

Abstract
Here, we review genomic target site selection during retroviral integration as a multistep process in which specific biases are introduced at each level. The first asymmetries are introduced when the virus takes a specific route into the nucleus. Next, by co‐opting distinct host cofactors, the integration machinery is guided to particular chromatin contexts. As the viral integrase captures a local target nucleosome, specific contacts introduce fine‐grained biases in the integration site distribution. In vivo, the established population of proviruses is subject to both positive and negative selection, thereby continuously reshaping the integration site distribution. By affecting stochastic proviral expression as well as the mutagenic potential of the virus, integration site choice may be an inherent part of the evolutionary strategies used by different retroviruses to maximise reproductive success.
Keywords: cofactor, HIV‐1, integration, latency, MLV, retrovirus, viral vector
Abbreviations
- ET
extraterminal
- HIV‐1
human immunodeficiency virus type‐1
- HTLV‐1
human T lymphotropic virus type 1
- IBD
integrase binding domain
- IN
integrase
- INSTI
integrase strand transfer inhibitor
- LTR
long terminal repeat
- MLV
murine leukaemia virus
- NPC
nuclear pore complex
- PFV
prototype foamy virus
- PIC
preintegration complex
- SIR
superinfection resistance
- tDNA
target DNA
Introduction
One of the defining events in the retroviral lifecycle is insertion of the reverse transcribed viral RNA genome into a host‐cell chromosome. This step, catalysed by the viral integrase protein (IN) establishes a stably integrated version of the virus, referred to as the provirus. The provirus provides a lasting template for viral gene expression, and its activity drives production of progeny virions by the infected cell. As the provirus constitutes an integral part of the genome, its fate is intimately linked to that of the infected cell: retroviruses persist in a host for the lifetime of the infected cells or their progeny.
Retroviral integration is not a random process: different retroviral genera (alpha‐ through epsilon‐, lenti‐ and spumaretroviruses) favour distinct chromatin environments for integration. Lentiviruses, such as the Human Immunodeficiency Virus Type‐1 (HIV‐1) for instance show a preference to integrate into actively transcribed regions 1. Conversely, gammaretroviruses such as the Murine Leukaemia Virus (MLV) target active promoter‐proximal as well as distal enhancer elements 2, 3, 4, 5. The deltaretrovirus Human T Lymphotropic Virus Type 1 (HTLV‐1) integrates more frequently in or near transcriptionally active areas 6, 7. Tree‐based clustering results of retroviruses based on their integration preferences generally map well onto the retroviral phylogenetic tree 6. This deeply rooted evolutionary link suggests that integration site selection is part of the strategy employed by retroviruses to maximise their replicative fitness.
The advent of next‐generation sequencing methods to identify integration sites 8, the resolution of crystal structures of the integration machinery (the intasome complex) 9, 10 as well as the identification of various host‐encoded integration cofactors 11, 12, 13, 14 allow us to paint a detailed picture of the retroviral integration site selection process. In this review essay, we attempt to put together the pieces required to do just that. We provide a detailed view of the known determinants of the integration site distribution and argue that target site selection can indeed be subject to evolutionary optimisation through natural selection. Finally, we discuss how the integration site distribution is further shaped by selection forces in the host. Most of the work in the field has focused on HIV and MLV owing to their relevance as a human pathogen and gene therapy vector, respectively. In the latter case, some of the implications of retroviral integration site selection have already become painfully clear. Treatment with gammaretroviral vectors in gene therapy clinical trials has led to clonal expansion and leukaemia development due to viral Long Terminal Repeat (LTR)‐driven activation of proto‐oncogenes (i.e. insertional mutagenesis) in a subset of patients 15, 16. Also in HIV‐infected patients, similar processes have recently become evident 17, 18, 19. Exploiting the extensive body of research on HIV and MLV, we will mainly discuss integration site selection as it occurs for lenti‐ and gammaretroviruses. Nevertheless, many of the mechanisms discussed here generalise to all retroviruses.
Nuclear entry route, an early determinant of viral integration site selection
The infectious cycle of retroviruses kicks off when specific receptors on a host cell are engaged by the viral envelope (Env) protein (Fig. 1A). These contacts culminate in the fusion of viral and cellular membranes and the concomitant release of the viral core into the cytoplasm. On its way towards the nucleus, the viral RNA genome is reverse transcribed into a double‐stranded DNA copy. The preintegration complex (PIC), consisting of the viral DNA associated with a specific set of viral and host proteins, then reaches the nuclear envelope. To overcome this physical barrier and gain access to the host‐cell chromatin, retroviruses have evolved different strategies. Taking distinct paths into the nucleus implies that the first chromatin environments generally encountered by various retroviral PICs are also different. If integration takes place quickly after nuclear entry, as results seem to suggest, the result is a first bias in the integration site distribution 20, 21, 22, 23.
Figure 1.
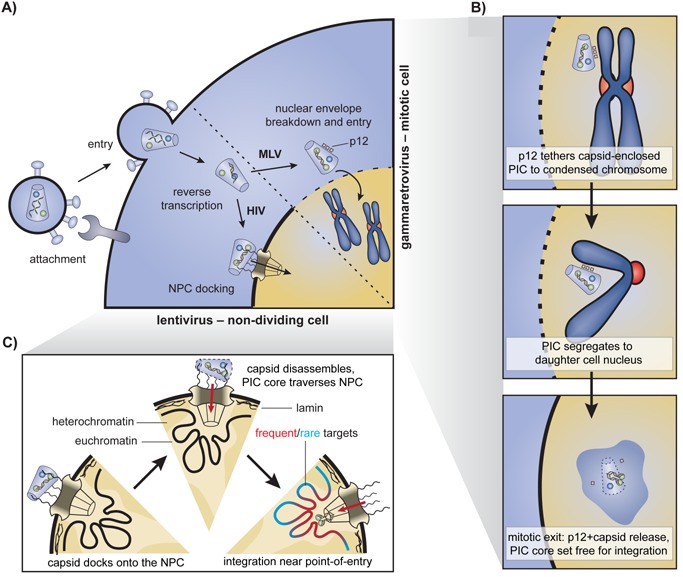
Early stages of retroviral replication and nuclear entry. A: During attachment, the viral envelope engages specific host receptors leading to membrane fusion and entry of the viral core into the cytoplasm. The viral RNA genome is reverse transcribed into a dsDNA copy as the complex is trafficked towards the nucleus. Gammaretroviral preintegration complexes (PICs) wait for mitosis and nuclear envelope disassembly to access the chromatin. Lentiviral PICs traverse a nuclear pore complex (NPC). B: In gammaretroviruses, p12 stabilises the capsid and tethers the PIC to a condensed chromosome during mitosis. The complex segregates to one of the daughter cells. Upon exit from mitosis, the nuclear envelope reassembles, p12 is released and the capsid uncoats. The MLV intasome complex is set free and integration is likely to occur quickly afterwards, in the vicinity. C: In lentiviruses, nuclear import and integration are tightly coupled. The viral capsid core docks onto the NPC. Completion of reverse transcription may promote uncoating of the docked capsid. The released PIC core engages nucleoporins and import factors to gain access to the nucleoplasm. Integration occurs on a first‐come, first served basis.
Tethering of the gammaretroviral capsid to mitotic chromatin
Gammaretroviruses, such as MLV only infect dividing cells. They encode an accessory p12 protein in the group‐specific antigen (gag) gene 24. On the one hand, p12 interacts with and stabilises the capsid shell of the PIC through its N‐terminal portion, preventing untimely uncoating and ensuring completion of reverse transcription 24, 25. When the infected cell enters mitosis and the nuclear envelope breaks down, the C‐terminal region binds to a condensed chromosome, effectively tethering the capsid‐associated PIC to the chromatin (Fig. 1A and B) 26. The complex segregates along with the chromosomes to one of the daughter cells. Upon exit from mitosis, the nuclear envelope reassembles and p12 is released, orchestrating capsid uncoating 27. The MLV intasome, associated with various host and viral factors, is set free in the nucleoplasm (Fig. 1B). If integration happens shortly after the intasome first encounters chromatin, near the point of release, then site selection may be influenced by the chromatin‐binding preferences of p12. Abolishing the MLV p12‐chromatin interaction by mutagenesis blocks viral replication, and can be rescued by introducing other chromatin tethers into the mutant p12. While the preference of the rescued viruses for enhancers did not differ from wild type, some subtle variations in the distributions were observed 27. Conceivably, similar broad chromatin recognition profiles of p12 and the other tethers may hide p12's effect on site selection.
In conclusion, p12 is clearly required for integration per se. However, it does not appear to induce major biases in integration site selection, potentially owing to its broad chromatin recognition. As we will see, host cofactors are acting together with IN downstream of p12 to further pin down the gammaretroviral integration site 12, 13, 14.
