to the onset of these PKD conditions, such as those which encode the proteins polycystin‐1, polycystin‐2, polaris, cystin, and inversin that normally localize to the primary cilia of nephron epithelial cells (Yoder, Hou, & Guay‐Woodford, 2002). Investigations into the pathologies of various PKD conditions unveiled the connections between proper primary cilia function and the developmental and pathophysiological consequences in the kidney. While the existence of primary cilia in the kidney has been known for a long time, the significance of these organelles has since then been reevaluated.
In comparison, the role of motile cilia, namely MCCs, is vastly understudied in nephrogenesis. MCCs are present and regulate fluid flow in the pronephros of lower vertebrates, such as fish and amphibians (Kramer‐Zucker et al., 2005; Li et al., 2014; Liu et al., 2007; Ma & Jiang, 2007; Marra & Wingert, 2016; Wang et al., 2013). As previously discussed, the pronephros of fish and amphibians serves as a functioning kidney within the embryo, which is distinct from mammalian species (Dressler, 2006). The formation of MCCs has been studied in the pronephric kidney by several research groups, which is discussed in subsequent sections of this review. In contrast, MCCs have not been found in healthy adult mammalian nephrons. Interestingly, however, MCCs have been documented anecdotally in the fetal kidney of humans, and in biopsies from patients with various diseased states of the kidney as well, based on the their distinctive “9 + 2” microtubule structure (Duffy & Suzuki, 1968; Hassan & Subramanyan, 1995; Katz & Morgan, 1984; Ong & Wagner, 2005). Detailed knowledge about the emergence, distribution, and duration of MCCs during human fetal development remains scarce within the current medical literature. Among the tissue samples from kidney disease patients, MCCs were noted on proximal tubular epithelia (Hassan & Subramanyan, 1995; Ong & Wagner, 2005). Similar to the lack of information about MCCs in the developing human kidney, though, voids in our knowledge about these cells during renal disease include the timing of their appearance and distribution in other locales.
With the rejuvenated outlook as to the role of nonmotile cilia in the developing kidney due to the discoveries of mutations in proteins associated with primary cilia and their connections to PKD, it is worth analyzing the potential role of motile cilia during kidney organogenesis and in an assortment of renal disease states. Although our current knowledge and understanding of the mammalian kidney does not provide a definitive explanation as to why these purported MCCs arise on tubular epithelial cells in cases of disease, one current hypothesis is that the presence of motile cilia represents a reversion to a more primitive state. In this view, the motile cilia are formed to help relieve low blood pressure and regulate filtration by facilitating nephron fluid flow (Duffy & Suzuki, 1968; Katz & Morgan, 1984; Kramer‐Zucker et al., 2005; Ong & Wagner, 2005; Zimmerman, 1971).
3. When ciliogenesis met nephrogenesis
Understanding more about when and where cilia are present during kidney development is the first step toward elucidating their role(s) in the process. Several studies have made observations about the appearance and distribution of primary cilia during human renal ontogeny (DeMartino & Zamboni, 1966; Saraga‐Babić et al., 2012; Zimmerman, 1971). The complex development of the human kidney begins at the beginning of the fourth week of gestation with the emergence and regression of the pronephros (Sadler, 2012). Between the fourth to sixth week, the pronephros is rapidly succeeded by mesonephros formation and the onset of metanephros development (Sadler, 2012). All of these renal tissues emerge from the anterior to the posterior end of the intermediate mesoderm (IM), which is located between the paraxial mesoderm and lateral plate mesoderm (Dressler, 2006; Sadler, 2012). There is a paucity of detailed information about the quantity and location(s) of cilia in the human pro‐ and mesonephros, likely due to the fast genesis and degeneration of these renal units, though the presence of cilia has been reported (DeMartino & Zamboni, 1966).
The metanephric kidney forms when the ureteric bud (UB) branches from the mesonephric duct and invades into the surrounding metanephric mesenchyme (MM) by approximately the fifth week of development (Sadler, 2012). Signaling molecules secreted from the UB tips then transform the MM cells into condensed MM aggregates (Dressler, 2006). However, in the sixth week of embryonic development, as these earliest stages of nephrogenesis take place in the metanephros, the presence of short primary cilia, based on a “9 + 0” microtubule structure and shown by strong α‐tubulin positivity, has been detected on the surface of nonproliferating UB epithelial cells, which will give rise to the collecting duct system (Saraga‐Babić et al., 2012). During the subsequent weeks of gestation, the UB undergoes intensive branching to generate several hundred thousand to as many as over 1 million nephrons along with an accompanying elegant collecting duct network (Dressler, 2009).
During nephrogenesis in the metanephros, the transition from spindle‐shaped MM cells toward an elongated and patterned nephron epithelium is known as a mesenchymal‐to‐epithelial transition (MET). Nephron progenitors undergo MET when the cap mesenchymal cells in the MM aggregate give rise to an organized renal vesicle (RV), a primitive epithelium with a basement membrane and a hollow lumen that is situated in close proximity to the UB stalk (Dressler, 2006). Formation of the RV is characterized by the establishment of an apical‐basal polarity and the expression of intercellular junctions (Dressler, 2006).
Remarkably, during human nephrogenesis, short primary cilia with the “9 + 0” pattern were observed as early as the RV stage in the developing primitive nephron, anchoring on the surface of the nonproliferating cells in the RV (Figure 2A) (Saraga‐Babić et al., 2012). Using antibodies against α‐tubulin to visualize cilia and Ki‐67 labeling to see proliferating mesenchymal cells, weak expression of primary cilia in the MM aggregates was also seen, while emergence of primary cilia became more robust starting from the RV stage in Ki‐67 negative epithelial cells (Saraga‐Babić et al., 2012). Expression of α‐tubulin continues in the nascent nephron as the RVs grow into comma and S‐shape forms (Figure 2B and C) (Saraga‐Babić et al., 2012). Presence of the ciliated cells overlaps formation of intercellular junctions and apical‐basal polarity, and is accompanied by formation of the nephric lumen (Saraga‐Babić et al., 2012). The existence of primary cilia on cells within RVs and during subsequent growth may suggest that cilia are playing important roles during nephrogenesis, such as renal progenitor proliferation, differentiation, patterning, cellular polarity establishment, and/or lumen formation.
When nephrogenesis is complete, the RV and its renal progenitors have differentiated and regionalized to give rise to a mature nephron with a distinct segmental pattern. A fully developed human nephron is comprised of a Bowman's capsule in the proximal end, which is linked to a segmented nephron tubule with distinct proximal and distal domains fused to the collecting duct system (Wingert & Davidson, 2008). Interestingly, as specification and patterning of the nephron progenitors takes place, the length of the primary cilia on cells lining the nephron lumen also exhibit significant growth, from 0.59 µm at the RV stage to 3.04 µm in fetal nephrons (Saraga‐Babić et al., 2012). Such findings suggest that the presence of primary cilia could play important roles in nephrogenesis and presumably establishment of fetal kidney functions later during embryogenesis.
