

Abstract
Patients with type 1 diabetes (T1D) who are recipients of pancreas transplants are believed to rarely develop T1D recurrence in the allograft if effectively immunosuppressed. We evaluated a cohort of 223 recipients of simultaneous pancreas–kidney allografts for T1D recurrence and its risk factors. With long‐term follow‐up, recurrence was observed in approximately 7% of patients. Comparing the therapeutic regimens employed in this cohort over time, lack of induction therapy was associated with recurrence, but this occurs even with the current regimen, which includes induction; there was no influence of maintenance regimens. Longitudinal testing for T1D‐associated autoantibodies identified autoantibody positivity, number of autoantibodies, and autoantibody conversion after transplantation as critical risk factors. Autoantibodies to the zinc transporter 8 had the strongest and closest temporal association with recurrence, which was not explained by genetically encoded amino acid sequence donor–recipient mismatches for this autoantigen. Genetic risk factors included the presence of the T1D‐predisposing HLA‐DR3/DR4 genotype in the recipient and donor–recipient sharing of HLA‐DR alleles, especially HLA‐DR3. Thus, T1D recurrence is not uncommon and is developing in patients treated with current immunosuppression. The risk factors identified in this study can be assessed in the transplant clinic to identify recurrent T1D and may lead to therapeutic advances.
Short abstract
This study demonstrates that recurrence of islet autoimmunity and type 1 diabetes is a significant cause of immune‐mediated endocrine pancreas graft function in immunosuppressed recipients of simultaneous pancreas‐kidney transplants, and defines risk factors for this condition.
Abbreviations
- AZA
azathioprine
- CyA
cyclosporine
- ESRD
end‐stage renal disease
- FK
tacrolimus
- GAD65
65‐kDa glutamic acid decarboxylase isoform
- GADA
GAD65 autoantibody
- HG
hyperglycemia
- HG‐T1DR
hyperglycemia with T1D recurrence
- HG‐PCR
hyperglycemia with pancreas chronic rejection
- HG‐UND
hyperglycemia of undetermined cause
- HR
hazard ratio
- IA‐2
insulinoma‐associated tyrosine phosphatase‐like protein
- IA‐2A
IA‐2 autoantibody
- MMF
mycophenolate mofetil
- NGT
normal glucose tolerance
- OKT3
anti‐CD3, muromonab
- PPV
positive predictive value
- SEM
standard error of the mean
- SPK
simultaneous pancreas–kidney
- T1D
type 1 diabetes
- ZnT8
zinc transporter 8
- ZnT8A
ZnT8 autoantibody
Introduction
Islet autoimmunity causes progressive loss of pancreatic beta cells, leading to impaired insulin secretion and the development of type 1 diabetes (T1D) 1. Several islet autoantigens are targeted by both cellular and humoral responses. Standardized assays measure autoantibodies to insulin, the 65‐kDa glutamic acid decarboxylase isoform (GAD65), the insulinoma‐associated tyrosine phosphatase‐like protein (IA‐2), and zinc transporter 8 (ZnT8). Autoantibodies are robust diagnostic and predictive T1D markers, with multiple autoantibodies conferring much higher risk than single autoantibody positivity 2, 3, 4, 5, 6, 7.
Simultaneous pancreas–kidney (SPK) transplantation restores insulin secretion and renal function in T1D patients with end‐stage renal disease (ESRD) 8. However, recipients may develop posttransplant diabetes, a broad clinical entity with multifactorial etiology including effects of immunosuppression, insulin resistance, weight gain, and others 9, 10. With advances in immunosuppression, acute rejection has become less prevalent and immunological failures are typically ascribed to chronic rejection 8. However, another potential cause of immunological failure is T1D recurrence 11, 12. A growing literature suggests that islet autoimmunity may become reactivated and affect the endocrine function of pancreas transplants 11, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29.
In this longitudinal study, we assessed T1D recurrence in a large cohort of 223 SPK recipients. We find that even with the current immunosuppression regimen, T1D recurrence is a common cause of posttransplant diabetes, no less frequent than diabetes resulting from pancreas chronic rejection. Further, we define immunological, genetic, and therapeutic risk factors for T1D recurrence. We show that monitoring islet autoimmunity and assessment of other risk factors help predict and correctly diagnose T1D recurrence.
Subjects and Methods
Subjects
We studied T1D patients with ESRD who received SPK transplants. All had no detectable c‐peptide response to a Sustacal/Boost test (Société des Produits Nestlé S.A., Vevey, Switzerland) before transplantation. All pancreas transplants were bladder‐drained with systemic venous effluent. We monitored urine amylase levels to aid in assessing pancreatic exocrine graft function and rejection; patients generally exhibit a stable range of amylase levels in units/hour (h) during a 12‐h overnight collection. A reduction in levels is suggestive of rejection. Between 1990 and 2013, 452 T1D patients underwent SPK transplantation at the University of Miami. The Institutional Review Board approved this study (#20053039) to evaluate islet autoimmunity. Participation was voluntary; subjects could withdraw at any time without affecting access to standard care. For some patients, outcome data and archived serum samples were evaluated under waiver of consent.
Patient classification according to pancreas graft outcomes
This analysis included 223 SPK recipients who had at least one year of post‐transplant follow‐up and for whom we could define overall autoantibody status prior to and after transplantation for the duration of the follow‐up. The 223 patients were transplanted between 1990 and 2012, with the last follow‐up sample collected in 2013. The demographic characteristics are shown in Table 1. The mean follow‐up was 6.2 ± standard deviation (SD) 4.1 years, range 1.0–22.6 years. Follow‐up started with the transplant and ended with the development of hyperglycemia or with the last sample obtained by December 2013 for those who were still normoglycemic. Patients were classified as normal glucose tolerant (NGT) if their HbA1c was ≤6.1% (the normal range in our laboratory is 4.5–6.1%) in the absence of insulin therapy or antidiabetic oral medications. Patients were categorized as having hyperglycemia (HG) if, on further follow‐up after posttransplant normalization of glucose metabolism, their HbA1c levels returned to the abnormal range and/or if they required antidiabetic therapy (oral agents, insulin, or both). We classified the HG group into three subgroups based on clinical features:
HG with T1D recurrence (HG‐T1DR): presence of classical diabetes symptoms with hyperglycemia requiring insulin therapy, severe loss of C‐peptide in the absence of rejection (stable urine amylase and serum creatinine levels);
HG with pancreas chronic rejection (HG‐PCR): hyperglycemia associated with loss of urine amylase; biopsy‐proven rejection in some instances;
HG with undetermined cause (HG‐UND): mild diabetes symptoms, typically not requiring insulin therapy, persisting C‐peptide secretion, often associated with obesity or significant weight gain; lack of clear signs of rejection.
Table 1.