Lentiviral capsid docking, uncoating and nuclear import
Lentiviruses have evolved the ability to traverse nuclear pore complexes (NPC), allowing them to infect non‐dividing, terminally differentiated cells. The viral capsid core is believed to dock onto the cytoplasmic side of the NPC through interactions with Nup358/RanBP2 (Fig. 1A and C) 28, 29. Termination of reverse transcription has been reported to promote uncoating of the docked capsid and release of the PIC 30. Through subsequent engagement of other nucleoporins such as Nup153 and Nup98‐Nup96, and import factors such as Transportin‐SR2 (TRN‐SR2, TNPO3), the released PIC gains access to the nucleoplasm 28, 31, 32.
The first nuclear subcompartment a lentiviral PIC encounters is the NPC‐associated chromatin. NPCs establish cone‐like heterochromatin‐exclusion zones at their nuclear baskets (reviewed in 33, 34, Fig. 1C). In contrast, amassment of heterochromatin into repressive lamin‐associated domains occurs adjacent to NPCs (Fig. 1C) 35.
Studies using a variety of fluorescence microscopy approaches have reported that the HIV PIC and integrated provirus preferentially localise to areas of euchromatin at the nuclear periphery 21, 22, 23. Furthermore, genes recurrently targeted for HIV integration are closely associated with NPCs 20. These results suggest that lentiviral integration site selection simply occurs in those regions of the chromatin that it encounters first as it enters the nucleus (Fig. 1C). This initial selection based on nuclear architecture may contribute to the lentiviral integration bias towards regions enriched in open chromatin marks.
The findings evince a tight coupling between lentiviral nuclear import and integration site choice. Indeed, depletion of the nuclear import factor TRN‐SR2 or NPC constituents Nup358/RanBP2, Nup153, Nup98‐Nup96 or Tpr altered the HIV integration site distribution, suggesting these proteins play a role in the nuclear import/integration pathway or in the maintenance of chromatin topology 29, 31, 36, 37, 38.
As the viral capsid affects the mode of nuclear import, it too has been shown to modulate site selection 28, 29, 36. When the entire HIV gag is replaced by its MLV counterpart, the hybrid virus integrates less into the gene‐dense environments underlying NPCs, likely reflecting an altered mode of nuclear entry 29. Moreover, several HIV capsid mutants that perturb binding to Nup358, Nup153 or cyclophilin A and/or alter capsid stability also affect integration site selection 28, 39, 40. The results further underscore the close connection between nuclear import and integration.
Host cofactors steer the PIC to specific chromatin environments
Upon entry or release of the PIC into the nucleus, IN hijacks host proteins to guide the complex to specific chromatin contexts. This likely represents an evolutionary ancient strategy as it is also adopted by LTR retrotranposons. In the case of lentiviruses, the co‐opted protein is Lens Epithelium‐Derived Growth Factor/p75 (LEDGF/p75) 11, 41. Gammaretroviral PICs on the other hand adopt Bromodomain and Extraterminal domain (BET) proteins 12, 13, 14.
LEDGF/p75 tethers lentiviral PICs to actively transcribed chromatin
LEDGF is a ubiquitously expressed chromatin reader encoded by the PSIP1 gene. It is a member of the Hepatoma‐Derived Growth Factor (HDGF) family, characterised by a conserved N‐terminal PWWP domain (named after its signature Pro‐Trp‐Trp‐Pro motif, Fig. 2A). The LEDGF PWWP specifically binds H3K36me3‐modified nucleosomes, a hallmark of active transcription 42, 43 (reviewed in 44). In contrast to the p52 splice variant, the p75 isoform harbours a C‐terminal integrase‐binding domain (IBD), which links it to a variety of cellular proteins. Aside from LEDGF/p75, only its close paralog HDGF‐Related Protein 2 (HRP‐2, HDGFRP2) carries both a PWWP and an IBD (Fig. 2A) 45.
Figure 2.
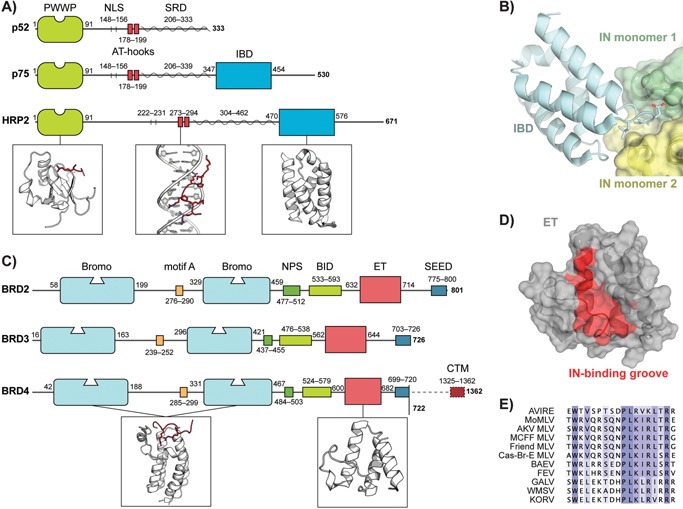
Lenti‐ and gammaretroviral INs interact with specific host chromatin readers. A: Domain overview of both the p52 and p75 splice variants of H. sapiens LEDGF and HRP‐2. Abbreviations: PWWP, Trp‐Pro‐Pro‐Trp; NLS, nuclear localization signal; AT‐hook, AT‐hook minor groove DNA binding motif; SRD, supercoiled‐DNA recognition region; IBD, integrase‐binding domain. Boxes highlight, from left to right, structures of a PWWP domain bound to a methylated histone peptide (red), an AT‐hook motif (red) bound into the DNA minor groove and the IBD. B: Interaction of the LEDGF/p75 IBD (cyan) and an IN catalytic core domain dimer (green and yellow). Although the CCD of IN is essential and sufficient for interaction with LEDGF/p75, a second interface exists involving an acidic patch on the IN N‐terminal domain and a complementary basic patch on the IBD 46, 125. C: Domain overview of H. sapiens bromo‐ and extraterminal domain containing (BET) proteins. Abbreviations: Bromo, bromodomain; motif A, ∼15 aa conserved motif that may contribute to chromatin binding; NPS, N‐terminal cluster of phosphorylation sites; BID, basic residue‐enriched interaction domain; ET, extraterminal domain; SEED, Ser/Glu/Asp‐rich C‐terminal cluster of phosphorylation sites; CTM, C‐terminal motif 66, 71, 126. Boxes highlight, from left to right, the structures of a bromodomain bound to a polyacetylated histone peptide (red) and the ET domain. D: Structure of the ET domain. Residues implicated in several interaction studies with MLV IN are coloured red and can be seen lining a cleft on the domain surface. E: Sequence alignment of the C‐terminal tails of various gammaretroviral INs showing strong conservation of a ∼16 aa BET ET domain binding motif. Abbreviations: AVIRE, Avian reticuloendotheliosis virus (UniProtKB accession: P03360); MoMLV, Moloney murine leukaemia virus (P03355); AKV MLV, AKV murine leukaemia virus (P03355); MCFF MLV, Mink cell focus‐forming murine leukaemia virus (P16103); Friend MLV, Friend murine leukaemia virus (P26809); Cas‐Br‐E MLV, Cas‐Br‐E murine leukaemia virus (P08361); BAEV, Baboon endogenous virus (P10272); FEV, Feline endogenous virus (P31792); GALV, Gibbon ape leukaemia virus (P21414); WMSV, Woolly monkey sarcoma virus (P03359); KORV, Koala retrovirus (Q9TTC1).
LEDGF/p75 was first picked up as the principal cellular binding partner of HIV IN by co‐immunoprecipitation 11. Later, mutagenesis, RNA interference, transdominant overexpression of the IBD and cellular knockout studies corroborated its role in HIV‐1 replication 46, 47, 48, 49, 50, 51, 52, 53, 54. Mutagenesis studies and X‐ray crystallography were employed to unravel the molecular details of the LEDGF/p75‐IN interaction (Fig. 2B) 48, 55, 56. The main interaction interface is formed between the IBD of LEDGF/p75 and the catalytic core domain of IN. In part by binding across the IN dimer interface, LEDGF/p75 modulates IN multimerisation and activity 11.
LEDGF/p75 depletion reduces HIV‐1 integration and shifts integration preferences away from active genes 53, 57, 58. In agreement with this, mapping LEDGF/p75 chromatin occupancy revealed that LEDGF/p75 associates with active regions proportional to their transcriptional output 59. Additionally, chimeric tethers, in which the N‐terminal chromatin‐binding portion of LEDGF/p75 has been replaced by chromatin reader modules of other proteins, shifted viral integration into regions normally recognised by the other protein 60, 61, 62. In the current model, LEDGF/p75 tethers the PIC to actively transcribed chromatin, biasing integration towards these regions (Fig. 3).
Figure 3.