Following the initiation of the first metanephric nephrons at the seventh and eighth week, the UB tips actively continue to undergo branching morphogenesis while simultaneously invading into the surrounding MM to induce increasing numbers of MM aggregates, which as described will each develop into RVs and primitive nephrons. The establishment of nephrons continues until approximately the thirty‐sixth week (Cukuranovic & Vlajkovic, 2005; Hinchcliffe, Howard, Chan, & van Velzen, 1991). Accompanied by the maturation of the first groups of nephrons, however, the human fetal metanephric kidney starts to function as early as the ninth week of the antenatal life (Cukuranovic & Vlajkovic, 2005). Throughout the induction of new nephrons, strong α‐tubulin expression could be detected in primary cilia of the collecting ducts, while moderate expression was seen in the MM ampulla and comma‐shaped body (Figure 2B and C) (Saraga‐Babić et al., 2012). Weak expression of α‐tubulin was also present in the newly induced metanephric cups (Saraga‐Babić et al., 2012). An accumulation of electron microscope data indicates that metanephric tubule samples from human fetuses ranging in age from thirteen to twenty‐two‐week olds have cilia with the “9 + 0” ultrastructure (Saraga‐Babić et al., 2012; Zimmerman, 1971). Until birth, renal tubular cells possess primary cilia in both nephrons and collecting ducts, with the exception of the intercalated cells that are interspersed along the collecting duct (Figure 2D) (Saraga‐Babić et al., 2012). While the observations compiled so far provide a substantial descriptive framework about the appearance of primary cilia, the presence of MCCs in the human kidney has only been reported in isolated case studies (Katz & Morgan, 1984) and therefore they remain an enigmatic feature of human renal development to explore in future studies.
4. Cilia: Antennas of kidney morphogenesis
Primary cilia function as sensory organelles essential for tissue homeostasis. To date, three putative roles have been proposed for the presence of primary cilia in the adult kidney nephrons: solute exchange, cellular signal transduction, and mechanical sensor of urine flow in the nephron (Rodat‐Despoix & Delmas, 2009). However, the presence of primary cilia in growing embryonic kidneys and the emerging evidence of renal developmental defects found in patients with ciliary disease argue that proper primary cilia function is indispensible for normal kidney organogenesis (Marshall & Nonaka, 2006; Poureetezadi & Wingert, 2016).
Conversely, the role of motile cilia in organogenesis, including kidney organ development, has been explored in lower vertebrates. Motile cilia of the zebrafish pronephros, spinal cord, and Kuppfer's vesicle (equivalent of the mammalian embryonic node) are required to regulate fluid flow in each of these organs (Kramer‐Zucker et al., 2005; Wang et al., 2013; Znosko et al., 2010). In the kidney and spinal cord, the absence of proper cilia formation causes back‐pressure resulting from a loss of fluid flow, where subsequent fluid accumulation could explain cyst formation and swelling (Kramer‐Zucker et al., 2005). Furthermore, MCCs of the zebrafish pronephros are required for proper kidney cell migration and morphogenesis as the kidney continues to grow with the developing animal (Wang et al., 2013).
4.1. Motile cilia regulated fluid flow and organogenesis
The mechanical forces generated from fluid flow are crucial for the development and morphogenesis of various tissues during organogenesis. For example, fluid shear stress produced by fluid flow in the tubular lumens has been indicated to have essential roles in regulating lumen diameter and morphology. In rat mesenteric blood vessels, flow‐induced vasculature remodeling has been documented (Tulis et al., 1998). Elevated blood flow under normal arterial pressure resulted in a time‐dependent increase in the lumen diameter of the artery. Meanwhile, arterial lumen expansion also involves active cell proliferation in the smooth muscles and the endothelial cells lining the artery, as they both show positive expression of cell proliferation markers (Tulis et al., 1998).
Moreover, the zebrafish embryonic pronephros and Kupffer's vesicle provide compelling evidence that cilia‐driven fluid flow can play crucial roles during normal tissue development (Kramer‐Zucker et al., 2005; Wang et al., 2013; Znosko et al., 2010). As previously stated, malfunctions of cilia structure or motility during organogenesis of these organs results in pronephric cyst formation, hydrocephalus and left‐right asymmetry defects in the embryo (Kramer‐Zucker et al., 2005). Examination of cilia anatomy revealed that cilia in all three of these locations are motile, suggesting that a crucial role of cilia function is to drive fluid flow (Kramer‐Zucker et al., 2005; Znosko et al., 2010). Particularly, obstruction of fluid flow in the zebrafish pronephros leads to fluid accumulation, epithelial distension, and cyst formation during kidney development (Kramer‐Zucker et al., 2005).
Contrary to nonmotile primary cilia found in mammalian kidneys, all cilia in zebrafish kidneys bear a “9 + 2” motile structure and undergo active rotational movements (Kramer‐Zucker et al., 2005). Beating cilia generate a corkscrew‐like wave in the lumen of the duct toward the cloaca (Kramer‐Zucker et al., 2005). It is believed that proper cilia structure and function are essential to initiate and maintain urinary flow in the pronephros, which starts at just 2 days post fertilization (dpf) (Kramer‐Zucker et al., 2005). Disruption of cilia function in IFT morphant embryos induced loss of fluid flow and subsequent development of kidney cysts (Kramer‐Zucker et al., 2005). Interestingly, during human fetal development, obstruction of fluid flow has also been identified to cause glomerular cyst formation, suggesting fluid flux as a driving force for both primitive and more advanced kidney development (Woolf et al., 2004).
MCC generated fluid flow in the pronephros of lower vertebrates is also required for morphogenic cell movement as the kidney structure becomes more complex (Wang et al., 2013). In zebrafish, the pronephric kidney is formed by 24 hours post‐fertilization (hpf), at which time morphogenesis events begin to remodel the two linear nephron tubules into more intricately arranged structures (Naylor & Davidson, 2016). Between 24 and 72 hpf, the characteristic linear tubules of the pronephros undergo movements such that their cranial aspects become convoluted, leading to a proximal tubule shape that is more similar to the mammalian nephron (Wingert et al., 2007). This morphogenesis process is driven by collective cell migration that is coincident with distal tubule proliferation as well (Vasilyev et al., 2009; Wang et al., 2013). Previous research has demonstrated that disrupted fluid flow through the kidney results in a loss of cell migration and proximal tubule convolution (Vasilyev et al., 2009; Wang et al., 2013) that is also associated with misexpression of various developmental factors in tubular epithelia (Gerlach & Wingert, 2014).
The genetic pathway directing MCC development has been studied in a variety of tissues found in vertebrate models, mainly the zebrafish pronephros, frog epidermis, and mammalian trachea. A discussion of the regulators of the MCC pathway demonstrated in the zebrafish pronephros, along with the well‐established, highly conserved MCC pathway, will succeed in the following subsections.
4.1.1. Using the zebrafish pronephros as a model to examine MCC development
Research in the zebrafish kidney has elucidated the interplay between the retinoic acid (RA) and Notch signaling pathways in MCC development (Li et al., 2014; Liu et al., 2007; Ma & Jiang, 2007; Marra & Wingert, 2016; Wang et al., 2013). In the zebrafish pronephros, there are at least two distinct types of polarized epithelial cells: (1) mono‐ciliated transporter cells and (2) MCCs (Figure 3) (Kramer‐Zucker et al., 2005; Li et al., 2014; Liu et al., 2007; Ma & Jiang, 2007; Marra & Wingert, 2016; Wang et al., 2013). The Notch signaling pathway mediates the fate choice between transporter cell and MCC. Cells expressing high levels of the Notch ligand Jagged2 interact with Notch3‐expressing neighbor cells, resulting in release of the Notch Intracellular Domain (NICD) into the nucleus and ultimate inhibition of ciliogenesis genes such as rfx2 and jagged2. Elegant studies have demonstrated that the inhibition of Notch by both genetic and chemically induced inactivation significantly increases the number and density of MCCs in the pronephros (Liu et al., 2007; Li et al., 2014; Ma & Jiang, 2007; Marra & Wingert, 2016). Thus, through lateral inhibition, the Notch signaling pathway promotes transporter cell identity and restricts MCC fate (Liu et al., 2007; Ma & Jiang, 2007).