Demographic and clinical features, overall and by analysis groups
| All patients | NGT | HG‐T1DR | HG‐PCR | HG‐UND | p value | |
|---|---|---|---|---|---|---|
| N | 223 | 176 | 17 | 10 | 20 | n/a |
| % of total (223) | n/a | 78.9 | 7.6 | 4.5 | 9 | n/a |
| % of HG (47) | n/a | n/a | 36.2 | 21.3 | 42.6 | n/a |
| Sex (F‐M) | 106–117 | 89–87 | 8–9 | 1–9 | 8–12 | p=0.14941 |
| White not Hispanic, n (%) | 117 (52.5) | 96 (54.5) | 9 (52.9) | 5 (50.0) | 7 (35.0) | p=0.30282 |
| White Hispanic, n (%) | 83 (37.2) | 63 (35.8) | 8 (47.1) | 3 (30.0) | 9 (45.0) | n/a |
| African‐American, n (%) | 22 (9.9) | 16 (9.1) | 0 | 2 (20.0) | 4 (20.0) | n/a |
| Other, n (%) | 1 (0.4) | 1 (0.6) | 0 | 0 | 0 | n/a |
| Age at T1D onset mean ± SD (years) | 14 ± 8.6 | 14.3 ± 8.4 | 13.9 ± 10.7 | 9.1 ± 5.3 | 14.7 ± 8.8 | p=0.3159a |
| Range (years) | 0–39 | 0–38 | 2–39 | 1–17 | 4–32 | n/a |
| Patients with no available data | 2 | 1 | 0 | 0 | 1 | n/a |
| T1D duration at transplantation mean ± SD (years) | 26.6 ± 8.6 | 27.5 ± 8.4 | 20.9 ± 10.0 | 27.6 ± 5.0 | 23.1 ± 7.8 | p = 0.0161 |
| Range (years) | 2–50 | 3–50 | 2–39 | 22–35 | 8–36 | n/a |
| Patients with no available data | 2 | 1 | 0 | 0 | 1 | n/a |
| Age at transplant mean ± SD (years) | 40.7 ± 8.0 | 41.8 ± 8.1 | 34.8 ± 6.6 | 36.6 ± 4.4 | 38.2 ± 6.3 | p = 0.0006 |
| Range (years) | 23–60 | 23–60 | 23–47 | 30–44 | 29–50 | n/a |
| Length of follow‐up mean ± SD (years) | 6.2 ± 4.1 | 6.2 ± 4.2 | 6.8 ± 3.8 | 4.5 ± 2.9 | 6.2 ± 4.4 | p = 0.5029* |
| Range (years) | 1.0–22.6 | 1.0–22.6 | 2.2–17.2 | 2.1–10.3 | 1.1–15.6 | n/a |
| Last HbA1c on follow‐up mean ± SD | 5.7 ± 0.7 | 5.5 ± 0.4 | 6.9 ± 0.9 | 6.9 ± 1.1 | 7.1 ± 0.8 | p < 0.0001 |
| Patients with no available data | 54 | 32 | 12 | 7 | 3 | n/a |
HG‐PCR, hyperglycemia with pancreas chronic rejection; HG‐T1DR, hyperglycemia with type 1 diabetes recurrence; NGT, normal glucose tolerance.
Overall p values of the chi‐square test are shown for independence between sex1 or ethnicity2 (excluding “other”) and group classification (NGT, HG‐T1DR, HG‐PCR, HG‐UND).
One‐ way analysis of variance (Global Kruskal–Wallis test).
Autoantibody testing of longitudinal samples
For each patient, we tested a pretransplant and multiple follow‐up serum samples. Patients had a mean 4.8 ± SD 3.1 samples tested per year of follow‐up (range 0.6–17.1 per year, only 5 patients had less than one sample/year tested on average). We used well‐established radioimmunoassays (RIA) to measure autoantibodies to the autoantigens GAD65 (GADA), IA‐2 (IA‐2A), and ZnT8 (ZnT8A) (Supplementary Methods). We did not measure insulin autoantibodies since antibodies to insulin are induced by insulin therapy in most patients, and these cannot be distinguished from autoantibodies.
Genetic typing
Donors and recipients were HLA typed, by standard serology and/or molecular techniques, at the time of transplantation. A subset of 19 donor–recipient pairs, from whom DNA was available from a pancreas transplant biopsy (donor) and blood (recipient), were typed at SCL30A8 locus, as described in Supplementary Methods.
Statistical analysis
Nonparametric comparisons of group differences, such as Mann–Whitney test and Kruskal–Wallis test (one‐way analysis of variance) were used for quantitative variables, applying Dunn's correction for multiple comparisons where appropriate. Categorical variables between groups were compared with the chi‐square and Fisher's exact tests. Kaplan–Meier curves were used to estimate hyperglycemia‐free survival; significant differences in survival distributions were assessed with the log‐rank test, unless otherwise noted, after verification of proportional hazard assumptions, with Bonferroni corrections applied for multiple comparisons. Two‐tailed tests of significance and a Type I error rate of 0.05 are used throughout. Statistical analysis was performed with either SAS 9.3 (SAS, Cary, NC) or GraphPad Prism 6.0 (Graphpad Software, La Jolla, CA).
Results
Development of posttransplant diabetes
Most patients (n = 176, 78.9%) were normoglycemic at the end of the follow‐up (mean follow‐up length=6.2 ± SD 4.2 years, range 1–22.6 years, Table 1). Forty‐seven patients (21.1%) developed posttransplant diabetes (HG group); 17 patients (7.6% overall, 36.2% of the HG group) had a clinical diagnosis of HG‐T1DR; this was confirmed by examination of a pancreas transplant biopsy in 15/17 (88%) patients, which demonstrated the presence of insulitis in 13/15 (87%) patients and significant loss of insulin staining in all (Table S1A) 11; thus, biopsy‐confirmed HG‐T1DR was found in 5.8% of our recipients (13/223). Although several of the patients had some biopsy evidence of coexisting pancreas rejection (Table S1B), this ranged from minimal to moderate in most with severe chronic rejection observed in a single patient (patient 5). Only 5 patients had coexisting laboratory findings suggestive of pancreas rejection given urine amylase levels <1000 units/h (patients 2, 6, 8, 9, and 11, Table S1B).
Ten patients (4.5% overall, 21.3% of the HG group) developed hyperglycemia with a clinical diagnosis of HG‐PCR. Twenty patients (9.0% overall and 42.6% of the HG group) developed hyperglycemia of undetermined cause (HG‐UND). The duration of follow‐up was comparable among NGT and HG groups (Table 1). The HG‐T1DR group had shorter T1D duration at transplantation compared to the NGT group (difference = 6.6 years, p = 0.0237). The patients who developed HG‐T1DR were significantly younger at transplantation than those in the NGT group (p = 0.0021). All HG groups had significantly higher HbA1c levels compared to NGT (HG‐T1DR, p = 0.0006; HG‐PCR, p = 0.0135; HG‐UND, p < 0.0001).