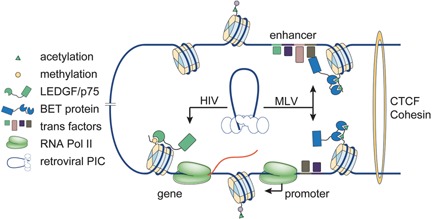
Host cofactors steer the PIC to specific chromatin environments. Example of retroviral integration in a chromatin contact domain with an active enhancer – promoter loop driving expression of a downstream gene. Retroviral PICs hijack distinct host proteins to steer themselves into specific chromatin environments. The lentiviral PIC is tethered by LEDGF/p75 to actively transcribed regions of the chromatin decorated with H3K36me3 marks. In the case of gammaretroviruses, the intasome recruits BET proteins recognizing hyperacetylated histones H3 and H4 at active promoter‐proximal as well as distal enhancer elements.
Upon LEDGF/p75 knockout, integration into active genes still occurs more frequently than statistically expected. Part of the remaining bias is ascribed to HRP‐2 63, 64. Likely owing to its lower affinity for HIV‐1 IN and a generally lower expression level, HRP‐2 only plays a role in HIV integration site targeting when LEDGF/p75 is absent 50, 54. Nevertheless, a substantial preference for active genes remains in doubly depleted cells, supporting the involvement of additional factors such as IN itself and nuclear topology 63, 64.
BET proteins direct gammaretroviral PICs to active enhancers
When p12 orchestrates capsid disassembly upon host cell mitotic exit, the core gammaretroviral PIC is released into the nucleoplasm. The intasome then recognises its specific chromatin reader cofactors, which tether the complex to active enhancers. While an initial yeast two‐hybrid screen revealed, among many others, the BET protein BRD2 as a potential IN interaction partner 65, it was not until 2013 that three groups independently identified BET proteins as bona fide gammaretroviral integration cofactors 12, 13, 14.
The BET protein family includes BRD2, ‐3 and ‐4, which are ubiquitously expressed and BRDT, which is restricted to the testes 66 (see 67 for a recent review). BET proteins are transcriptional co‐regulators characterised by one (plants) or two (fungi/animals) bromodomains at their N‐terminus, followed by an extraterminal (ET) domain (Fig. 2C). The dual bromodomain module bestows BET proteins with a preference for hyper‐acetylated tails of histone H3 and H4 68, 69, 70. The ET domain is a protein‐protein interaction hub, connecting BET proteins to several other coactivators 67.
The interaction between BET proteins and MLV IN involves the BET ET (Fig. 2D) domain and the flexible C‐terminal tail of IN 12, 71. A ∼16 aa motif, conserved specifically among gammaretroviruses is essential and sufficient for binding (Fig. 2E) 12, 72. Specifically, alanine substitution of a single conserved Trp residue in MLV IN (W390A) was sufficient to abolish the interaction 12, 73.
Support for an integration targeting role of BET proteins came from several directions. First, the BET protein chromatin‐binding profile showed remarkable overlap with MLV integration sites 12, 13. Second, BET knockdown or specific inhibition of BET chromatin tethering through the use of bromodomain inhibitors attenuated MLV, but not HIV replication and blocked integration of MLV‐derived viral vectors 12, 13, 14. When scrutinised under these conditions, integration shifted away from enhancer elements 13. Third, a hybrid tethering factor, containing the chromatin‐binding part of LEDGF/p75 and the MLV IN‐binding part of BRD4, redirected MLV integration into active transcription units, a pattern reminiscent of HIV integration 12. Taken together, these findings firmly established BET proteins as specific mediators of integration target site selection for gammaretroviruses (Fig. 3).
Hitting the spot: The viral integrase determines the precise insertion site
LEDGF/p75 and BET proteins, respectively, guide the lenti‐ and gammaretroviral PICs to distinct chromatin contexts (Fig. 3). Subsequently, IN selects the final site as the intasome recognises a target DNA (tDNA) strand and catalyses the cutting and joining reactions that fuse viral and host genomes.
Retroviral IN contains three structurally conserved domains, connected through flexible linkers (Fig. 4A; see 74 for a recent review). X‐ray crystal structures of the spumaviral Prototype Foamy Virus (PFV) intasome complex have boosted our understanding of the retroviral integration machinery 9, 10. The intasome consists of a dimer of IN dimers assembled on the two viral DNA LTR ends (Fig. 4C). After processing the viral DNA ends, the complex captures a tDNA duplex in a groove between its inner monomers (Fig. 4C, the target capture complex, TCC). In a reaction called strand transfer, the viral DNA ends are inserted 4–6 bp apart (depending on the retroviral species) into opposing strands of the tDNA 75. The remaining single‐strand gaps are repaired by host‐cell machinery, accordingly yielding 4–6 bp duplications flanking the provirus.
Figure 4.
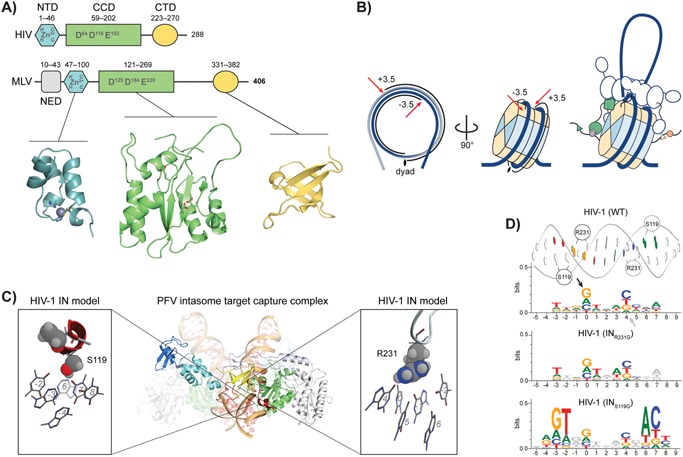
Integrase determines the precise insertion site. A: Domain structure of HIV‐1 and MLV IN. All retroviral INs contain three structurally conserved domains connected through flexible linkers: an N‐terminal Zn2+‐binding domain, a catalytic core domain harbouring the characteristic DD‐X35‐E triad motif to chelate two catalytic Mg2+ ions, and a C‐terminal SH3‐like domain (NTD, CCD and CTD, respectively). Spuma‐ and most likely gammaretroviral INs contain an additional NTD‐extension domain (NED). B: Retroviral integration into nucleosomal DNA. The first two panels show the integration hotspots at ± 3.5 turns of DNA from the nucleosome dyad axis. The right panel suggests a general model for retroviral integration. The intasome is tethered to one or several nucleosomes through a host cofactor. Through extensive interactions with both the targeted‐ and non‐targeted DNA gyre and several core histones (potentially sensing epigenetic marks), the intasome discriminates different nucleosomes and determines the final integration site. C: Structure of the PFV intasome target capture complex (central) consisting of a dimer of IN dimers assembled on the two viral DNA LTR ends. The complex is docked onto a target DNA strand (bottom). Boxes on either side highlight direct aa – tDNA base contacts in the complex using HIV‐1 numbering. Target DNA bases are numbered starting from the sites of strand transfer on both strands. D: Sequence logos of HIV‐1 integration sites of wild type (WT) and two variant viruses (INR231G and INS119G), representing their intasome footprints on the tDNA. Arrows denote the sites of strand transfer (position 0) on the plus (black) and minus (grey) strands. A B‐DNA representation of the tDNA with the different aa‐base contacts is placed on top and positions correspond directly to those in the sequence logo. Positions where base preferences are significantly different from wild type HIV‐1 are coloured 85, 86.
Nucleosomes are the natural target for integration and induce further biases
When bound to the intasome, the tDNA is considerably kinked in order to correctly position the scissile phosphodiester bonds in the two active sites 10. This structure is in agreement with early observations that integration is favoured at inherently bendable sites, and specifically on nucleosomal DNA 76, 77. Results from cellular integration site sequencing suggest that integration is directed into outward‐facing major grooves on nucleosomal DNA 78, 79.
Recent work confirms that the PFV intasome is able to stably capture and efficiently catalyse integration into mononucleosomes 80. Integration occurs preferentially at two possible, symmetric positions, ± 3.5 turns of the DNA helix (∼36 bp) away from the nucleosome dyad axis (Fig. 4B) 80. Cryo‐electron microscopy of the intasome‐nucleosome complex revealed an extensive interaction interface 80. The contacts with the integration‐targeted gyre of the nucleosomal DNA are similar to those observed previously in the PFV intasome TCC structure (Fig. 4C) 10, 80. The second gyre is cradled by one of the two IN dimer interfaces and an inner subunit interacts with the histone H2B C‐terminal helix (Fig. 4B). Additional contacts exist between the histone H2A N‐terminal tail and the C‐terminal domain of an inner subunit of the intasome and between the C‐terminal domains of the outer subunits and other parts of the nucleosome (Fig. 4B). These contacts may allow the intasome to decode epigenetic marks 80, a situation reminiscent of the distantly related chromovirus Ty3/Gypsy LTR retrotransposons which encode a chromodomain at their IN C‐terminus, allowing them to read histone modifications and steer integration 81. Indeed, mutagenesis of residues in contact with the second gyre of DNA or with H2B reduced the PFV bias towards gene poor, lamin‐associated heterochromatin 80.