Figure 3.
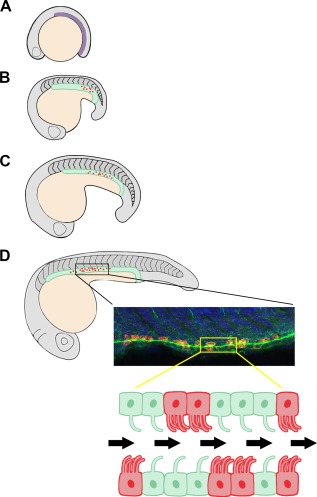
MCCs in zebrafish nephron development. (A) The renal progenitor field emerges at 10 hpf from the intermediate mesoderm, shown in purple. (B) By the 18–20 ss stage, a distinction between motile cilia (green) and mature MCCs (red) can be observed. (C) The MCC domain continues to expand as the embryo grows, but the “salt‐and‐pepper” pattern of MCCs remains. (D) At the 28 ss, the zebrafish pronephros is segmented. Mature MCCs are intercalated with mono‐ciliated transporter cells, with a dense expression pattern seen in the middle of the nephron. The confocal image is representative of wild‐type zebrafish pronephros where MCCs were labeled by florescent whole mount in situ hybridization to detect odf3b transcripts (red), cilia were labeled by α‐tubulin immunostaining (green), and nuclei were labeled with DAPI (blue). Below is a schematic of ciliated cells in the pronephros, where fluid flow is indicated by the black arrows. Abbreviations: hpf (hours post‐fertilization), ss (somite stage), MCC (multiciliated cell)
In contrast to the negative regulation provided by Notch signaling on the selection of MCC fate, the RA signaling pathway promotes MCC development from the renal progenitor pool (Figure 4) (Li et al., 2014; Marra & Wingert, 2016). Previous studies have demonstrated the importance of RA signaling in specifying proximal tubule identity in mesenchymal renal progenitors that give rise to the zebrafish pronephros (Cheng & Wingert, 2015; Wingert & Davidson, 2011; Wingert et al., 2007; Marra & Wingert, 2014), prior to the emergence of epithelial features of these cells indicating the onset of their maturation (McKee, Gerlach, Jou, Cheng, & Wingert, 2014; Gerlach & Wingert, 2014). Interestingly, RA inhibits the Notch signaling pathway through negative regulation of mecom (Figure 4), a transcription factor required for distal tubule fate and MCC inhibition (Li et al., 2014). After the addition of exogenous RA to the developing zebrafish embryo, the MCC number and domain are both greatly increased—changes that correlate with restricted expression of mecom (Li et al., 2014).
Figure 4.
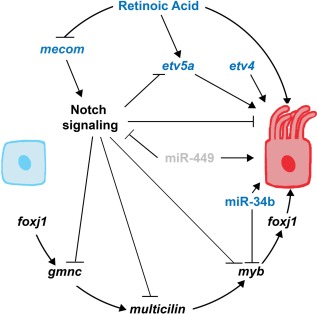
Integrated genetic pathway of MCC development. Genes that are presently known to be conserved across species are in black, those that have only been demonstrated in MCC development of the zebrafish pronephros are in blue, and grey indicates that the factor has not yet been shown in the zebrafish.
Furthermore, recent research has revealed that the closely related ETS transcription factors etv5a and etv4 are required for proper MCC development in the zebrafish pronephros (Figure 4) (Marra & Wingert, 2016). Knockdown of etv5a and etv4 alone resulted in a significant decrease in average MCC number, while combination knockdown of etv5a and etv4 together demonstrated a greater reduction of MCCs in the embryonic kidney at 24 hpf (Marra & Wingert, 2016). Interestingly, etv5a was found to be one factor that responds downstream of RA signaling to positively regulate MCC development (Figure 4), but the incomplete loss of MCCs in the absence of etv5a/etv4 suggests there are likely to be other RA targets in this pathway that need to be identified (Marra & Wingert, 2016). No detectable change was seen in the etv4 pronephros domain after alterations to the RA and Notch signaling pathways, suggesting that Etv4 may act non‐cell autonomously during MCC development (Marra & Wingert, 2016).
4.1.2. miRNA regulation of MCC development
The role for miRNA‐mediated molecular regulation of multiciliogenesis has only recently been appreciated (Marcet et al., 2011; Wang et al., 2013). In the zebrafish pronephros (and olfactory placode as well), miR‐34b is solely expressed in the MCCs, and miR‐34b knockdown resulted in the blockage of cilia bundle formation (Wang et al., 2013). Additionally, ciliary genes such as rfx2 were up‐regulated in miR‐34b morphants (Wang et al., 2013). Interestingly, the loss of miR‐34b did not affect mono‐ciliated cells (Wang et al., 2013). In the pronephros, miR‐34b functions to regulate and maintain myb expression (Figure 4). An optimal level of myb is required for normal multiciliogenesis, where both elevated expression of myb as well as depleted myb expression result in MCC defects and disorganization (Wang et al., 2013).
An additional group consisting of miR‐449a, miR‐449b, and miR‐449c, known collectively as miR‐449, has been demonstrated to regulate MCC development in both the frog epidermis and human airway mucociliary epithelial cells (HAECs) (Marcet et al., 2011). In both tissues, miR‐449 directly represses the Delta/Notch signaling pathway to promote multiciliogenesis (Figure 4) (Marcet et al., 2011).
4.1.3. Conserved developmental pathway between vertebrate species
In addition to the zebrafish pronephros, elegant studies in the frog epidermis and mammalian trachea have uncovered a core regulatory pathway of highly conserved genes that interact with the Notch signaling pathway to mediate multiciliogenesis (Stubbs et al., 2012; Tan et al., 2013; Zhou et al., 2015). Similar to the inhibitory role observed in MCCs of the zebrafish pronephros, the Notch signaling pathway has been implicated as a repressor of MCC fate across the aforementioned vertebrate species and tissues (Figure 4) (Li et al., 2014; Liu et al., 2007; Ma & Jiang, 2007; Marra & Wingert, 2016; Stubbs, Vladar, Axelrod, & Kitner, 2012; Tan et al., 2013; Zhou et al., 2015).
Positive regulators of multiciliogenesis include gmnc, multicilin, myb, and foxj1 (Figure 4) (Stubbs et al., 2012; Tan et al., 2013; Zhou et al., 2015). Recently, research in the zebrafish pronephros and frog epidermis has elucidated the role of gmnc as a master regulator of MCC development (Figure 4). As such, the Notch signaling pathway is antagonistic toward the Gmnc protein, and in the zebrafish kidney Gmnc responds to FoxJ1a activity to promote multiciliogenesis (Figure 4) (Zhou et al., 2015). Downstream of gmnc is highly conserved multicilin, whose protein (MCDIAS, MCI, IDAS, or MCIN depending on species) regulates myb (Figure 4) (Stubbs et al., 2012; Tan et al., 2013). Studies in the mouse lung, mouse tracheal epithelial cell (MTECs) culture, and frog epidermis have demonstrated that foxj1 is downstream of myb (Figure 4) (Tan et al., 2013). Although MCC development is initially repressed in the absence of myb, multiciliogenesis does recover in myb knockdowns, suggesting that another factor(s) downstream of multicilin is responsible (Tan et al., 2013).