Influence of immunosuppression on the development of T1D recurrence
We investigated whether immunosuppression, which has evolved over time, influences the development of T1D recurrence (Table 2). Using Kaplan–Meier estimates of survival, we evaluated hazard ratios with log‐rank tests of significance.T1D recurrence was more common in patients who did not receive induction therapy (Group B, Figure 1, hazard ratio [HR] = 6.324, p = 0.0036) compared to Group D where induction was achieved with thymoglobulin and anti‐CD25; these groups received the same maintenance therapy. Group B also had a higher incidence of T1D recurrence compared to Groups D + E, which included patients treated with the current regimen (since year 2000, induction with thymoglobulin and anti‐CD25 and maintenance with steroids, tacrolimus [FK], and either rapamycin or mycophenolate mofetil [MMF]; Figure 1, HR = 6.058, p = 0.0011). There was no effect associated with rapamycin or MMF. Induction with anti‐CD3, muromonab (OKT3) (Group A) resulted in comparable survival with the current regimen and lower T1D recurrence risk compared to the no‐induction regimen (Group B, p = 0.0530).
Table 2.
Patient groups according to immunosuppression regimens
| Group | Period | Patients (n) | Induction therapy | Maintenance therapy | Years of follow‐up (mean ± SEM) (range) | HG‐T1DR | Years from Tx to HG‐T1DR (mean ± SEM) (range) |
|---|---|---|---|---|---|---|---|
| A | 6/13/1990–5/6/1997 | 16 | OKT3 (n = 16) | CyA (n = 4) or FK (n = 12), AZA (n = 14) or MMF (n = 2), steroids | 9.12 ± 1.20 (2.78–19.07) | 4 | 10.79 ± 2.16 (8.18–17.18) |
| B | 8/2/1997–6/14/2000 | 19 | No induction (n = 11) | FK, MMF, steroids (n = 19) | 7.12 ± 1.11 (4.81–7.93) | 5 | 5.81 ± 0.58a (2.78–19.07) |
| C | Anti‐CD25 (n = 8) | 7.21 ± 1.77 (2.25–14.07) | 1 | 2.25 (na) | |||
| D | 9/6/2000–9/30/2013 | 108 | Thymo + Anti‐CD25 (n = 108) | FK, MMF, steroids (n=55) | 5.51 ± 0.47 (0.94–11.52) | 3 | 7.07 ± 1.52 (4.09–9.08) |
| E | FK, rapamycin, steroids (n=53) | 4.22 ± 0.38 (0.99–10.66) | 2 | 2.38 ± 1.10 (1.28–3.48) | |||
| D+E | FK, MMF, or rapamycin, steroids (n = 108) | 4.88 ± 0.31 (0.94–11.52) | 5 | 5.19 ± 1.46 (1.28–9.08) | |||
| Total | 143 | 15 | |||||
Immunosuppression regimens used at our center, inclusive dates, number of patients in each group, and development of HG‐T1DR. This analysis does not include 80 patients who developed hyperglycemia following rejection or for undetermined cause, and/or who had significant changes in maintenance immunosuppression on follow‐up and would be difficult to assign to a treatment group; the single patient treated with Minnesota anti‐lymphocyte globulin (MALG) was not included. Data are shown for 143 patients: 119 had no changes to maintenance therapy on follow‐up, 24 patients had only early changes (within 6 months) in maintenance drugs and afterwards remained on the same regimens; 128 patients had normal glucose tolerance by the end of follow‐up and 15 patients had developed HG‐T1DR.
Time from transplantation to diagnosis of HG‐T1DR was significantly shorter for Group B when compared to Group A, p = 0.0159; no other comparisons showed statistical differences.
HG‐T1DR, hyperglycemia with type 1 diabetes recurrence; OKT3, anti‐CD3, muromonab; anti‐CD25, daclizumab or basiliximab; CyA, cyclosporine; FK, tacrolimus; AZA, azathiprine; MMF, mycofenilate mofetil.
Figure 1.
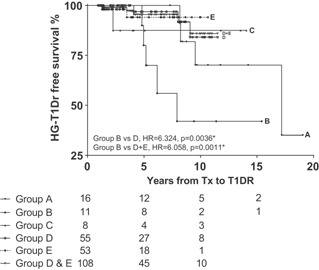
Effects of immunosuppression on type 1 diabetes recurrence. Kaplan–Meier analysis of HG‐T1DR‐free survival according to the immunosuppression regimens used. Regimens used in each of the groups are shown in Table 2. Group B had a higher incidence of HG‐T1DR compared to Group D (HR = 6.324, p = 0.0036*) and Group D+E (HR = 6.058, p = 0.0011*). Group B and Group C did not differ (HR = 2.674, p = 0.3458), similar to Group A vs. Group B (HR = 3.23, p = 0.0530), Group C (HR = 1.106, p = 0.9292), and Group D+E (HR = 1.49, p = 0.4856). There was no difference in the incidence of HG‐T1DR between Group D and Group E (HR = 1.178, p = 0.8538). *Significant after correction for multiple comparisons. HG‐T1DR, hyperglycemia with type 1 diabetes recurrence.
Autoantibody status and patterns defined by pre‐ and posttransplant analysis
We assessed T1D‐associated autoantibodies in our cohort to investigate whether autoantibodies are associated with T1D recurrence. Patients were classified as autoantibody‐negative (Figure 2) if they had no autoantibodies pretransplant and on follow‐up (n = 97); this group included a small number of patients (n = 28/223, 12.5%) with sporadic autoantibody positivity, at very low levels and inconsistent for autoantibody type, who similar to the autoantibody‐negative group showed no association with hyperglycemia (not shown). Patients who were positive pretransplant but converted to negative soon after transplantation (n = 19) were also considered autoantibody‐negative overall. On posttransplant follow‐up (Table S2, Figure 2), the proportions of autoantibody‐negative and autoantibody‐positive patients changed little (pre‐ to posttransplant, 57–52% negative, 43–48% positive). However, the determination of pre‐ and posttransplant autoantibody status allowed us to appreciate changes and thus define two patterns of autoantibody positivity, persistence, and conversion. Patients who were autoantibody‐positive pretransplant and remained positive without acquiring additional autoantibodies were classified as having autoantibody persistence (n = 57, 25.6%). Patients who were autoantibody‐negative pretransplant and became autoantibody‐positive on follow‐up, or if positive pretransplant later acquired additional autoantibodies, were defined as having an autoantibody conversion pattern (n = 50, 22.4%).
Figure 2.
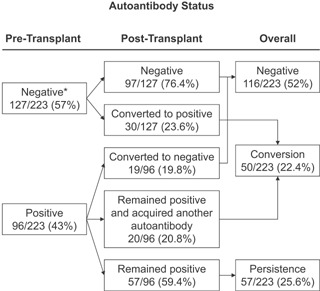
Autoantibody patterns. Autoantibody status pre‐ and posttransplant, based on which we determined overall autoantibody patterns (negative, conversion, persistence) in 223 SPK recipients.*Eight patients could not be tested for ZnT8A pretransplant but were always negative on follow‐up and were assumed to be negative for this analysis of overall autoantibody status. SPK, simultaneous pancreas–kidney.