In order to avoid steric clashes, the intasome ‘lifts’ the DNA from the surface of the nucleosome (Fig. 4B). As a result, sequences yielding less stable nucleosomes appear to be better targets for capture by the intasome. Histone variants may influence integration site preferences in a similar fashion. The histone H2A L1‐loop directly underlies the integration hotspots and shows considerable variation, resulting in differential stability of nucleosomes containing certain H2A variants 80, 82. Asymmetry of the nucleosomal DNA sequence may also be expected to bias towards one of the preferred sites.
On a side note, nucleosomal capture may explain the site‐specificity of HIV‐1 integration events observed in Alu elements 17. Alu elements contribute to nucleosome positioning in the primate genome, with each dimeric Alu element having two fixed nucleosome slots 83, 84. The first of these positions a nucleosome in such a manner that the integration hotspot in the Alu element precisely overlaps one of the two hotspots on the nucleosome.
Intasome—tDNA base contacts induce integration site sequence preferences
Molecular recognition between retroviral intasome and nucleosome biases the integration site distribution. Two recent studies employed the structure of the PFV intasome TCC to predict and modify IN – tDNA contacts in the context of HIV‐1 infection (Fig. 4C) 85, 86. Two IN aa – tDNA base contacts have been discerned in the retroviral intasome, and these directly induce palindromic sequence preferences owing to the intasome central dyad axis (Fig. 4C and D) 10, 86. Using HIV‐1 numbering, the first is IN119, which projects directly into the tDNA minor groove, contacting base pairs at positions −2 and −3, as numbered from the sites of strand transfer (position 0). IN231 in HIV‐1 is the second site: it extends into the tDNA major groove and interacts with base pairs 0 and −1. IN119 is a highly polymorphic position in HIV‐1 IN that varies amongst retroviruses as well. Position IN231 is conserved in lentiviruses, but the loop in which it is located varies considerably among retroviruses. Different amino acids at these two positions yield distinct local sequence biases for viral integration 85, 86. These residues additionally modulate central tDNA bending: where interactions at IN231 can be envisioned to act as a tether, pulling the tDNA into the active sites, bulky aa at IN119 push it down at the sides, requiring stronger distortion in the centre 86.
IN119 and IN231 directly shape nucleotide preferences at tDNA positions −4 to 0 (Fig. 4D). At the site of strand transfer, retroviruses bias against T as its methyl group sterically hinders the transesterification reaction 10. Finally, as the tDNA is kinked most profoundly between the two sites of strand transfer (positions 1–3) and no aa – base contacts are established in this region, intrinsic bending potential dictates the nucleotide preferences 10, 85, 86. Depending on the size of the integration stagger, the deformation may be spread over 2–4 bp. In PFV with its 4 bp stagger, the centre of the integration site is preferentially occupied by flexible pyrimidine – purine dinucleotides 10. In the case of HIV and other retroviruses with a 5 bp stagger, minor groove compressible WWW trinucleotides such as AAA, AAT or TAA and their reverse complements dominate positions 1–3 of the integration site consensus (Fig. 4D) 86, 87.
In conclusion, capture of a target duplex by the retroviral intasome directly imposes additional restraints on the integration site, yielding fine‐grained biases in the distribution.
Local DNA contacts affect genome‐wide integration site distribution
IN – tDNA base contacts in the retroviral intasome provide mechanistic insights into the local sequence preferences for viral integration. HIV‐1 variants INS119G and INR231G however, additionally direct integration into globally less gene‐dense regions when compared to the wild‐type INS119‐R231 virus 86. The INS119G and INR231G variants exhibit wild‐type catalytic activity but alter tDNA bending requirements and consequently may reduce the shape and/or electrostatic compatibility with certain nucleosomes 86. These variants may prefer integration into the weakly bound nucleosomes present in GC‐poor regions, which are generally also gene poor. Interestingly, INS119G and INR231G viruses were associated with a more rapid disease progression in a subset of antiretroviral naive, HIV‐1 subtype C chronically infected participants of the Sinikithemba cohort 86. Additional data are required though to corroborate the observed correlation between altered intasome target site selection in these variants and viral fitness or pathogenesis.
Aside from local preferences, IN contact variants with different parts of the nucleosome can affect large‐scale biases in the integration site distribution. These global effects have now been demonstrated for HIV‐1 86 and PFV 80 and are expected to generalise to other retroviruses.
Selection pressure can directly impinge on intasome target specificity
The prevalence of IN119 and IN231 variants differs among HIV subtypes, implying that integration preferences differ slightly among subtypes 86. Additionally, HIV‐1 IN119 polymorphisms are subject to selection pressure from different sources. INS119R for instance was recently found to be an immune escape variant selected for by the HLA‐C*05 allele 88. Similarly, several HIV‐1 IN119 variants change prevalence in INSTI‐untreated versus ‐treated patients, and they have been described as secondary resistance mutations 89, 90, 91. IN119, thus, represents a position of close interaction between immune and drug selection pressure on the one hand and HIV‐1 integration site targeting on the other. While these examples are specific to HIV‐1, it is clear that any selection pressure acting on positions homologous to HIV‐1 IN119/231 in other retroviruses will influence intasome target site selection.
Proviral transcriptional activity determines the fate of the infection
At this point, the retrovirus has become an integral part of the host genome. Proviral transcriptional activity determines the ensuing course of the infection, and reshapes the integration site distribution in the population of infected cells. The activity of the viral promoter shortly after infection is probabilistically determined by a variety of cellular transcription factors binding their cognate recognition sequences in the retroviral LTR 92 (Fig. 5). Additionally, LTR activity depends on the chromatin environment 93, 94. Together, these factors result in a range of intrinsic basal transcriptional states for the provirus (Fig. 5).
Figure 5.
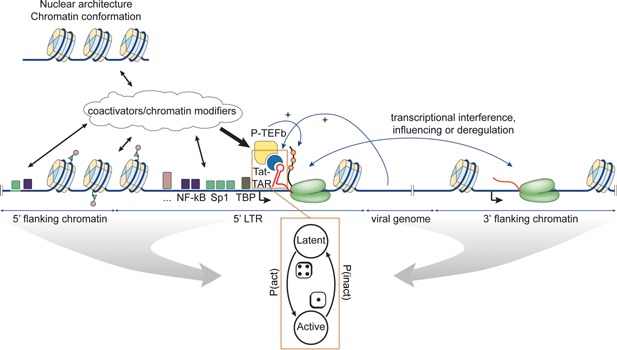
Simplified model of HIV‐1 proviral transcription. The chromatin context, both in cis and in trans, together with the presence of various LTR‐binding transcription factors, probabilistically determine intrinsic proviral activity through various other coactivators and chromatin modifiers. When the viral trans‐activator of transcription (Tat) accumulates beyond a critical threshold, it binds the trans‐activation response (TAR) RNA stem‐loop at the 5′ end of nascent viral transcripts and recruits P‐TEFb. P‐TEFb phosphorylates RNA Pol II as well as its pausing factors enabling productive elongation. Amplification of stochastic viral expression by this Tat positive feedback creates a robust latency switch. By influencing the intrinsic LTR activity, the chromatin context and hence integration site selection influences the parameters of the switch, resulting in different probabilities for the ON and OFF states. Recognizing viral latency as a bet hedging strategy, integration site selection holds important consequences for virus evolution.
Spuma‐, lenti‐, delta‐ and epsilonretroviruses encode their own transcriptional master regulators, while alpha‐, beta‐ and gammaretroviruses rely more on host transcription factors. The HIV trans‐activator of transcription (Tat) protein amplifies stochastic expression from the LTR promoter and establishes a positive feedback loop (Fig. 5, 95, reviewed in 96). The architecture of this regulatory circuit (and perhaps those of other retroviral genera) creates a phenotypic bifurcation: the provirus can be transcriptionally active or quiescent. In the first case, progeny virions are rapidly produced, whereas in the second case, the provirus enters a potentially long‐lived latent state. Simulations of Tat protein levels in cells suggest stochastic switching between the two states (transcriptional shutdown or reactivation of the provirus) is possible under a wide array of conditions 97. The latently infected cells constitute a reservoir for the virus and represent a major hurdle towards obtaining a functional HIV‐1 cure. As will be discussed in the last section, recent reports suggest that some latently infected cells are actively proliferating and contribute to the reservoir under antiretroviral treatment 18, 19.
It is worth mentioning the phenomenon of superinfection resistance (SIR), in which an infected cell becomes resistant to superinfection by a similar type of virus. SIR has been described in several groups of viruses. As virus‐encoded proteins are usually responsible, SIR is directly related to integration and transcription in retroviruses. Often, viral (or host‐acquired) Env expression is found to interfere with infection, simply by occupying the cellular receptors for viral entry 98. The spumaviral Bet gene (Between‐env‐and‐LTR‐1‐and‐2) contributes to SIR and inhibits expression of the viral transcriptional master regulator (Tas) to maintain the original latently infected cell 98. In the case of HIV‐1, the mechanisms of SIR are not well understood 98. Nevertheless, SIR is far from absolute. Both in HIV‐1 and HTLV‐1 infected patients, evidence suggests a small percentage of the infected cells may harbour multiple proviruses 99, 100. In such cases, superinfection and viral recombination during the next round could be of evolutionary significance 101.