Although demonstrated in MCC development of the zebrafish pronephros, the interaction between RA and Notch signaling has yet to be determined in other multiciliated tissues across vertebrate species. However, because the inhibitory role of Notch is strongly conserved between species, it will be enlightening to further investigate the role(s) of RA in multiciliogenesis in other cell type contexts. It is also of interest to discern the presence and role of RA targets that have been documented in the pronephros, such as etv5a (Marra & Wingert, 2016) and mecom (Li et al., 2014), to assess if they play fundamental roles in multiciliated tissues in other vertebrate species or represent unique aspects of the renal MCC lineage. While our recent work places etv5a downstream of RA, elucidating the relationship of etv4 with RA requires additional inquiry. The genetic tractability of the zebrafish model provides an especially valuable launching point for research about the core MCC pathway and these possibly distinguishing aspects of MCC genesis in different tissues. For example, in the zebrafish pronephros, MCC number was not completely lost in the absence of etv5a and etv4, while treatment with exogenous RA and inhibition of Notch in etv5a morphants increased MCC density (Marra & Wingert, 2016). These data suggest that another factor(s) works downstream of the RA and Notch pathways to promote MCC development in kidney cells. As stated above, it is also highly probable that other genes act downstream of multicilin in multiciliogenesis of various vertebrate tissues. Continued use of the zebrafish can identify such regulatory factors of multiciliogenesis.
4.2. Primary cilia as a mechanosensor of fluid flow
Whereas it is intuitive to imagine the antenna‐like cilia protruding out into the nephron lumen and bending as fluid contents flow through the nephron tubules, the molecular mechanisms that primary cilia apply to transduce flow information into an intracellular cascade of signaling pathways, which regulate cellular differentiation and patterning crucial for organogenesis and morphogenesis, are poorly understood. However, using both in vitro and in vivo systems, increasing evidence has indicated that primary cilia in the kidney serve as a mechanosensory organelle in response to urinary flux. Applying an in vitro culture system of renal collecting duct cells, a changing flow rate on the apical side of MDCK cells resulted in cilia deflection (Praetorius & Spring, 2001). Surprisingly, primary cilia responded to mechanical flow with a significant increase in intracellular calcium (Ca2+) levels through extracellular Ca2+ influx. This calcium signal was then amplified by the release of Ca2+ from intracellular sources, possibly through inositol triphosphate receptors (IP3P)‐dependent stores. Ca2+ signals stimulated by fluid flow spread to neighboring cells as a Ca2+ wave through gap junctions, probably mediated by IP3 or other intercellular messengers (Praetorius & Spring, 2001).
A breakthrough finding on how primary cilia dysfunction could lead to abnormal kidney development and cyst formation came from the documentation that polycystin‐1 (PC1) and polycystin‐2 (PC2) proteins, encoded respectively by the pkd1 and pkd2 genes that are mutated in human PKD disorders, co‐localize in the primary cilia of kidney epithelium (Nauli et al., 2003). PC1 is a G‐protein‐binding protein regulating voltage‐gated Ca2+ and potassium (K+) channels. Coupled with PC1, PC2 functions as a Ca2+‐permeable cation channel. In the primary cilia, PC1 may sense the bending of the cilia generated by fluid flow and function as a mechano‐fluid stress sensor, as primary cilia cells isolated from pkd1‐deficient mice failed to increase Ca2+ influx in response to fluid flow. PC1 and PC2 seem to act in concert with ryanodine receptors to transduce extracellular mechanical stimuli into an intracellular Ca2+ signaling response in kidney epithelial cells. Local increase in the cytosolic Ca2+ level initiated by primary cilia bending resulting from sensing of the fluid movement could then regulate numerous subcellular activities that contribute to various cellular behaviors accounting for kidney development (Nauli et al., 2003).
In contrast, PC1‐dependent intracellular Ca2+ increase may be accomplished by intramembrane proteolysis of a C‐terminal tail (CTT) domain of the PC1 protein located at its last 200 amino acids, and translocation of this CTT domain into the nuclei (Chauvet et al., 2004). Nuclear translocation of the PC1 CTT domain has been observed in renal epithelial cells of wild‐type mice after kidney tubular injury and those of mice with ciliary dysfunction (Chauvet et al., 2004). With respect to the potential relationship between Polycystin function and flow sensing, alterations in flow stimuli or cilia function correlate with the release of the CTT of PC1 in vivo. Under physical conditions, primary cilia responding to urinary flow may cause a chain of cellular events and lead to nuclear translocation of CTT, which will in turn initiate a series of cellular responses to the mechanical stimuli by interacting with proteins and transcription factors that regulate proliferation and apoptosis (Chauvet et al., 2004; Merrick et al., 2014).
4.3. Connection between cilia and PKD
An increasing body of evidence suggests that mutations in protein components localized to the primary cilia are a major cause of PKD in human infants. The hallmark of PKD conditions is the formation of abnormal accumulation of fluids, or cysts, within the kidney (Figure 5). These cysts can form along the nephron proper and/or collecting tubules. Discoveries about PKD pathogenesis have provided knowledgeable insights about how primary cilia contribute to early stages of nephron formation and renal morphogenesis, which facilitate a better understanding of the complexity and importance of primary cilium as an essential structure regulating proper kidney development and function. Mutations in the pkd1 loci encoding PC1 are the most common cause of ADPKD in humans (Lu et al., 1997). The onset of ADPKD induces progressive formation of epithelial cysts invading healthy kidney tissues, eventually leading to renal failure. The renal cysts formed in utero increase in size and number throughout the affected person's lifetime, and this process is accompanied by increased rates of both proliferation and apoptosis of the renal epithelial cells (Boletta et al., 2000).
Figure 5.
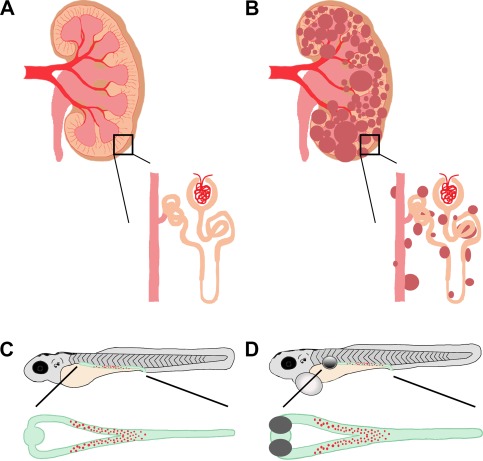
PKD in mammals and zebrafish. (A, B) Schematic of a healthy, adult mammalian kidney with enlarged nephron (A) compared to a diseased, polycystic adult mammalian kidney and enlarged nephron (B). The collecting duct system is in pink, nephron tubule is in orange, and cysts are in dark red. (C, D) Drawing of zebrafish larvae where the kidney is highlighted in green with red MCCs. (C) A healthy zebrafish larva with an enlarged, dorsal view of a healthy embryonic kidney. (D) Schematic of an unhealthy zebrafish larva, with bilateral kidney cysts (grey circle) and pericardial edema (white circle). Enlarged is a dorsal view of the pronephros, showing the bilateral kidney cysts (grey) as well as distended tubular lumen (green) compared to the wild‐type zebrafish embryonic kidney.