Pretransplant autoantibody status does not predict future pancreas graft endocrine function
Pretransplant, 43% of the patients had at least one T1D‐associated autoantibody, despite having developed T1D on average two decades earlier (Tables 1 and S2); GADA and IA‐2 autoantibody (IA‐2A) were more common than ZnT8A, but their prevalence did not significantly differ among NGT and HG groups (GADA p = 0.1391, IA‐2A p = 0.7044, ZnT8A p = 0.4818). Kaplan–Meier estimates of HG‐free survival did not significantly differ by pretransplant autoantibody status (Figure S1), even considering number of autoantibodies (Figure S2).
Posttransplant autoantibody positivity, autoantibody number, and conversion are risk factors for HG‐T1DR
Table S2 reports the pre‐ and posttransplant prevalence of each autoantibody according to group. Figure 3A shows higher prevalence posttransplant for all autoantibodies only in the HG‐T1DR group. To appropriately examine this dramatic difference, we conducted a paired analysis of the proportion of patients who converted from negative pretransplant to positive on follow‐up, which again was increased only in the HG‐T1DR group (GADA, p = 1.884 × 10−9; IA‐2A, p = 9.391 × 10−8; ZnT8A, p = 1.682 × 10−6) (Figure 3B). On follow‐up, autoantibody positivity was specifically associated with increased risk of HG‐T1DR (HR = 14.53, p = 0.0005) (Figure 4A) but not with the risk of HG‐PCR and HG‐UND (Figure S3). Multiple autoantibodies were associated with increased risk of HG‐T1DR (Figure 4B) but not with HG‐PCR and HG‐UND (Figure S4). Table S3 describes specificity and sensitivity according to number of autoantibodies. Figure 5 shows the prevalence of autoantibody conversion, persistence, and negativity for each group. The prevalence of conversion was higher in the HG‐T1DR group (p < 0.0001 vs. negative and persistence); conversion was not associated with HG‐UND and HG‐PCR. Survival analysis confirmed that autoantibody conversion confers increased risk of HG‐T1DR (Figure 4C; HR = 27.11, p<0.0001, conversion vs. negativity; HR = 15.57, p<0.0003, conversion vs. persistence). There was no effect of autoantibody conversion on HG‐UND or HG‐PCR (Figure S5). Compared to negativity, persistence did not influence risk of hyperglycemia in any of the HG groups (Figure 4C and Figure S5). Of 57 patients with autoantibody persistence, all had GADA or IA‐2A persistence, and none had persistent ZnT8A. None of 9 patients with ZnT8A pretransplant had persisting levels as all became negative on follow‐up. Two such patients reacquired ZnT8A on follow‐up as a single autoantibody and were still NGT after 10 years of follow‐up. A third patient converted back to ZnT8A positivity, as well as to positivity for GADA and IA‐2A, and developed HG‐UND 1.9 years after transplantation. Thus, all patients with ZnT8A positivity posttransplant had autoantibody conversions. The specific association of autoantibody positivity and conversion with risk of T1D recurrence was also observed in similar analysis for each of the autoantibodies (data not shown).
Figure 3.
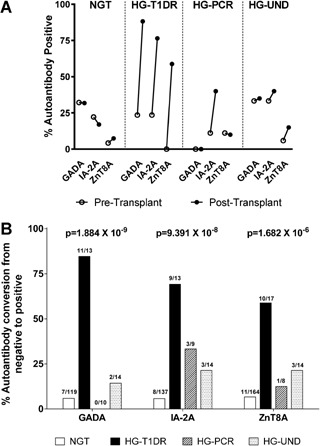
Pre‐ and posttransplant autoantibody prevalence and conversion. (A) Pre‐ and posttransplant prevalence of GADA, IA‐2A, and ZnT8A in 223 SPK patients according to group. (B) Prevalence of conversion from autoantibody negative to positive for GADA, IA‐2A, and ZnT8A in 223 SPK patients according to group. P values were calculated using paired Fisher's exact test. For both panels, ZnT8A data are shown for 215 patients since pretransplant ZnT8A could not be assessed in eight patients. GADA, 65‐kDa glutamic acid decarboxylase isoform autoantibody; HG‐PCR, hyperglycemia with pancreas chronic rejection; HG‐T1DR, hyperglycemia with type 1 diabetes recurrence; HG‐UND, hyperglycemia of undetermined cause; ZnT8A, ZnT8 autoantibody.
Figure 4.
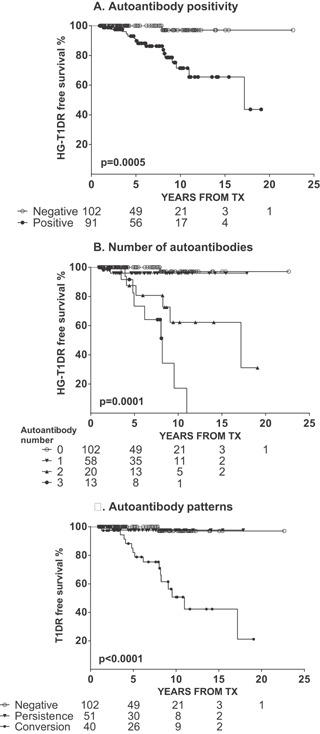
Kaplan–Meier analysis of HG‐T1DR‐free survival according to autoantibodies on follow‐up. (A) Autoantibody positivity: positive versus negative, HR = 14.53, p = 0.0005; (B) number of autoantibodies: overall, p = 0.0001; 1 versus 0, HR = 3.53, p = 0.2672; 2 versus 0, HR = 14.60, p = 0.0005*; 3 versus 0, HR = 53.88, p < 0.0001*; 2 versus 1, HR = 5.65, p = 0.0131**; 3 versus 1, HR = 17.04, p<0.0001*; 3 versus 2, HR = 3.00, p = 0.0261**; p value *was or **was not significant after correction for multiple comparisons. (C) Autoantibody conversion:overall, p<0.0001; persistence versus negative, HR = 1.88, p = 0.6486; conversion versus negative, HR = 27.11, p < 0.0001*; conversion versus persistence, HR = 15.57, p = 0.0003*; *significant p values after correction for multiple comparisons. HG‐T1DR, hyperglycemia with type 1 diabetes recurrence.
Figure 5.
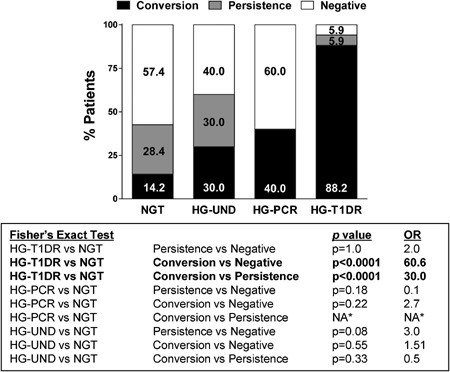
Prevalence of autoantibody conversion, persistence, and negativity in the groups studied. Results of statistical comparisons are shown in the text box. *There were no patients in the HG‐PCR group with autoantibody persistence. HG‐PCR, hyperglycemia with pancreas chronic rejection; HG‐T1DR, hyperglycemia with type 1 diabetes recurrence; NGT, normal glucose tolerance.