The chromatin environment influences proviral activity
Several studies of chromatin position effects on gene expression from the HIV‐1 LTR or active promoters showed that variability in the integration site affects transcriptional kinetics and can result in up to ∼1,000‐fold differences in expression level 93, 94, 102, 103. Besides cis‐effects, also long‐range topological interactions are known to affect proviral activity (Fig. 5) 104, 105. At least for HIV‐1 and HTLV‐1, proviral (and host gene) transcription is affected by the orientation of the integrated retrovirus through transcriptional interference 7, 106, 107, 108.
Proviruses integrated in the same genomic neighbourhood in different cells tend to have the same activation status, corroborating the existence of local features influencing latency 109. Nevertheless, the precise nature of these (epi‐) genomic features remains to be identified 108, 109, 110. For HTLV‐1, specific transcription factor binding sites and the relative position of the nearest host gene as well as its relative orientation have been linked to proviral expression 108.
The chromatin environment determines the threshold level of transcription factors required to induce LTR‐driven gene expression (Fig. 5) 111. Although the study was performed with HIV‐1, chromatin likely modulates transcription factor binding and activation in similar ways on all retroviral LTRs. Taken together, the results support the notion that intrinsic LTR activity is governed by the chromatin environment in combination with the available transcription factors (Fig. 5).
Integration site targeting is subject to natural selection: Latency and bet hedging
The Tat circuit is optimised to amplify stochastic fluctuations in gene expression 92, 95, 112. Tat feedback is sufficient to induce a robust, probabilistic latency switch independent of cellular activation state 97. In agreement with the hardwiring of this switch, the latency programme was proposed to represent a bet hedging strategy. Bet hedging is an evolutionary strategy based on stochastic phenotype switching that optimises survival in fluctuating or unfavourable environments. In the case of HIV‐1, a stochastic latency switch may allow the virus to maximise transmission by reducing extinction during mucosal infections while maintaining sufficiently high‐plasma viral loads later on 113.
While the switch itself is always present, LTR activity, determined by transcription factors and chromatin environment, directly modulates the probabilities of a provirus being in the ON or the OFF states, and hence the population of latent and productively infected cells 97. As it affects the probabilistic parameters of the latency switch, the integration site represents an inherent part of the viral bet hedging strategy and can be subject to direct evolutionary optimisation. For instance, consistent integration at permissive or repressive sites results in more proviruses in the ON or OFF state, respectively. Both scenarios are selected against due to a reduced transmissibility 113.
Not all retroviruses encode a positive transcriptional feedback loop. Nevertheless, similar bet hedging strategies may be adopted by other genera besides lentiviruses. Proviral transcription is inherently noisy, potentially magnified by the formation of a gene loop joining 5′ LTR promoter and 3′ LTR poly(A) signal 114, 115. Such loops are also observed at various cellular promoters where they impose transcriptional directionality and allow efficient recycling of the RNA Pol II machinery 115. Intrinsic transcriptional bursting from a retroviral LTR, modulated in frequency and size by the chromatin context and the presence of cognate host transcription factors, may provide enough variability to create repressed and permissive proviral states. Even without a positive feedback loop, integration site selection could thus be part of an evolutionary optimised bet hedging strategy. In this case, the phenotypic switching parameters are likely to be more dependent on host‐transcription factors and the proviral chromatin context. The MLV preference for strong enhancer elements for instance may be an adaptation to guarantee close interactions with the transcriptional machinery 4, 5.
Of note, theory predicts that modulating the latency switch to result in a latent provirus in 90–95% of infected cells would push the basic reproduction number of HIV below one, resulting in an unsustainable infection 113. This may prove to be a viable alternative to shock‐and‐kill strategies and could be envisioned using Tat antagonists 116, 117, but also with modulators of integration site targeting, chromatin or transcription factors.
The fact that LEDGF/p75 depletion hampers HIV‐1 replication 54, 64, is in line with a fitness advantage through integration site selection. However, as LEDGF/p75 affects IN oligomerisation as well as integration site targeting, it is difficult to uncouple these contributions. MLV‐derived viral vectors deficient for interaction with BET proteins suffer no or only a marginal reduction in transduction efficiency in vitro 73, 118. The effect of these mutants on multiple round viral replication (in vivo) however, remains to be determined.
Selective forces in the host shape the final integration site distribution
In vivo, selection forces continuously reshape the integration site landscape. Integration events at loci that support high expression will probabilistically lead to more active proviruses 93, 94, 109. In turn, these cells are likely to be weeded out by the immune system or die due to viral cytopathic effects 119. In the case of HTLV‐1, negative selection on the integration site distribution is dominant during chronic infection 7. Reciprocally, infected cells with a growth advantage may increase in number, for instance due to an oncogenic integration event, antigen stimulation or homeostatic proliferation 18, 19, 120, 121.
BET proteins steer gammaretroviral integration into strong promoter‐proximal as well as distal enhancer elements 4, 5. In both cases insertional mutagenesis can lead to oncogene activation and ultimately malignant transformation of the host cell 122. Insertion of an LTR in a promoter can easily be envisioned to result in transcriptional deregulation. Distal enhancers are brought into contact with promoters through DNA looping (Fig. 3) 123. Integration of an LTR enhancer in a distal regulatory region can similarly lead to activation of the target promoter via these three‐dimensional interactions, as commonly observed among oncogenic MLV integrations 122.
Recent studies have found that also HIV integration can lead to clonal expansion 17, 18, 19. HIV integration into specific cancer‐associated genes may occasionally yield a cellular survival advantage, promoting expansion and viral persistence. At least in one case a prominent clone has been found to perpetuate the infectious viral reservoir by producing replication‐competent virus in sufficient quantities to cause viraemia 18. It remains to be determined whether this finding turns out to be a general one and not all expanded clones harbour defective viruses 17, 18, 19.
Driving proliferation of the host cell may be an inherent part of the general retroviral evolutionary strategy. While some non‐acutely transforming viruses encode their own oncogenes (e.g. HTLV Tax and HBZ), others cause transformation of the host cell through insertional mutagenesis. The efficiency of cellular transformation differs considerably between retroviruses, in agreement with varying contributions to the strategies of different retroviral genera. Logically, the mutagenic potential of a retrovirus relies, at least in part, on its specific integration site biases. Integration site selection therefore contributes to this strategy as well.
Considering the potential of retroviruses to drive host cell proliferation together with stochastic viral gene expression, one may envision a reservoir of latently infected, clonally expanding cells, carrying genetically identical virus that stochastically pops up (blips). In agreement with this, genetically identical HIV variants have been reported to emerge after long‐term suppressive antiretroviral therapy 124.
Conclusions: Site matters
It has become clear that retroviral integration site selection is an intricate multistep process that starts with the mode of nuclear entry of the viral PIC (Fig. 6). Similar to the ancestral LTR retrotransposons, retroviruses co‐opt host factors to guide the viral integration machinery to specific chromatin environments (Fig. 6). Lentiviruses grab hold of the chromatin reader LEDGF/p75, directing integration into actively transcribed regions. Likewise, gammaretroviruses target enhancers by adopting BET proteins as chaperones for their integrase. Both LEDGF/p75 and BET proteins are chromatin readers, and have ample binding sites across the genome. The invading PIC therefore likely encounters an environment suitable for integration soon after it enters the nucleus. Tethered to specific chromatin regions, the intasome captures a nucleosome and catalyses the cutting and joining reactions fusing the viral and host genomes, to pin down the final integration site (Fig. 6). Direct contacts of the intasome with target DNA bases, the intrinsic bendability of the sequence, and contacts with the rest of the nucleosome further bias the integration site distribution (Fig. 6).
Figure 6.
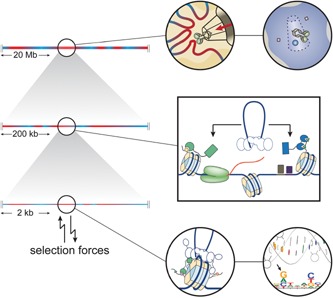
Overview of retroviral integration site targeting. Integration is believed to occur relatively quickly after the retroviral integration machinery encounters the chromatin. As a result, taking a specific route into the nucleus generates a first coarse‐grained bias in the integration site distribution (top). In this still extensive chromatin environment, further biases are introduced as the viral PIC hijacks host cofactors tethering it to well defined, functional features of the chromatin (middle). Tethered more locally to the chromatin, the intasome itself needs to capture a nucleosomal target DNA strand, catalyse the insertion process, and hence determine the final insertion points (bottom). Specific contacts between intasome and nucleosome as well as target DNA sequence considerations result in further biases in the integration site distribution. Finally, in infected hosts, selection forces continue to reshape the established population of proviruses.