Supporting a major role of primary cilia in PKD, genetic screens in zebrafish have also linked defects in genes encoding components of IFT particles involved in cilia formation to PKD conditions in zebrafish larvae, which are manifested by cyst formation in the pronephros (Figure 5) (Drummond et al., 1998; Sun et al., 2004). In a murine model of pkd1 inactivation, it has been shown that proper PC1 function is crucial for tubular elongation and maturation during final stages of renal morphogenesis (Lu et al., 1997). In this study, animals with homozygous inactivated pkd1 exhibited normal phases of renal development though day E14, including unaltered UB branching, mesenchymal condensation, and comma‐ and S‐shaped body formations (Lu et al., 1997). Abnormal cyst formation was first observed around day E15, when nephron tubules normally undergo progressive lengthening and maturation, and nephron segmentation is finalized. Homozygous pkd1−/− mice died in utero with severe cystic diseases (Lu et al., 1997).
As renal cyst formation is accompanied by both accelerated epithelial proliferation and apoptosis, it is suggested that PC1 may function to regulate both pathways during kidney development. Indeed, expression of full‐length human pkd1 in MDCK cells prohibited their growth and induced resistance to cell death (Boletta et al., 2000). Overexpression of human pkd1 also resulted in spontaneous tubulogenesis in MDCK cells, whereas control cells showed cyst formation in this in vitro cell culture system (Boletta et al., 2000). As such, it was hypothesized that PC1 functions to inhibit proliferation of epithelial cells in developing kidneys, allowing them to proceed toward terminal differentiation without inducing apoptosis (Boletta et al., 2000).
Inversin, which also exhibits a unique subcellular localization to primary cilium of renal tubular epithelial cells, acts as a molecular switch between canonical and noncanonical Wnt signaling pathways (Simons et al., 2005). During early kidney development, induction of MM and cell proliferation in branching morphogenesis depends on canonical Wnt signaling (Perantoni, 2003). However, at later developmental stages, persistent activation of Wnt/β‐catenin results in cyst formation. It was shown that Inversin functions to inhibit the canonical Wnt pathway from continuous activity by targeting cytoplasmic Dishevelled for degradation (Simons et al., 2005). Importantly, fluid flow induced upregulation of Inversin expression was seen in ciliated tubular epithelial cells, which seemed to be essential for regulating this molecular switch between Wnt signaling pathways during kidney development (Simons et al., 2005). Therefore, early urine production during embryogenesis could be an upstream mechanical signal to turn down canonical Wnt signaling. As such, this signal transduced by localizing Inversin to the primary cilia enabled spatial recognition of the developing kidney to maintain a proper tubular geometry as nephron branching and morphogenesis take place (Simons et al., 2005).
Another example involves mutations in the gene encoding Fibrocystin/Polyductin (FPC) resulting in ARPKD, a monogenic disorder found in 1/20,000 newborns with a mortality of 30% (Wang et al., 2007). In normal human kidneys, FPC was found localized to the basal body of primary cilia and at the plasma membrane of the distal tubule epithelial cells (Wang et al., 2007). Basolateral membrane localization of FPC in kidney tubules indicates it may function as an adhesive molecule and may have a role in cell‐matrix interactions (Wang et al., 2007). As defects in extracellular protein could also lead to cystic kidney disease (Shannon, Patton, Harvey, & Miner, 2006), dynamic subcellular localization of FPC in the primary cilia and plasma membrane suggests potential roles of FPC in tubulogenesis and cystogenesis. More importantly, FPC colocalized with PC2 at the plasma membrane and on the primary cilium (Nauli et al., 2003; Yoder et al., 2002). Disruption of the FPC‐PC2 complex at the primary cilium blocked flow‐induced Ca2+ response with an impaired mechanosensation of fluid flow, indicating interactions between FPC and the polycystin complex are essential for proper mechanotransduction function of renal primary cilia (Wang et al., 2007). Future studies aiming at discoveries of new primary cilia components could lead to better understandings on the mechanisms of cilium‐mediated mechanosensory signaling, which play crucial roles in kidney organogenesis and PKD conditions.
Interestingly, mutations in the genes described above cause kidney cysts and other defects in ciliated tissues of the zebrafish (Figure 5C and D). Traditionally associated with primary cilia, pkd2 is expressed in tissues associated with MCCs in the zebrafish, and knockdown of pkd2 produced kidney cysts and other phenotypes associated with Pkd2−/− mice (Bisgrove, Snarr, Emrazian, & Yost, 2005; Pennekamp et al., 2002; Sun et al., 2004; Wu et al., 1998, 2000). In addition to forward genetic approaches to study PKD, a number of zebrafish mutants with kidney cyst formation have been pulled out of reverse genetic screens. In these mutants, genes ift57, ift172, pk1, and swt, whose mammalian homologs have been associated with PKD, are responsible for cilia defects in the zebrafish pronephros (Drummond et al., 1998; Cao, Park, & Sun, 2010; Sun et al., 2004; Sullivan‐Brown et al., 2008; van Rooijen et al., 2008). Importantly, the heterogeneity of cilia defects seen in the aforementioned zebrafish mutants and the varied location and presence of pronephric cysts nicely mirrors that which is seen in mammalian PKD (Figure 5), suggesting that a core set of cilia‐associated genes is conserved across species, and shared between primary and motile cilia. The simplicity and accessibility of the zebrafish pronephros thus presents a useful tool to study ciliogenesis and its association with kidney development.
5. Conclusion
Anchored on the apical surface of the nephron tubular epithelium, primary cilia serve as a sensory organelle in the kidney, transferring information from the tubular lumen to the epithelial interior. These microscopic cellular antennae convert luminal environmental information into intracellular signaling cascades to control cellular responses by sensing urine flow. In the kidney, cellular signals transduced by primary cilia are involved in multiple cellular processes, including epithelial proliferation, differentiation, and cytoskeletal organization, while defects in this organelle cause severe cystic kidney disease characterized by epithelial abnormalities affecting proper kidney development. Primary cilia bear a complex structure composed of complicated protein complexes, such as cation channels sensing mechanical stress and ligand receptors binding to extracellular signaling molecules at the ciliary membrane and basal body. The essential roles of primary cilia in kidney organogenesis are emphasized by the fact that crucial signaling pathways for tubulogenesis and nephron induction are highly regulated by protein complexes localized to the primary cilia. Meanwhile, increasing evidence from studies of PKD has linked proper cilia function to normal kidney organogenesis. Comprehensive understanding of how primary cilia contribute to direct nephrogenesis and affect kidney function will provide useful insights on kidney organogenesis, and may facilitate better knowledge about the pathogenesis of PKD and other kidney diseases.
Similarly, building on our understanding of the development and integration of MCCs and motile cilia during kidney organogenesis in lower vertebrates can provide insight to their function in the onset of developmental disorders and disease in the kidney. Specifically, the abnormal presence of MCCs has been documented in cases of mammalian kidney disease. With our current understanding, it is difficult to explain this phenomenon. However, by using advantageous vertebrate model organisms such as the zebrafish, we can continue to identify and explore possible reasons as to why kidney cells of higher vertebrates can revert, as it would seem, to a more primitive state.
Acknowledgments
We thank the staffs of the Department of Biological Sciences and the Center for Zebrafish Research at the University of Notre Dame for their dedication and care of our zebrafish aquarium. Finally, we thank the members of our lab for their support, discussions, and insights about this work. This work was supported in part by the following grant to R. A. W. (R01DK100237). The work of A. N. M. was supported in part by the National Science Foundation Graduate Research Fellowship (No. DGE‐1313583).