Autoantibody patterns in 17 patients with T1D recurrence
The autoantibody patterns of the 17 HG‐T1DR patients are shown in Table S1C. Most HG‐T1DR patients had autoantibody conversions and/or multiple autoantibodies. The average time from transplantation to the first autoantibody conversion was 3.92 ± SD 3.16 years (range 0.49–11.52 years). The mean time from the first autoantibody conversion to HG‐T1DR was 2.69 ± SD 2.06 years (range 0–5.85 years). We compared time from transplant to conversion, from conversion HG‐T1DR, and duration of positivity, for each autoantibody (Figure 6); time from transplant to conversion was similar, but time from ZnT8A conversion to HG‐T1DR (mean 0.66 ± SEM 0.34 years) was shorter compared to IA‐2A (mean 3.36 ± SEM 0.59 years, p = 0.0004) and to GADA (mean 2.27 ± SEM 0.75 years, p = 0.053). The duration of ZnT8A positivity (mean 2.03 ± SEM 0.43 years) was shorter compared to IA‐2A (mean 8.54 ± SEM 1.71 years, p=0.0007) and GADA (mean 5.87 ± SEM 1.73 years, p = 0.030).
Figure 6.
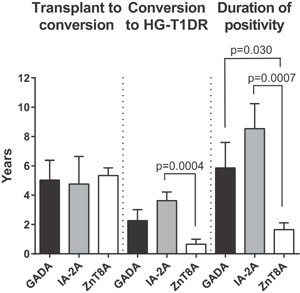
Assessment of time elapsed from transplant to autoantibody conversion, from conversion to diagnosis, and duration of autoantibody positivity in patients with T1D recurrence. Data shown as mean ± SEM years. P values of statistically significant comparisons are shown. Data for each patient are presented in Table S1C. GADA, 65‐kDa glutamic acid decarboxylase isoform autoantibody; HG‐T1DR, hyperglycemia with type 1 diabetes recurrence; IA‐2A, IA‐2 autoantibody; ZnT8A, ZnT8 autoantibody.
Analysis of donor–recipient SLC30A8 gene mismatch
ZnT8 is a cation efflux protein involved in the insulin secretory pathway 30, 31 and is encoded by the SLC30A8 gene. SLC30A8 polymorphisms affect the amino acid sequence at residue 325, an essential determinant of antigenicity 32. We investigated whether a donor–recipient genetic mismatch at this locus was associated with ZnT8A and T1D recurrence in 19 donor–recipient pairs (Table S4). Among 10 donor–recipient pairs with a mismatched SLC30A8 genotype, 5 had a mismatch introducing a donor allele not present in the recipient. Comparing to matched or partially mismatched donors, or both, we found no significant differences in the prevalence of T1D recurrence and ZnT8A.
Influence of HLA types and donor–recipient HLA sharing on the development of autoantibody conversion and T1DR
We analyzed the frequency of HLA‐A, ‐B, and ‐DR alleles associated with T1D risk 33. HG‐T1DR patients had increased frequency of the high‐risk DR3/DR4 genotype (52.9%) compared to NGT patients (24.4%, p = 0.02, odds ratio [OR] = 3.5, Figure 7A). By Kaplan–Meier analysis, this genotype was associated with higher risk of autoantibody conversion (HR = 1.8, p = 0.0488) and T1D recurrence (HR = 2.40, p=0.055) (Figure S6A). Seven HG‐T1DR patients and 9 NGT patients had both conversion and HLA‐DR3/DR4 versus 0 HG‐T1DR and 79 NGT patients who were autoantibody negative and did not carry this genotype (p<0.0001, OR = 125.5). Thus, having both autoantibody conversion and HLA‐DR3/DR4 conferred much higher risk than having neither, than HLA‐DR3/DR4 (OR = 3.5, p = 0.02) or autoantibody conversion alone (OR = 60.6, p < 0.0001, Figure 5). When examining donor–recipient HLA sharing, class II HLA‐DR alleles (any allele, and DR3) were shared at higher frequency in the HG‐T1DR group compared to NGT (any DR, p = 0.002; DR3, p = 0.003, Figure 7B). Survival analysis provides further evidence of an effect of HLA sharing on the development of T1D recurrence (sharing any HLA‐DR, p = 0.0075, HR = 4.57, sharing HLA‐DR3, p = 0.0421, HR = 2.4, Figure S6D and F).
Figure 7.
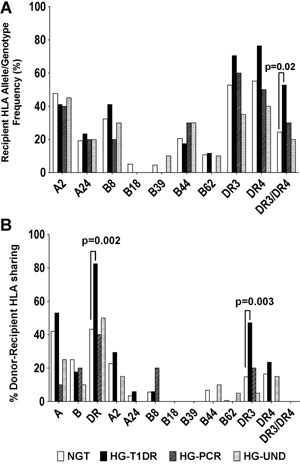
Influence of HLA on development of type 1 diabetes recurrence. (A) Frequencies of T1D‐predisposing HLA variants among simultaneous pancreas–kidney recipients, according to group (HLA‐DR3/DR4, normal glucose tolerance [NGT] versus hyperglycemia with type 1 diabetes recurrence [HG‐T1DR], p = 0.02; RR = 2.16; OR = 3.5; positive predictive value [PPV] = 0.53; sensitivity = 0.17; specificity = 0.94). (B) Frequency of donor–recipient sharing for the HLA types shown. Statistical differences were found for sharing of any HLA‐DR (NGT versus HG‐T1DR, p = 0.002; RR = 3.2; OR = 6.14; PPV = 0.57; sensitivity = 0.97; specificity = 0.16) and of HLA‐DR3 (NGT versus HG‐T1DR, p = 0.003; RR = 1.6; OR = 5.1; PPV = 0.85; sensitivity = 0.94; specificity = 0.23). For both panels, significant p values are shown calculated with the Fisher's exact test.
Discussion
T1D recurrence in pancreas transplant recipients is considered rare. Sibley et al 34 examined 100 pancreas grafts from the 1978 to 1986 period. There were no cases of T1D recurrence among fully immunosuppressed recipients of pancreas grafts from deceased donors. Instead, T1D recurrence was observed in nine identical twins or HLA‐identical siblings who received no or minimal immunosuppression for living‐related grafts 13, 14, 15. Later studies contributed evidence that recurrence of islet autoimmunity may occur regardless of HLA matching 18, 24 and despite immunosuppression 11, 12, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 35. We initially reported 11 three patients in whom T1D recurrence was characterized by 1 hyperglycemia requiring insulin with impaired insulin secretion; 2 seroconversion of autoantibodies years prior to recurrence; 3 circulating autoreactive T cells around the time of diagnosis, often with simultaneous detection in pancreas transplant and associated lymph nodes; 4 insulitis and/or beta cell loss in the pancreas transplant biopsy, with no or minimal rejection; and 5 lack of laboratory evidence of rejection (unchanged urine amylase and serum creatinine levels).