Upon establishment of the provirus, the available transcription factors and the chromatin context probabilistically affect the course of the infection. Importantly, by modulating the probabilities of switching between proviral ON and OFF states, the integration site selection processes of retroviruses may be part of a bet hedging strategy and could have been optimised through natural selection. Finally, in vivo, the integration site distribution is continuously reshaped by selection processes such as immune pressure and the viral cytopathic effect, but also by clonal expansion, either passively or actively due to insertional mutagenesis (Fig. 6).
Acknowledgements
Financial support was provided by grants from the KU Leuven Research Fund, Research Foundation – Flanders (FWO), IAP BelVir (BelSpo), FP7 CHAARM, the Eranet EUREKA and the Creative and Novel Ideas in HIV Research Program (CNIHR), through a supplement to the University of California at San Francisco (UCSF) Center For AIDS Research funding (P30 AI027763). The latter funding was made possible by collaborative efforts of the Office of AIDS Research, the National Institute of Allergy and Infectious Diseases and the International AIDS Society.
The authors have declared no conflict of interest.
References
- 1. Schröder ARW, Shinn P, Chen H, Berry C, et al. 2002. HIV‐1 integration in the human genome favors active genes and local hotspots. Cell 110: 521–9. [DOI] [PubMed] [Google Scholar]
- 2. Wu X, Li Y, Crise B, Burgess SM. 2003. Transcription start regions in the human genome are favored targets for MLV integration. Science 300: 1749–51. [DOI] [PubMed] [Google Scholar]
- 3. Mitchell RS, Beitzel BF, Schröder ARW, Shinn P, et al. 2004. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol 2: E234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lafave MC, Varshney GK, Gildea DE, Wolfsberg TG, et al. 2014. MLV integration site selection is driven by strong enhancers and active promoters. Nucleic Acids Res 42: 4257–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. De Ravin SS, Su L, Theobald N, Choi U, et al. 2014. Enhancers are major targets for murine leukemia virus vector integration. J Virol 88: 4504–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Derse D, Crise B, Li Y, Princler G, et al. 2007. Human T‐cell leukemia virus type 1 integration target sites in the human genome: comparison with those of other retroviruses. J Virol 81: 6731–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gillet NA, Malani N, Melamed A, Gormley N, et al. 2011. The host genomic environment of the provirus determines the abundance of HTLV‐1‐infected T‐cell clones. Blood 117: 3113–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang GP, Ciuffi A, Leipzig J, Berry CC, et al. 2007. HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res 17: 1186–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hare S, Gupta SS, Valkov E, Engelman A, et al. 2010. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 464: 232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maertens GN, Hare S, Cherepanov P. 2010. The mechanism of retroviral integration from X‐ray structures of its key intermediates. Nature 468: 326–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cherepanov P, Maertens G, Proost P, Devreese B, et al. 2003. HIV‐1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J Biol Chem 278: 372–81. [DOI] [PubMed] [Google Scholar]
- 12. De Rijck J, de Kogel C, Demeulemeester J, Vets S, et al. 2013. The BET family of proteins targets moloney murine leukemia virus integration near transcription start sites. Cell Rep 5: 886–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sharma A, Larue RC, Plumb MR, Malani N, et al. 2013. BET proteins promote efficient murine leukemia virus integration at transcription start sites. Proc Natl Acad Sci USA 110: 12036–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gupta SS, Maetzig T, Maertens GN, Sharif A, et al. 2013. Bromo‐ and extraterminal domain chromatin regulators serve as cofactors for murine leukemia virus integration. J Virol 87: 12721–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hacein‐Bey‐Abina S, Garrigue A, Wang GP, Soulier J, et al. 2008. Insertional oncogenesis in 4 patients after retrovirus‐mediated gene therapy of SCID‐ X1. J Clin Invest 118: 3132–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hacein‐Bey‐Abina S, Kalle von C, Schmidt M, McCormack MP, et al. 2003. LMO2‐associated clonal T cell proliferation in two patients after gene therapy for SCID‐ X1. Science 302: 415–9. [DOI] [PubMed] [Google Scholar]
- 17. Cohn LB, Silva IT, Oliveira TY, Rosales RA, et al. 2015. HIV‐1 integration landscape during latent and active infection. Cell 160: 420–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maldarelli F, Wu X, Su L, Simonetti FR, et al. 2014. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 345: 179–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wagner TA, McLaughlin S, Garg K, Cheung CYK, et al. 2014. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 345: 570–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marini B, Kertesz‐Farkas A, Ali H, Lucic B, et al. 2015. Nuclear architecture dictates HIV‐1 integration site selection. Nature 521: 227–31. [DOI] [PubMed] [Google Scholar]
- 21. Albanese A, Arosio D, Terreni M, Cereseto A. 2008. HIV‐1 pre‐integration complexes selectively target decondensed chromatin in the nuclear periphery. PLoS One 3: e2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burdick RC, Hu W‐S, Pathak VK. 2013. Nuclear import of APOBEC3F‐labeled HIV‐1 preintegration complexes. Proc Natl Acad Sci USA 110: E4780–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Di Primio C, Quercioli V, Allouch A, Gijsbers R, et al. 2013. Single‐cell imaging of HIV‐1 provirus (SCIP). Proc Natl Acad Sci USA 110: 5636–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wight DJ, Boucherit VC, Nader M, Allen DJ, et al. 2012. The gammaretroviral p12 protein has multiple domains that function during the early stages of replication. Retrovirology 9: 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wight DJ, Boucherit VC, Wanaguru M, Elis E, et al. 2014. The N‐terminus of murine leukaemia virus p12 protein is required for mature core stability. PLoS Pathog 10: e1004474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Elis E, Ehrlich M, Prizan‐Ravid A, Laham‐Karam N, et al. 2012. P12 tethers the murine leukemia virus pre‐integration complex to mitotic chromosomes. PLoS Pathog 8: e1003103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schneider WM, Brzezinski JD, Aiyer S, Malani N, et al. 2013. Viral DNA tethering domains complement replication‐defective mutations in the p12 protein of MuLV Gag. Proc Natl Acad Sci USA 110: 9487–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, et al. 2011. HIV‐1 capsid‐cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog 7: e1002439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ocwieja KE, Brady TL, Ronen K, Huegel A, et al. 2011. HIV integration targeting: a pathway involving Transportin‐3 and the nuclear pore protein Ran BP2. PLoS Pathog 7: e1001313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Arhel NJ, Souquere‐Besse S, Munier S, Souque P, et al. 2007. HIV‐1 DNA Flap formation promotes uncoating of the pre‐integration complex at the nuclear pore. EMBO J 26: 3025–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Di Nunzio F, Fricke T, Miccio A, Valle‐Casuso JC, et al. 2013. Nup153 and Nup98 bind the HIV‐1 core and contribute to the early steps of HIV‐1 replication. Virology 440: 8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Christ F, Thys W, De Rijck J, Gijsbers R, et al. 2008. Transportin‐SR2 imports HIV into the nucleus. Curr Biol 18: 1192–202. [DOI] [PubMed] [Google Scholar]
- 33. Sood V, Brickner JH. 2014. Nuclear pore interactions with the genome. Curr Opin Genet Dev 25: 43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pascual‐Garcia P, Capelson M. 2014. Nuclear pores as versatile platforms for gene regulation. Curr Opin Genet Dev 25: 110–7. [DOI] [PubMed] [Google Scholar]
- 35. Guelen L, Pagie L, Brasset E, Meuleman W, et al. 2008. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453: 948–51. [DOI] [PubMed] [Google Scholar]