A. N. Marra and Y. Li contributed equally to this work.
References
- Berbari, N. F. , O'Connor, A. K. , Haycraft, C. J. , & Yoder, B. K. (2009). The primary cilium as a complex signaling center. Current Biology, 19, R526–R535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgrove, B. W. , Snarr, B. S. , Emrazian, A. , & Yost, H. J. (2005). Polaris and Polycystin‐2 in dorsal forerunner cells and Kupffer's vesicle are required for specification of the zebrafish left‐right axis. Developmental Biology, 287, 274–288. [DOI] [PubMed] [Google Scholar]
- Bisgrove, B. W. , & Yost, H. J. (2006). The roles of cilia in developmental disorders and disease. Development, 133, 4131–4143. [DOI] [PubMed] [Google Scholar]
- Boletta, A. , Qian, F. , Onuchic, L. F. , Bhunia, A. K. , Phakdeekitcharoen, B. , Hanaoka, … Germino, G. G. (2000). Polycystin‐1, the gene product of PKD1, induces resistance to apoptosis and spontaneous tubulogenesis in MDCK cells. Molecular Cell, 6, 1267–1273. [DOI] [PubMed] [Google Scholar]
- Bouchard, M. , Souabni, A. , Mandler, M. , Neubuser, A. , & Busslinger, M. (2002). Nephric lineage specification by Pax2 and Pax8. Genes & Development, 16, 2958–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, E. R. , & Wallingford, J. B. (2014). Multiciliated cells. Current Biology, 24, R973–R982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvet, J. P. (2002). Cilia in PKD—Letting it all hang out. Journal of the American Society of Nephrology, 13, 2614–2616. [DOI] [PubMed] [Google Scholar]
- Cao, Y. , Park, A. , & Sun, Z. (2010). Intraflagellar transport proteins are essential for cilia formation and for planar cell polarity. Journal of the American Society of Nephrology, 21, 1326–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho‐Santos, Z. , Azimzadeh, J. , Pereira‐Leal, J. B. , & Bettencourt‐Dias, M. (2011). Evolution: Tracing the origins of centrioles, cilia, and flagella. Journal of Cell Biology, 194, 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapin, H. C. , & Caplan, M. J. (2010). The cell biology of polycystic kidney disease. Journal of Cell Biology, 191, 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvet, V. , Tian, X. , Husson, H. , Grimm, D. H. , Wang, T. , Hiesberger, T. , … Caplan M. J. (2004). Mechanical stimuli induce cleavage and nuclear translocation of the polycystin‐1 C terminus. Journal of Clinical Investigation, 114, 1433–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, C. N. , & Wingert, R. A. (2015). Nephron proximal tubule patterning and corpuscles of Stannius formation are regulated by the sim1a transcription factor and retinoic acid in zebrafish. Developmental Biology, 399, 100–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choksi, S. P. , Lauter, G. , Swoboda, P. , & Roy, S. (2014). Switching on cilia: Transcriptional networks regulating ciliogenesis. Development, 141, 1427–1441. [DOI] [PubMed] [Google Scholar]
- Costantini, F. , & Kopan, R. (2010). Patterning a complex organ: Branching morphogenesis and nephron segmentation in kidney development. Developmental Cell, 18, 698–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan, M. J. , Gladwin, M. T. , & Shelhamer, J. H. (2001). Disorders of ciliary motility. American Journal of the Medical Sciences, 321, 3–10. [DOI] [PubMed] [Google Scholar]
- Cukuranovic, R. , & Vlajkovic, S. (2005). Age related anatomical and functional characteristics of human kidney. Facta Universitatis: Medicine and Biology, 12, 61–69. [Google Scholar]
- Davis, E. E. , Brueckner, M. , & Katsanis, N. (2006). The emerging complexity of the vertebrate cilium: New functional roles for an ancient organelle. Developmental Cell, 11, 9–19. [DOI] [PubMed] [Google Scholar]
- Deane, J. A. , & Ricardo, S. D. (2007). Polycystic kidney disease and the renal cilium. Nephrology, 12, 559–564. [DOI] [PubMed] [Google Scholar]
- Deane, J. A. , & Ricardo, S. D. (2012). Emerging roles for renal primary cilia in epithelial repair. International Review of Cell and Molecular Biology, 293, 169–193. [DOI] [PubMed] [Google Scholar]
- DeMartino, S. , & Zamboni, L. (1966). A morphological study of mesonephros of the human embryo. Journal of Ultrastructure Research, 16, 399–427. [DOI] [PubMed] [Google Scholar]
- Desgrange, A. , & Cereghini, S. (2015). Nephron patterning: Lessons from Xenopus, zebrafish and mouse studies. Cells, 4, 483–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressler, G. R. (2006). The cellular basis of kidney development. Annual Review of Cell and Developmental Biology, 22, 509–529. [DOI] [PubMed] [Google Scholar]
- Dressler, G. R. (2009). Advances in early kidney specification, development and patterning. Development, 136, 3863–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond, I. A. , Majumdar, A. , Hentschel, H. , Elger, M. , Solnica‐Krezel, L. , Schier, A. F. , … Fishman, M. C. (1998). Early development of the zebrafish pronephros and analysis of mutations affecting pronephric function. Development, 125, 4655–4667. [DOI] [PubMed] [Google Scholar]
- Duffy, J. L. , & Suzuki, Y. (1968). Ciliated human renal proximal tubular cells. Observations in three cases of hypercalcemia. American Journal of Pathology, 53, 609–616. [PMC free article] [PubMed] [Google Scholar]
- Gerlach, G. F. , & Wingert, R. A. (2013). Kidney organogenesis in the zebrafish: Insights into vertebrate nephrogenesis and regeneration. Wiley Interdisciplinary Reviews: Developmental Biology, 2, 559–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach, G. F. , & Wingert, R. A. (2014). Zebrafish pronephros tubulogenesis and epithelial identity maintenance are reliant on the polarity proteins Prkc iota and zeta. Developmental Biology, 396, 183–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan, M. O. , & Subramanyan, S. (1995). Ciliated renal tubular cells in crescentic glomerulonephritis. Ultrastructural Pathology, 19, 201–203. [DOI] [PubMed] [Google Scholar]
- Hinchcliffe, S. A. , Sargent, P. H. , Howard, C. V. , Chan, Y. F. , & van Velzen, D. (1991). Human intrauterine renal growth expressed in absolute number of glomeruli assessed by the disector method and Cavalieri principle. Laboratory Investigation, 64, 777–784. [PubMed] [Google Scholar]
- Jain, R. , Pan, J. , Driscoll, J. A. , Wisner, J. W. , Huang, T. , Gunsten, S. P. , … Brody, S. L. (2010). Temporal relationship between primary and motile ciliogenesis in airway epithelial cells. American Journal of Respiratory Cell and Molecular Biology, 43, 731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James, R. G. , & Schultheiss, T. M. (2003). Patterning of the avian intermediate mesoderm by lateral plate and axial tissues. Developmental Biology, 253, 109–124. [DOI] [PubMed] [Google Scholar]
- Jang, D. G. , Chae, H. , Shin, J. C. , Park, I. Y. , Kim, M. , & Kim, Y. (2011). Prenatal diagnosis of autosomal recessive polycystic kidney disease by molecular genetic analysis. Journal of Obstetrics and Gynaecology Research, 37, 1744–1747. [DOI] [PubMed] [Google Scholar]