Our study represents the largest evaluation of islet autoimmunity and T1D recurrence in SPK recipients. About one third developed posttransplant diabetes. The three categories of posttransplant diabetes had relatively similar frequency. T1D recurrence was shown in 7.6% of the patients, or 36.2% of the HG group; the diagnosis was validated by biopsy demonstrating insulitis and/or beta cell loss in most patients (15/17) (Table S1A), and thus the prevalence of T1D recurrence confirmed by biopsy was 5.8%. T1D recurrence usually developed on average 6.8 years after transplantation (Table 1), with a wide range of 2.2 to 17.2 years, suggesting that the process can be quite chronic. Some patients also had biopsy evidence of rejection, which was most often minimal to moderate, and only five had decreased urine amylase levels, suggesting that recurrent islet autoimmunity and rejection may at times coexist.
We assessed whether T1D recurrence could have been affected by immunosuppression regimens used over time. We could not identify major effects of maintenance therapy. A small group of patients who did not receive induction therapy experienced higher incidence of T1D recurrence compared to those who had induction; regimens were not compared in a randomized trial and patients were enrolled in different time periods, yet the findings suggest that induction may protect from recurrence, possibly by depleting autoreactive T cells present at transplantation, which could include memory autoreactive T cells that can be reactivated on follow‐up 36, 37. Importantly, most (157/223, 70%) of the patients studied had been treated with our current immunosuppressive protocol 38, 39, 40.T1D recurrence has continued to occur with the current regimen with an overall prevalence of about 4.6% during the 2000–2013 period. Therefore, current immunosuppression may not prevent recurrence in all patients.
We examined whether T1D‐associated autoantibodies are risk factors for T1D recurrence in our SPK cohort. Earlier but smaller studies examined GADA, IA‐2A, islet cell antibodies and/or insulin autoantibodies in pancreas transplant recipients and reported associations with graft failure, albeit in the absence of biopsy data or analysis of autoreactive T cell responses 22, 23, 24, 25, 29. Similar to these earlier studies, we find that pretransplant autoantibody positivity does not predict future pancreas graft endocrine function 25, 29. In contrast, posttransplant autoantibody positivity, multiple autoantibodies, and especially autoantibody conversion were strong predictors of T1D recurrence, similar to the associations reported in prospective studies of relatives 5, 7. These associations were specific for T1D recurrence, and no associations were found with posttransplant diabetes when this had an undetermined cause or was ascribed to chronic pancreas rejection; a report suggested that pretransplant GADA could be relevant to acute rejection 41. Persistence of autoantibodies did not confer increased risk. While previous studies have noted associations of autoantibodies with graft failure 25, 29, our study is the first to link autoantibodies specifically to biopsy‐confirmed T1D recurrence, and in our own ongoing studies these are being linked to autoimmune T cell responses (Table S1A) 11, 42.
We also evaluated ZnT8A in our SPK recipients. So far, ZnT8A in transplantation have been studied only in a small cohort of 25 recipients of solitary pancreas transplants 28, in whom they predicted loss of pancreas graft function. In our study, ZnT8A were only detected with the high‐risk conversion pattern. Similar to prospective studies of relatives and patients 43, 44, ZnT8A were the last autoantibodies to become positive on follow‐up, positivity was short lived, and their appearance was much closer to the development of diabetes symptoms (0.66 years vs. 2.27 and 3.63 years for GADA and IA‐2A, respectively). Thus, ZnT8A positivity raises the concern that diabetes recurrence may occur in the near future. As the number of autoantibodies was associated with higher risk (Figure 4B), ZnT8A should be used with GADA and IA‐2A testing to identify SPK patients at risk of T1D recurrence.
We examined whether genetic factors could be identified that increase the risk of T1D recurrence. While limited by the small number of informative pairs, our study does not support a donor–recipient mismatch at the SLC30A8 locus as a risk factor for T1D recurrence and ZnT8A. In contrast, we found associations with HLA genes. Given the well‐known associations of certain HLA types with T1D 33, we analyzed the frequency of HLA‐A, ‐B, and ‐DR alleles in the SPK recipients as well as HLA sharing with their respective donors. The high‐risk HLA‐DR3/DR4 heterozygous genotype was associated with T1D recurrence; HG‐T1DR SPK patients carried this genotype twice as often as NGT recipients. No association was found with either HLA‐DR4 or HLA‐DR3 alleles, suggesting that T1D recurrence is more common in those with the highest HLA risk. Patients with T1D recurrence also shared HLA‐DR alleles twice as frequently with their donors, an association largely sustained by increased sharing of HLA‐DR3. The HLA‐DR3/DR4 genotype in the recipient and HLA‐DR sharing with the donor were also associated with autoantibody conversions. Importantly, having both autoantibody conversion and HLA‐DR3/DR4 was associated with twice as high the risk of T1D recurrence compared to autoantibody conversion alone. Given the limited number of cases available, we did not apply the Bonferroni correction to the multiple HLA comparisons, yet our data suggest that HLA genes may be important risk factors for the recurrence of islet autoimmunity and T1D in SPK recipients.
We conclude that T1D recurrence is not an uncommon cause of posttransplant diabetes. Our studies identify several risk factors for this condition that could be assessed in transplantation clinics. Autoantibody conversion precedes diabetes symptoms by a few years and is a strong risk factor, especially in those with the HLA‐DR3/DR4 high‐risk genotype. Because ZnT8A positivity equates to autoantibody conversion, it confers high risk of recurrence and typically predicts an imminent diagnosis. Thus, ZnT8A can aid in staging progression of T1D recurrence, a process that in many patients proceeds slowly but will eventually lead to loss of graft endocrine function. The field will benefit from larger studies, to expand and replicate these findings, and possibly identify a sufficient number of patients in whom new therapies can be meaningfully evaluated, given that current immunosuppression cannot prevent T1D recurrence in all patients.
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Supporting information
Supplemental Materials and Methods
Figure S1: Kaplan–Meier analysis of hyperglycemia (HG)‐free survival according to pretransplant autoantibody status, by HG groups.
Figure S2: Kaplan–Meier analysis of hyperglycemia (HG)‐free survival according to pretransplant autoantibody number, by HG groups.
Figure S3: Kaplan–Meier analysis of hyperglycemia‐free survival according to autoantibody status on follow‐up, for hyperglycemia with pancreas chronic rejection (HG‐PCR) and hyperglycemia of undetermined cause (HG‐UND) groups.
Figure S4: Kaplan–Meier analysis of hyperglycemia‐free survival according to autoantibody number on follow‐up, for hyperglycemia with pancreas chronic rejection (HG‐PCR) and hyperglycemia of undetermined cause (HG‐UND) groups.
Figure S5: Kaplan–Meier analysis of hyperglycemia‐free survival according to autoantibody patterns on follow‐up, for hyperglycemia with pancreas chronic rejection (HG‐PCR) and hyperglycemia of undetermined cause (HG‐UND) groups.
Figure S6: Kaplan–Meier analysis of autoantibody conversion and HG‐T1DR free survival according to recipient HLA genotype and donor‐recipient HLA‐sharing.
Table S1A: Clinical, genetic, immunological, and biopsy data for 17 patients with HG‐T1DR.
Table S1B: Assessment of rejection in 17 patients with hyperglycemia with type 1 diabetes recurrence.