- 36. Koh Y, Wu X, Ferris AL, Matreyek KA, et al. 2013. Differential effects of human immunodeficiency virus type 1 capsid and cellular factors nucleoporin 153 and LEDGF/p75 on the efficiency and specificity of viral DNA integration. J Virol 87: 648–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lelek M, Casartelli N, Pellin D, Rizzi E, et al. 2015. Chromatin organization at the nuclear pore favours HIV replication. Nat Commun 6: 6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Krull S, Dörries J, Boysen B, Reidenbach S, et al. 2010. Protein Tpr is required for establishing nuclear pore‐associated zones of heterochromatin exclusion. EMBO J 29: 1659–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Price AJ, Jacques DA, McEwan WA, Fletcher AJ, et al. 2014. Host cofactors and pharmacologic ligands share an essential interface in HIV‐1 capsid that is lost upon disassembly. PLoS Pathog 10: e1004459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matreyek KA, Yücel SS, Li X, Engelman A. 2013. Nucleoporin NUP153 phenylalanine‐glycine motifs engage a common binding pocket within the HIV‐1 capsid protein to mediate lentiviral infectivity. PLoS Pathog 9: e1003693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cherepanov P. 2007. LEDGF/p75 interacts with divergent lentiviral integrases and modulates their enzymatic activity in vitro. Nucleic Acids Res 35: 113–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Eidahl JO, Crowe BL, North JA, McKee CJ, et al. 2013. Structural basis for high‐affinity binding of LEDGF PWWP to mononucleosomes. Nucleic Acids Res 41: 3924–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Nuland R, van Schaik FM, Simonis M, van Heesch S, et al. 2013. Nucleosomal DNA binding drives the recognition of H3K36‐methylated nucleosomes by the PSIP1‐PWWP domain. Epigenetics Chromatin 6: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Qin S, Min J. 2014. Structure and function of the nucleosome‐binding PWWP domain. Trends Biochem Sci 39: 536–47. [DOI] [PubMed] [Google Scholar]
- 45. Cherepanov P, Devroe E, Silver PA, Engelman A. 2004. Identification of an evolutionarily conserved domain in human lens epithelium‐derived growth factor/transcriptional co‐activator p75 (LEDGF/p75) that binds HIV‐1 integrase. J Biol Chem 279: 48883–92. [DOI] [PubMed] [Google Scholar]
- 46. Maertens G, Cherepanov P, Pluymers W, Busschots K, et al. 2003. LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV‐1 integrase in human cells. J Biol Chem 278: 33528–39. [DOI] [PubMed] [Google Scholar]
- 47. Llano M, Delgado S, Vanegas M, Poeschla EM. 2004. Lens epithelium‐derived growth factor/p75 prevents proteasomal degradation of HIV‐1 integrase. J Biol Chem 279: 55570–7. [DOI] [PubMed] [Google Scholar]
- 48. Emiliani S, Mousnier A, Busschots K, Maroun M, et al. 2005. Integrase mutants defective for interaction with LEDGF/p75 are impaired in chromosome tethering and HIV‐1 replication. J Biol Chem 280: 25517–23. [DOI] [PubMed] [Google Scholar]
- 49. De Rijck J, Vandekerckhove L, Gijsbers R, Hombrouck A, et al. 2006. Overexpression of the lens epithelium‐derived growth factor/p75 integrase binding domain inhibits human immunodeficiency virus replication. J Virol 80: 11498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Llano M, Saenz DT, Meehan A, Wongthida P, et al. 2006. An essential role for LEDGF/p75 in HIV integration. Science 314: 461–4. [DOI] [PubMed] [Google Scholar]
- 51. Vandekerckhove L, Christ F, Van Maele B, De Rijck J, et al. 2006. Transient and stable knockdown of the integrase cofactor LEDGF/p75 reveals its role in the replication cycle of human immunodeficiency virus. J Virol 80: 1886–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hombrouck A, De Rijck J, Hendrix J, Vandekerckhove L, et al. 2007. Virus evolution reveals an exclusive role for LEDGF/p75 in chromosomal tethering of HIV. PLoS Pathog 3: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shun M‐C, Raghavendra NK, Vandegraaff N, Daigle JE, et al. 2007. LEDGF/p75 functions downstream from preintegration complex formation to effect gene‐specific HIV‐1 integration. Genes Dev 21: 1767–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schrijvers R, De Rijck J, Demeulemeester J, Adachi N, et al. 2012. LEDGF/p75‐independent HIV‐1 replication demonstrates a role for HRP‐2 and remains sensitive to inhibition by LEDGINs. PLoS Pathog 8: e1002558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cherepanov P, Ambrosio ALB, Rahman S, Ellenberger T, et al. 2005. Structural basis for the recognition between HIV‐1 integrase and transcriptional coactivator p75. Proc Natl Acad Sci USA 102: 17308–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cherepanov P, Sun Z‐YJ, Rahman S, Maertens G, et al. 2005. Solution structure of the HIV‐1 integrase‐binding domain in LEDGF/p75. Nat Struct Mol Biol 12: 526–32. [DOI] [PubMed] [Google Scholar]
- 57. Ciuffi A, Llano M, Poeschla E, Hoffmann C, et al. 2005. A role for LEDGF/p75 in targeting HIV DNA integration. Nat Med 11: 1287–9. [DOI] [PubMed] [Google Scholar]
- 58. Marshall HM, Ronen K, Berry C, Llano M, et al. 2007. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS ONE 2: e1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. De Rijck J, Bartholomeeusen K, Ceulemans H, Debyser Z, et al. 2010. High‐resolution profiling of the LEDGF/p75 chromatin interaction in the ENCODE region. Nucleic Acids Res 38: 6135–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ferris AL, Wu X, Hughes CM, Stewart C, et al. 2010. Lens epithelium‐derived growth factor fusion proteins redirect HIV‐1 DNA integration. Proc Natl Acad Sci USA 107: 3135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gijsbers R, Ronen K, Vets S, Malani N, et al. 2010. LEDGF hybrids efficiently retarget lentiviral integration into heterochromatin. Mol Ther 18: 552–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Silvers RM, Smith JA, Schowalter M, Litwin S, et al. 2010. Modification of integration site preferences of an HIV‐1‐based vector by expression of a novel synthetic protein. Hum Gene Ther 21: 337–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schrijvers R, Vets S, De Rijck J, Malani N, et al. 2012. HRP‐2 determines HIV‐1 integration site selection in LEDGF/p75 depleted cells. Retrovirology 9: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang H, Jurado KA, Wu X, Shun M‐C, et al. 2012. HRP2 determines the efficiency and specificity of HIV‐1 integration in LEDGF/p75 knockout cells but does not contribute to the antiviral activity of a potent LEDGF/p75‐binding site integrase inhibitor. Nucleic Acids Res 40: 11518–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Studamire B, Goff SP. 2008. Host proteins interacting with the Moloney murine leukemia virus integrase: multiple transcriptional regulators and chromatin binding factors. Retrovirology 5: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Paillisson A, Levasseur A, Gouret P, Callebaut I, et al. 2007. Bromodomain testis‐specific protein is expressed in mouse oocyte and evolves faster than its ubiquitously expressed paralogs BRD2, ‐3, and ‐4. Genomics 89: 215–23. [DOI] [PubMed] [Google Scholar]
- 67. Belkina AC, Denis GV. 2012. BET domain co‐regulators in obesity, inflammation and cancer. Nat Rev Cancer 12: 465–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Morinière J, Rousseaux S, Steuerwald U, Soler‐López M, et al. 2009. Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature 461: 664–8. [DOI] [PubMed] [Google Scholar]
- 69. Leroy G, Chepelev I, Dimaggio PA, Blanco MA, et al. 2012. Proteogenomic characterization and mapping of nucleosomes decoded by Brd and HP1 proteins. Genome Biol 13: R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Filippakopoulos P, Picaud S, Mangos M, Keates T, et al. 2012. Histone recognition and large‐scale structural analysis of the human bromodomain family. Cell 149: 214–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Larue RC, Plumb MR, Crowe BL, Shkriabai N, et al. 2014. Bimodal high‐affinity association of Brd4 with murine leukemia virus integrase and mononucleosomes. Nucleic Acids Res 42: 4868–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Aiyer S, Swapna GVT, Malani N, Aramini JM, et al. 2014. Altering murine leukemia virus integration through disruption of the integrase and BET protein family interaction. Nucleic Acids Res 42: 5917–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ashkar El S, De Rijck J, Demeulemeester J, Vets S, et al. 2014. BET‐independent MLV‐based vectors target away from promoters and regulatory elements. Mol Ther Nucleic Acids 3: e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cherepanov P, Maertens GN, Hare S. 2011. Structural insights into the retroviral DNA integration apparatus. Curr Opin Struct Biol 21: 249–56. [DOI] [PubMed] [Google Scholar]
- 75. Hare S, Maertens GN, Cherepanov P. 2012. 3'‐processing and strand transfer catalysed by retroviral integrase in crystallo. EMBO J 31: 3020–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pryciak PM, Varmus HE. 1992. Nucleosomes, DNA‐binding proteins, and DNA sequence modulate retroviral integration target site selection. Cell 69: 769–80. [DOI] [PubMed] [Google Scholar]
- 77. Pruss D, Bushman FD, Wolffe AP. 1994. Human immunodeficiency virus integrase directs integration to sites of severe DNA distortion within the nucleosome core. Proc Natl Acad Sci USA 91: 5913–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Roth SL, Malani N, Bushman FD. 2011. Gammaretroviral integration into nucleosomal target DNA in vivo. J Virol 85: 7393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Benleulmi MS, Matysiak J, Henriquez DR, Vaillant C, et al. 2015. Intasome architecture and chromatin density modulate retroviral integration into nucleosome. Retrovirology 12: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Maskell DP, Renault L, Serrao E, Lesbats P, et al. 2015. Structural basis for retroviral integration into nucleosomes. Nature 523: 366–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gao X, Hou Y, Ebina H, Levin HL. 2008. Chromodomains direct integration of retrotransposons to heterochromatin. Genome Res 18: 359–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Shaytan AK, Landsman D, Panchenko AR. 2015. Nucleosome adaptability conferred by sequence and structural variations in histone H2A‐H2B dimers. Curr Opin Struct Biol 32C: 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Englander EW, Howard BH. 1995. Nucleosome positioning by human Alu elements in chromatin. J Biol Chem 270: 10091–6. [DOI] [PubMed] [Google Scholar]