- Jin, H. , & Nachury, M. V. (2009). The BBSome. Current Biology, 19, R472–R473. [DOI] [PubMed] [Google Scholar]
- Katz, S. M. , & Morgan, J. J. (1984). Cilia in the human kidney. Ultrastructural Pathology, 6, 285–294. [DOI] [PubMed] [Google Scholar]
- Kolb, R. J. , Woost, P. G. , & Hopfer, U. (2004). Membrane trafficking of angiotensin receptor type‐1 and mechanochemical signal transduction in proximal tubule cells. Hypertension, 44, 352–359. [DOI] [PubMed] [Google Scholar]
- Kozminski, K. G. , Johnson, K. A. , Forscher, P. , & Rosenbaum, J. L. (1993). A motility in the eukaryotic flagellum unrelated to flagellar beating. Proceedings of the National Academy of Sciences United States of America, 90, 5519–5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozminski, K. G. , Beech, P. L. , & Rosenbaum, J. L. (1995). The Chlamydomonas kinesin‐like protein FLA10 is involved in motility associated with the flagellar membrane. Journal of Cell Biology, 131, 1517–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer‐Zucker, A. G. , Olale, F. , Haycraft, C. J. , Yoder, B. K. , Schier, A. F. , & Drummond, I. A. (2005). Cilia‐driven fluid flow in the zebrafish pronephros, brain and Kupffer's vesicle is required for normal organogenesis. Development, 132, 1907–1921. [DOI] [PubMed] [Google Scholar]
- Li, Y. , & Wingert, R. A. (2013). Regenerative medicine for the kidney: Stem cell prospects & challenges. Clinical and Translational Medicine, 2, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Cheng, C. N. , Verdun, V. A. , & Wingert, R. A. (2014). Zebrafish nephrogenesis is regulated by interactions between retinoic acid, mecom, and Notch signaling. Developmental Biology, 386, 111–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little, M. H. , & McMahon, A. P. (2012). Mammalian kidney development: Principles, progress, and projections. Cold Spring Harbor Perspectives in Biology, 4, a008300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Narendra, P. , Kramer‐Zucker, A. , & Drummond, I. A. (2007). Notch signaling controls the differentiation of transporting epithelia and multiciliated cells in the zebrafish pronephros. Development, 134, 1111–1122. [DOI] [PubMed] [Google Scholar]
- Lu, W. , Peissel, B. , Babakhanlou, H. , Pavlova, A. , Geng, L. , Fan, X. , … Zhou, J. (1997). Perinatal lethality with kidney and pancreas defects in mice with a targeted Pkd1 mutation. Nature Genetics, 17, 179–181. [DOI] [PubMed] [Google Scholar]
- Ma, M. , & Jiang, Y. J. (2007). Jagged2a‐Notch signaling mediates cell fate choice in the zebrafish pronephric duct. PLoS Genetics, 3, e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcet, B. , Chevalier, B. , Luxardi, G. , Corauz, C. , Zaragosi, L.‐E. , Cibois, … Barbry, P. (2011). Control of vertebrate multiciliogenesis by miR‐449 through direct repression of the Delta/Notch pathway. Nature Cell Biology, 13, 693–699. [DOI] [PubMed] [Google Scholar]
- Marra, A. N. , & Wingert, R. A. (2014). Roles of Iroquois transcription factors in kidney development. Seminars in Cell and Developmental Biology, 3, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra, A. N. , & Wingert, R. A. (2016). Epithelial cell fate in the nephron tubule is mediated by the ETS transcription factors etv5a and etv4 during zebrafish kidney development. Developmental Biology, 411, 231–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall, W. F. , & Rosenbaum, J. L. (2001). Intraflagellar transport balances continuous turnover of outer doublet microtubules: Implications for flagellar length control. Journal of Cell Biology, 155, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall, W. F. , & Nonaka, S. (2006). Cilia: Tuning in to the cell's antenna. Current Biology, 16, R604–R614. [DOI] [PubMed] [Google Scholar]
- Mauch, T. J. , Yang, G. , Wright, M. , Smith, D. , & Schoenwolf, G. C. (2000). Signals from trunk paraxial mesoderm induce pronephros formation in chick intermediate mesoderm. Developmental Biology, 220, 62–75. [DOI] [PubMed] [Google Scholar]
- McCampbell, K. K. , & Wingert, R. A. (2012). Renal stem cells: fact or science fiction? Biochemical Journal, 444, 153–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee, R. , Gerlach, G. F. , Jou, J. , Cheng, C. N. , & Wingert, R. A. (2014). Temporal and spatial expression of tight junction genes during zebrafish pronephros development. Gene Expression Patterns, 16, 104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick, D. , Bertuccio, C. A. , Chapin, H. C. , Lal, M. , Chauvet, V. , & Caplan, M. J. (2014). Polycystin‐1 cleavage and the regulation of transcriptional pathways. Pediatric Nephrology, 29, 505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauli, S. M. , Alenghat, F. J. , Luo, Y. , Williams, E. , Vassilev, P. , Li, X. , … Zhou, J. (2003). Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nature Genetics, 33, 129–137. [DOI] [PubMed] [Google Scholar]
- Naylor, R. W. , & Davidson, A. J. (2016). Pronephric tubule formation in zebrafish: morphogenesis and migration. Pediatric Nephrology, [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Ong, A. C. M. , & Wagner, B. (2005). Detection of proximal tubular motile cilia in a patient with renal sarcoidosis associated with hypercalcemia. American Journal of Kidney Diseases, 45, 1096–1099. [DOI] [PubMed] [Google Scholar]
- Pennekamp, P. , Karcher, C. , Fischer, A. , Schweickert, A. , Skryabin, B. , Horst, J. , … Dworniczak, B. (2002). The ion channel plycystin‐2 is required for left‐right axis determination in mice. Current Biology, 12, 938–943. [DOI] [PubMed] [Google Scholar]
- Perantoni, A. O. (2003). Renal development: Perspectives on a Wnt‐dependent process. Seminars in Cell & Developmental Biology, 14, 201–208. [DOI] [PubMed] [Google Scholar]
- Poureetezadi, S. J. , & Wingert, R. A. (2016). Little fish, big catch: Zebrafish as a model for kidney disease. Kidney International, 89, 1204–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praetorius, H. A. , & Spring, K. R. (2001). Bending the MDCK cell primary cilium increases intracellular calcium. Journal of Membrane Biology, 184, 71–79. [DOI] [PubMed] [Google Scholar]
- Reimschuessel, R. (2001). A fish model of renal regeneration and development. ILAR Journal, 42, 285–291. [DOI] [PubMed] [Google Scholar]
- Rodat‐Despoix, L. , & Delmas, P. (2009). Ciliar functions in the nephron. Pflugers Arch, 458, 179–187. [DOI] [PubMed] [Google Scholar]
- Romagnani, P. , Lasagni, L. , & Remuzzi, G. (2013). Renal progenitors: An evolutionary conserved strategy for kidney regeneration. Nature Reviews Nephrology, 9, 137–146. [DOI] [PubMed] [Google Scholar]
- Sadler, T. W. ( 2012). Langman's medical embryology (12th ed.). Philadelphia, PA: Lippincott Williams & Wilkins. [Google Scholar]