Table S1C: Autoantibody status, duration of positivity, and time to events in 17 patients with hyperglycemia with type 1 diabetes recurrence.
Table S2: Pre‐ and posttransplant autoantibody prevalence.
Table S3: Specificity and sensitivity for single and multiple autoantibodies for the development of type 1 diabetes recurrence.
Table S4: SLC30A8 locus DNA‐typing data for 19 simultaneous pancreas–kidney donor–recipient pairs.
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01 DK070011, R01 DK052068), the JDRF (17‐2011‐594, 17‐2012‐3), the American Diabetes Association (RA‐1‐09‐RA‐413), the John C. Hench Foundation, and by the Diabetes Research Institute Foundation, Hollywood, Florida. We are thankful to our research nurses, Lissett Tueros, Lois Hanson, and Sandra Flores. Ami Charmley at the Benaroya Research Institute performed the SLC30A8 typing. We are indebted to our patients for their participation in the study.
[The copyright line for this article was changed on 22 September, 2016 after original online publication.]
Contributor Information
G. W. Burke, III, Email: gburke@med.miami.edu.
A. Pugliese, Email: apuglies@med.miami.edu
References
- 1. Pugliese A. The multiple origins of type 1 diabetes. Diabet Med 2013; 30: 135–146. [DOI] [PubMed] [Google Scholar]
- 2. Verge CF, Gianani R, Kawasaki E, Yu L, Pietropaolo M, Chase HP, et al. Number of autoantibodies (against insulin, GAD or ICA512/IA2) rather than particular autoantibody specificities determines risk of type I diabetes. J Autoimmun 1996; 9: 379–83. [DOI] [PubMed] [Google Scholar]
- 3. Verge CF, Gianani R, Kawasaki E, et al. Prediction of type I diabetes in first‐degree relatives using a combination of insulin, GAD, and ICA512bdc/IA‐2 autoantibodies. Diabetes 1996; 45: 926–933. [DOI] [PubMed] [Google Scholar]
- 4. Krischer JP, Cuthbertson DD, Yu L, et al. Screening strategies for the identification of multiple antibody‐positive relatives of individuals with type 1 diabetes. J Clin Endocrinol Metab 2003; 88: 103–108. [DOI] [PubMed] [Google Scholar]
- 5. Vehik K, Beam CA, Mahon JL, et al. Development of autoantibodies in the TrialNet Natural History Study. Diabetes Care 2011; 34: 1897–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sosenko JM, Skyler JS, Palmer JP, et al. A longitudinal study of GAD65 and ICA512 autoantibodies during the progression to type 1 diabetes in Diabetes Prevention Trial‐Type 1 (DPT‐1) participants. Diabetes Care 2011; 34: 2435–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ziegler AG, Rewers M, Simell O, et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 2013; 309: 2473–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. White SA, Shaw JA, Sutherland DE. Pancreas transplantation. Lancet 2009; 373: 1808–1817. [DOI] [PubMed] [Google Scholar]
- 9. Dean PG, Kudva YC, Larson TS, Kremers WK, Stegall MD. Posttransplant diabetes mellitus after pancreas transplantation. Am J Transplant 2008; 8: 175–182. [DOI] [PubMed] [Google Scholar]
- 10. Neidlinger N, Singh N, Klein C, et al. Incidence of and risk factors for posttransplant diabetes mellitus after pancreas transplantation. Am J Transplant 2010; 10: 398–406. [DOI] [PubMed] [Google Scholar]
- 11. Vendrame F, Pileggi A, Laughlin E, et al. Recurrence of type 1 diabetes after simultaneous pancreas‐kidney transplantation, despite immunosuppression, is associated with autoantibodies and pathogenic autoreactive CD4 T‐cells. Diabetes 2010; 59: 947–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gruessner RW, Pugliese A, Reijonen HK, et al. Development of diabetes mellitus in living pancreas donors and recipients. Expert Rev Clin Immunol 2011; 7: 543–551. [DOI] [PubMed] [Google Scholar]
- 13. Sutherland DE, Sibley R, Xu XZ, et al. Twin‐to‐twin pancreas transplantation: Reversal and reenactment of the pathogenesis of type I diabetes. Trans Assoc Am Physicians 1984; 97: 80–87. [PubMed] [Google Scholar]
- 14. Sibley RK, Sutherland DE, Goetz F, Michael AF. Recurrent diabetes mellitus in the pancreas iso‐ and allograft: a light and electron microscopic and immunohistochemical analysis of four cases. Lab Invest 1985; 53: 132–144. [PubMed] [Google Scholar]
- 15. Sutherland DE, Goetz FC, Sibley RK. Recurrence of disease in pancreas transplants. Diabetes 1989; 38(Suppl 1): 85–87. [DOI] [PubMed] [Google Scholar]
- 16. Nakhleh RE, Gruessner RW, Swanson PE, et al. Pancreas transplant pathology: A morphologic, immunohistochemical, and electron microscopic comparison of allogeneic grafts with rejection, syngeneic grafts, and chronic pancreatitis. Am J Surg Pathol 1991; 15: 246–256. [PubMed] [Google Scholar]
- 17. Santamaria P, Nakhleh RE, Sutherland DE, Barbosa JJ. Characterization of T lymphocytes infiltrating human pancreas allograft affected by isletitis and recurrent diabetes. Diabetes 1992; 41: 53–61. [DOI] [PubMed] [Google Scholar]
- 18. Bosi E, Bottazzo GF, Secchi A, et al. Islet cell autoimmunity in type I diabetic patients after HLA‐mismatched pancreas transplantation. Diabetes 1989; 38(Suppl 1): 82–84. [DOI] [PubMed] [Google Scholar]
- 19. Dieterle CD, Hierl FX, Gutt B, et al. Insulin and islet autoantibodies after pancreas transplantation. Transpl Int 2005; 18: 1361–1365. [DOI] [PubMed] [Google Scholar]
- 20. Lohmann T, Klemm T, Geissler F, et al. Islet cell‐specific autoantibodies as potential markers for recurrence of autoimmune type 1 diabetes after simultaneous pancreas‐kidney transplantation. Transplant Proc 2002; 34: 2249–2250. [DOI] [PubMed] [Google Scholar]
- 21. Sundkvist G, Tyden G, Karlsson FA, Bolinder J. Islet autoimmunity before and after pancreas transplantation in patients with type I diabetes mellitus [letter] [In Process Citation]. Diabetologia 1998; 41: 1532–1533. [DOI] [PubMed] [Google Scholar]
- 22. Esmatjes E, Rodriguez‐Villar C, Ricart MJ, et al. Recurrence of immunological markers for type 1 (insulin‐dependent) diabetes mellitus in immunosuppressed patients after pancreas transplantation. Transplantation 1998; 66: 128–131. [DOI] [PubMed] [Google Scholar]
- 23. Thivolet C, Abou‐Amara S, Martin X, et al. Serological markers of recurrent beta cell destruction in diabetic patients undergoing pancreatic transplantation. Transplantation 2000; 69: 99–103. [DOI] [PubMed] [Google Scholar]
- 24. Petruzzo P, Andreelli F, McGregor B, et al. Evidence of recurrent type I diabetes following HLA‐mismatched pancreas transplantation. Diabetes Metab 2000; 26: 215–218. [PubMed] [Google Scholar]