- 84. Tanaka Y, Yamashita R, Suzuki Y, Nakai K. 2010. Effects of Alu elements on global nucleosome positioning in the human genome. BMC Genomics 11: 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Serrao E, Krishnan L, Shun M‐C, Li X, et al. 2014. Integrase residues that determine nucleotide preferences at sites of HIV‐1 integration: implications for the mechanism of target DNA binding. Nucleic Acids Res 42: 5164–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Demeulemeester J, Vets S, Schrijvers R, Madlala P, et al. 2014. HIV‐1 integrase variants retarget viral integration and are associated with disease progression in a chronic infection cohort. Cell Host Microbe 16: 651–62. [DOI] [PubMed] [Google Scholar]
- 87. Satchwell SC, Drew HR, Travers AA. 1986. Sequence periodicities in chicken nucleosome core DNA. J Mol Biol 191: 659–75. [DOI] [PubMed] [Google Scholar]
- 88. Brockman MA, Chopera DR, Olvera A, Brumme CJ, et al. 2012. Uncommon pathways of immune escape attenuate HIV‐1 integrase replication capacity. J Virol 86: 6913–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Armenia D, Vandenbroucke I, Fabeni L, Van Marck H, et al. 2012. Study of genotypic and phenotypic HIV‐1 dynamics of integrase mutations during raltegravir treatment: a refined analysis by ultra‐deep 454 pyrosequencing. J Infect Dis 205: 557–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ceccherini‐Silberstein F, Malet I, Fabeni L, Dimonte S, et al. 2010. Specific HIV‐1 integrase polymorphisms change their prevalence in untreated versus antiretroviral‐treated HIV‐1‐infected patients, all naive to integrase inhibitors. J Antimicrob Chemother 65: 2305–18. [DOI] [PubMed] [Google Scholar]
- 91. Hachiya A, Ode H, Matsuda M, Kito Y, et al. 2015. Natural polymorphism S119R of HIV‐1 integrase enhances primary INSTI resistance. Antiviral Res 119: 84–8. [DOI] [PubMed] [Google Scholar]
- 92. Burnett JC, Miller‐Jensen K, Shah PS, Arkin AP, et al. 2009. Control of stochastic gene expression by host factors at the HIV promoter. PLoS Pathog 5: e1000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Dar RD, Razooky BS, Singh A, Trimeloni TV, et al. 2012. Transcriptional burst frequency and burst size are equally modulated across the human genome. Proc Natl Acad Sci USA 109: 17454–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Singh A, Razooky B, Cox CD, Simpson ML, et al. 2010. Transcriptional bursting from the HIV‐1 promoter is a significant source of stochastic noise in HIV‐1 gene expression. Biophys J 98: L32–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Weinberger LS, Burnett JC, Toettcher JE, Arkin AP, et al. 2005. Stochastic gene expression in a lentiviral positive‐feedback loop: HIV‐1 Tat fluctuations drive phenotypic diversity. Cell 122: 169–82. [DOI] [PubMed] [Google Scholar]
- 96. Romani B, Engelbrecht S, Glashoff RH. 2010. Functions of Tat: the versatile protein of human immunodeficiency virus type 1. J Gen Virol 91: 1–2. [DOI] [PubMed] [Google Scholar]
- 97. Razooky BS, Pai A, Aull K, Rouzine IM, et al. 2015. A hardwired HIV latency program. Cell 160: 990–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Nethe M, Berkhout B, van der Kuyl AC. 2005. Retroviral superinfection resistance. Retrovirology 2: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Josefsson L, King MS, Makitalo B, Brännström J, et al. 2011. Majority of CD4+ T cells from peripheral blood of HIV‐1‐infected individuals contain only one HIV DNA molecule. Proc Natl Acad Sci USA 108: 11199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Cook LB, Melamed A, Niederer H, Valganon M, et al. 2014. The role of HTLV‐1 clonality, proviral structure, and genomic integration site in adult T‐cell leukemia/lymphoma. Blood 123: 3925–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Simon‐Loriere E, Holmes EC. 2011. Why do RNA viruses recombine? Nat Rev Microbiol 9: 617–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Jordan A, Defechereux P, Verdin E. 2001. The site of HIV‐1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. EMBO J 20: 1726–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Akhtar W, de Jong J, Pindyurin AV, Pagie L, et al. 2013. Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell 154: 914–27. [DOI] [PubMed] [Google Scholar]
- 104. Dieudonné M, Maiuri P, Biancotto C, Knezevich A, et al. 2009. Transcriptional competence of the integrated HIV‐1 provirus at the nuclear periphery. EMBO J 28: 2231–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lusic M, Marini B, Ali H, Lucic B, et al. 2013. Proximity to PML nuclear bodies regulates HIV‐1 latency in CD4+ T cells. Cell Host Microbe 13: 665–77. [DOI] [PubMed] [Google Scholar]
- 106. Han Y, Lin YB, An W, Xu J, et al. 2008. Orientation‐dependent regulation of integrated HIV‐1 expression by host gene transcriptional readthrough. Cell Host Microbe 4: 134–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Lenasi T, Contreras X, Peterlin BM. 2008. Transcriptional interference antagonizes proviral gene expression to promote HIV latency. Cell Host Microbe 4: 123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Melamed A, Laydon DJ, Gillet NA, Tanaka Y, et al. 2013. Genome‐wide determinants of proviral targeting, clonal abundance and expression in natural HTLV‐1 infection. PLoS Pathog 9: e1003271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Sherrill‐Mix S, Lewinski MK, Famiglietti M, Bosque A, et al. 2013. HIV latency and integration site placement in five cell‐based models. Retrovirology 10: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Dahabieh MS, Ooms M, Brumme C, Taylor J, et al. 2014. Direct non‐productive HIV‐1 infection in a T‐cell line is driven by cellular activation state and NFκB. Retrovirology 11: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Miller‐Jensen K, Dey SS, Pham N, Foley JE, et al. 2012. Chromatin accessibility at the HIV LTR promoter sets a threshold for NF‐κB mediated viral gene expression. Integr Biol (Camb) 4: 661–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Weinberger LS, Dar RD, Simpson ML. 2008. Transient‐mediated fate determination in a transcriptional circuit of HIV. Nat Genet 40: 466–70. [DOI] [PubMed] [Google Scholar]
- 113. Rouzine IM, Weinberger AD, Weinberger LS. 2015. An evolutionary role for HIV latency in enhancing viral transmission. Cell 160: 1002–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Perkins KJ, Lusic M, Mitar I, Giacca M, et al. 2008. Transcription‐dependent gene looping of the HIV‐1 provirus is dictated by recognition of pre‐mRNA processing signals. Mol Cell 29: 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Hebenstreit D. 2013. Are gene loops the cause of transcriptional noise? Trends Genet 29: 333–8. [DOI] [PubMed] [Google Scholar]
- 116. Mousseau G, Clementz MA, Bakeman WN, Nagarsheth N, et al. 2012. An analog of the natural steroidal alkaloid cortistatin A potently suppresses Tat‐dependent HIV transcription. Cell Host Microbe 12: 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Mousseau G, Kessing CF, Fromentin R, Trautmann L, et al. 2015. The Tat inhibitor didehydro‐Cortistatin A prevents HIV‐1 reactivation from latency. MBio 6: 15–e00465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Roth MJ. 1991. Mutational analysis of the carboxyl terminus of the Moloney murine leukemia virus integration protein. J Virol 65: 2141–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Coffin JM. 1995. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science 267: 483–9. [DOI] [PubMed] [Google Scholar]
- 120. Chomont N, El‐Far M, Ancuta P, Trautmann L, et al. 2009. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med 15: 893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Niederer HA, Bangham CRM. 2014. Integration site and clonal expansion in human chronic retroviral infection and gene therapy. Viruses 6: 4140–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Babaei S, Akhtar W, de Jong J, Reinders M, et al. 2015. 3D hotspots of recurrent retroviral insertions reveal long‐range interactions with cancer genes. Nat Commun 6: 6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Rao SSP, Huntley MH, Durand NC, Stamenova EK, et al. 2014. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159: 1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kearney MF, Spindler J, Shao W, Yu S, et al. 2014. Lack of detectable HIV‐1 molecular evolution during suppressive antiretroviral therapy. PLoS Pathog 10: e1004010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Hare S, Shun M‐C, Gupta SS, Valkov E, et al. 2009. A novel co‐crystal structure affords the design of gain‐of‐function lentiviral integrase mutants in the presence of modified PSIP1/LEDGF/p75. PLoS Pathog 5: e1000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Wu S‐Y, Lee A‐Y, Lai H‐T, Zhang H, et al. 2013. Phospho switch triggers brd4 chromatin binding and activator recruitment for gene‐specific targeting. Mol Cell 49: 843–57. [DOI] [PMC free article] [PubMed] [Google Scholar]