- Saraga‐Babić, M. , Vukojević, K. , Bočina, I. , Drnašin, K. , & Saraga, M. (2012). Ciliogenesis in normal human kidney development and post‐natal life. Pediatric Nephrology, 27, 55–63. [DOI] [PubMed] [Google Scholar]
- Satlin, L. M. , Sheng, S. , Woda, C. B. , & Kleyman, T. R. (2001). Epithelial Na+ channels are regulated by flow. American Journal of Physiology ‐ Renal Physiology, 280, F1010–F1018. [DOI] [PubMed] [Google Scholar]
- Saxen, L. ( 1987). Organogenesis of the kidney. Cambridge, UK: Cambridge University Press. [Google Scholar]
- Shannon, M. B. , Patton, B. L. , Harvey, S. J. , & Miner, J. H. (2006). A hypomorphic mutation in the mouse laminin alpha5 gene causes polycystic kidney disease. Journal of the American Society of Nephrology, 17, 1913–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shawlot, W. , & Behringer, R. R. (1995). Requirement for Lim1 in head‐organizer function. Nature, 374, 425–430. [DOI] [PubMed] [Google Scholar]
- Simons, M. , Gloy, J. , Ganner, A. , Bullerkotte, A. , Bashkurov, M. , Krönig, C. , … Walz, G. (2005). Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nature Genetics, 37, 537–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla, V. , & Reiter, J. F. (2006). The primary cilium as the cell's antenna: Signaling at a sensory organelle. Science, 313, 629–633. [DOI] [PubMed] [Google Scholar]
- Stubbs, J. L. , Vladar, E. K. , Axelrod, J. D. , & Kitner, C. (2012). Multicilin promotes centriole assembly and ciliogenesis during multiciliated cell differentiation. Nature Cell Biology, 14, 140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan‐Brown, J. , Schottenfeld, J. , Okabe, N. , Hostetter, C. L. , Serluca, F. C. , Thiberge, S. Y. , & Burdine, R. D. (2008). Zebrafish mutations affecting cilia motility share similar cystic phenotypes and suggest a mechanism of cyst formation that differs from pkd2 morphants. Developmental Biology, 314, 261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Z. , Amsterdam, A. , Pazour, G. J. , Cole, D. G. , Miller, M. S. , & Hopkins, N. (2004). A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development, 131, 4085–4093. [DOI] [PubMed] [Google Scholar]
- Tan, F. E. , Vladar, E. K. , Ma, L. , Fuentealba, L. C. , Hoh, R. , Espinoza, F. H. , … Krasnow, M. A. (2013). Myb promotes centriole amplification and later steps of the multiciliogenesis program. Development, 140, 4277–4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulis, D. A. , Unthank, J. L. , & Prewitt, R. L. (1998). Flow‐induced arterial remodeling in rat mesenteric vasculature. American Journal of Physiology, 274, H874–H882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooijen, E. , Giles, R. H. , Voest, E. E. , van Rooijen, C. , Schulte‐Merker, S. , & van Eeden, F. J. (2008). LRRC50, a conserved ciliary protein implicated in plycystic kidney disease. Journal of the American Society of Nephrology, 19, 1128–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilyev, A. , Liu, Y. , Mudumana, S. , Mangos, S. , Lam, P. Y. , Majumdar, A. , … Drummond, I.A. (2009). Collective cell migration drives morphogenesis of the kidney nephron. PLoS Biology, 7, e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincensini, L. , Blisnick, T. , & Bastin, P. (2011). 1001 model organisms to study cilia and flagella. Biology of the Cell, 103, 109–130. [DOI] [PubMed] [Google Scholar]
- Vize, P. D. , Woolf, A. S. , & Bard, J. B. L. (2003). The kidney from normal development to congenital diseases (pp. 1–519). London: Academic Press. [Google Scholar]
- Wang, L. , Fu, C. , Fan, H. , Du, T. , Dong, M. , Chen, Y. , … Zhou, Y. (2013). miR‐34b regulates multiciliogenesis during organ formation in zebrafish. Development, 140, 2755–2764. [DOI] [PubMed] [Google Scholar]
- Wang, S. , Zhang, J. , Nauli, S. M. , Li, X. , Starremans, P. G. , Luo, Y. , Roberts, K. A. , & Zhou, J. (2007). Fibrocystin/polyductin, found in the same protein complex with polycystin‐2, regulates calcium responses in kidney epithelia. Molecular and Cellular Biology, 27, 3241–3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber, W. A. , & Lee, J. (1975). Fine structure of mammalian renal cilia. Anatomical Record, 182, 339–343. [DOI] [PubMed] [Google Scholar]
- Wingert, R. A. , Selleck, R. , Yu, J. , Song, H. , Chen, Z. , Song, A. , … Davidson, A. J. (2007). The cdx genes and retinoic acid control the positioning and segmentation of the zebrafish pronephros. PLoS Genetics, 3, e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingert, R. A. , & Davidson, A. J. (2008). The zebrafish pronephros: A model to study nephron segmentation. Kidney International, 73, 1120–1127. [DOI] [PubMed] [Google Scholar]
- Wingert, R. A. , & Davidson, A. J. (2011). Zebrafish nephrogenesis involves dynamic spatiotemporal expression changes in renal progenitor and essential signals from retinoic acid and irx3b. Developmental Dynamics, 240, 2011–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf, A. S. , Price, K. L. , Scambler, P. J. , & Winyard, P. J. (2004). Evolving concepts in human renal dysplasia. Journal of the American Society of Nephrology, 15, 998–1007. [DOI] [PubMed] [Google Scholar]
- Worthington, W. C., Jr. , & Cathcart, R. S., III. (1963). Ependymal cilia: Distribution and activity in the adult human brain. Science, 139, 221–222. [DOI] [PubMed] [Google Scholar]
- Wu, G. D. , Agati, V. , Cai, Y. , Markowitz, G. , Park, J. H. , Reynolds, D. M. , … Somlo, S. (1998). Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell, 93, 177–188. [DOI] [PubMed] [Google Scholar]
- Wu, G. , Markowitz, G. S. , Li, L. D. , Agati, V. D. , Factor, S. M. , Geng, L. , … Somolo, S. (2000). Cardiac defects and renal failure in mice with targeted mutations in Pkd2. Nature Genetics, 24, 75–78. [DOI] [PubMed] [Google Scholar]
- Yoder, B. K. , Hou, X. , & Guay‐Woodford, L. M. (2002). The polycystic kidney disease proteins, polycystin‐1, polycystin‐2, polaris, and cystin, are co‐localized in renal cilia. Journal of the American Society of Nephrology, 13, 2508–2516. [DOI] [PubMed] [Google Scholar]
- Yoder, B. K. (2007). Role of primary cilia in the pathogenesis of polycystic kidney disease. Journal of the American Society of Nephrology, 18, 1381–1388. [DOI] [PubMed] [Google Scholar]
- Zhou, F. , Narasimhan, V. , Shboul, M. , Chong, Y. L. , Reversade, B. , & Roy, S. (2015). Gmnc is a master regulator of the multiciliated cell differentiation program. Current Biology, 25, 3267–3273. [DOI] [PubMed] [Google Scholar]
- Zimmerman, H. D. (1971). Cilia in the fetal kidney of man. Beiträge zur Pathologie, 143, 227–240. [PubMed] [Google Scholar]
- Znosko, W. A. , Yu, S. , Thomas, K. , Molina, G. A. , Li, C. , Tsang, W. , … Tsang, M. (2010). Overlapping functions of Pea3 ETS transcription factors in FGF signaling during zebrafish development. Developmental Biology, 342, 11–25. [DOI] [PMC free article] [PubMed] [Google Scholar]