- 25. Braghi S, Bonifacio E, Secchi A, Di Carlo V, Pozza G, Bosi E. Modulation of humoral islet autoimmunity by pancreas allotransplantation influences allograft outcome in patients with type 1 diabetes. Diabetes 2000; 49: 218–224. [DOI] [PubMed] [Google Scholar]
- 26. Ishida‐Oku M, Iwase M, Sugitani A, et al. A case of recurrent type 1 diabetes mellitus with insulitis of transplanted pancreas in simultaneous pancreas‐kidney transplantation from cardiac death donor. Diabetologia 2010; 53: 341–345. [DOI] [PubMed] [Google Scholar]
- 27. Assalino M, Genevay M, Morel P, Demuylder‐Mischler S, Toso C, Berney T. Recurrence of type 1 diabetes after simultaneous pancreas‐kidney transplantation in the absence of GAD and IA‐2 autoantibodies. Am J Transplant 2012; 12: 492–495. [DOI] [PubMed] [Google Scholar]
- 28. Occhipinti M, Lampasona V, Vistoli F, et al. Zinc transporter 8 autoantibodies increase the predictive value of islet autoantibodies for function loss of technically successful solitary pancreas transplant. Transplantation 2011; 92: 674–677. [DOI] [PubMed] [Google Scholar]
- 29. Martins LS, Henriques AC, Fonseca IM, et al. Pancreatic autoantibodies after pancreas‐kidney transplantation—do they matter? Clin Transplant 2014; 28: 462–469. [DOI] [PubMed] [Google Scholar]
- 30. Chimienti F, Devergnas S, Pattou F, et al. In vivo expression and functional characterization of the zinc transporter ZnT8 in glucose‐induced insulin secretion. J Cell Sci 2006; 119: 4199–4206. [DOI] [PubMed] [Google Scholar]
- 31. Davidson HW, Wenzlau JM, O'Brien RM. Zinc transporter 8 (ZnT8) and beta cell function. Trends Endocrinol Metabol TEM 2014; 25: 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wenzlau JM, Liu Y, Yu L, et al. A common nonsynonymous single nucleotide polymorphism in the SLC30A8 gene determines ZnT8 autoantibody specificity in type 1 diabetes. Diabetes 2008; 57: 2693–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Noble JA, Erlich HA. Genetics of type 1 diabetes. Cold Spring Harb Perspect Med 2012; 2: a007732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sibley RK, Sutherland DE. Pancreas transplantation: An immunohistologic and histopathologic examination of 100 grafts. Am J Pathol 1987; 128: 151–170. [PMC free article] [PubMed] [Google Scholar]
- 35. Tyden G, Reinholt FP, Sundkvist G, Bolinder J. Recurrence of autoimmune diabetes mellitus in recipients of cadaveric pancreatic grafts. N Engl J Med 1996; 335: 860–863. [DOI] [PubMed] [Google Scholar]
- 36. Monti P, Heninger AK, Bonifacio E. Differentiation, expansion, and homeostasis of autoreactive T cells in type 1 diabetes mellitus. Curr Diab Rep 2009; 9: 113–118. [DOI] [PubMed] [Google Scholar]
- 37. Huurman VA, Hilbrands R, Pinkse GG, et al. Cellular islet autoimmunity associates with clinical outcome of islet cell transplantation. PLoS One 2008; 3: e2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Burke GW, Ciancio G, Olson L, Roth D, Miller J. Ten‐year survival after simultaneous pancreas/kidney transplantation with bladder drainage and tacrolimus‐based immunosuppression. Transplant Proc 2001; 33: 1681–1683. [DOI] [PubMed] [Google Scholar]
- 39. Burke GW, III., Kaufman DB, Millis JM, et al. Prospective, randomized trial of the effect of antibody induction in simultaneous pancreas and kidney transplantation: Three‐year results. Transplantation 2004; 77: 1269–1275. [DOI] [PubMed] [Google Scholar]
- 40. Ciancio G, Sageshima J, Chen L, et al. Advantage of rapamycin over mycophenolate mofetil when used with tacrolimus for simultaneous pancreas kidney transplants: Randomized, single‐center trial at 10 years. Am J Transplant 2012; 12: 3363–3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ringers J, van der Torren CR, van de Linde P, et al. Pretransplantation GAD‐autoantibody status to guide prophylactic antibody induction therapy in simultaneous pancreas and kidney transplantation. Transplantation 2013; 96: 745–752. [DOI] [PubMed] [Google Scholar]
- 42. Laughlin E, Burke G, Pugliese A, Falk B, Nepom GT. Recurrence of autoreactive antigen‐specific CD4+ T cells in autoimmune diabetes after pancreas transplantation. Clin Immunol 2008; 128: 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wenzlau JM, Juhl K, Yu L, et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci USA 2007; 104: 17040–17045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wenzlau JM, Walter M, Gardner TJ, et al. Kinetics of the post‐onset decline in zinc transporter 8 autoantibodies in type 1 diabetic human subjects. J Clin Endocrinol Metab 2010; 95: 4712–4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Materials and Methods
Figure S1: Kaplan–Meier analysis of hyperglycemia (HG)‐free survival according to pretransplant autoantibody status, by HG groups.
Figure S2: Kaplan–Meier analysis of hyperglycemia (HG)‐free survival according to pretransplant autoantibody number, by HG groups.
Figure S3: Kaplan–Meier analysis of hyperglycemia‐free survival according to autoantibody status on follow‐up, for hyperglycemia with pancreas chronic rejection (HG‐PCR) and hyperglycemia of undetermined cause (HG‐UND) groups.
Figure S4: Kaplan–Meier analysis of hyperglycemia‐free survival according to autoantibody number on follow‐up, for hyperglycemia with pancreas chronic rejection (HG‐PCR) and hyperglycemia of undetermined cause (HG‐UND) groups.
Figure S5: Kaplan–Meier analysis of hyperglycemia‐free survival according to autoantibody patterns on follow‐up, for hyperglycemia with pancreas chronic rejection (HG‐PCR) and hyperglycemia of undetermined cause (HG‐UND) groups.
Figure S6: Kaplan–Meier analysis of autoantibody conversion and HG‐T1DR free survival according to recipient HLA genotype and donor‐recipient HLA‐sharing.
Table S1A: Clinical, genetic, immunological, and biopsy data for 17 patients with HG‐T1DR.
Table S1B: Assessment of rejection in 17 patients with hyperglycemia with type 1 diabetes recurrence.
Table S1C: Autoantibody status, duration of positivity, and time to events in 17 patients with hyperglycemia with type 1 diabetes recurrence.
Table S2: Pre‐ and posttransplant autoantibody prevalence.
Table S3: Specificity and sensitivity for single and multiple autoantibodies for the development of type 1 diabetes recurrence.
Table S4: SLC30A8 locus DNA‐typing data for 19 simultaneous pancreas–kidney donor–recipient pairs.