

Abstract
Dinuclear gold complexes have the ability to interact with one or more substrates in a dual‐activation mode, leading to different reactivity and selectivity than their mononuclear relatives. In this contribution, this difference was used to control the catalytic properties of a gold‐based catalytic system by site‐isolation of mononuclear gold complexes by selective encapsulation. The typical dual‐activation mode is prohibited by this catalyst encapsulation, leading to typical behavior as a result of mononuclear activation. This strategy can be used as a switch (on/off) for a catalytic reaction and also permits reversible control over the product distribution during the course of a reaction.
Keywords: catalyst encapsulation, catalytic switch, dual catalysis, gold catalysis, supramolecular chemistry
There is a growing interest in transition‐metal catalysis in confined spaces as the approach provides an additional tool to control selectivity and activity in catalysis.1 For example, it has been demonstrated that the encapsulation of rhodium complexes in a hemispherical porphyrin assembly results in catalysts with increased activity and unprecedented branched selectivity in the hydroformylation of terminal and internal alkenes.2 In addition, the encapsulation of transition‐metal complexes in preformed cavities through weak interactions can lead to unexpected reactivity: M4L6 anionic tetrahedral capsules, for instance, can host cationic organometallic catalysts in their hydrophobic cavities,3 thereby inducing substrate selectivity.4 Moreover, reaction rates5 and product distribution6 can be greatly affected by catalyst encapsulation. Interestingly, many catalytic reactions operate through a dinuclear mechanism7 or deactivate via a dinuclear pathway;8, 9 by encapsulation of a transition metal complex, such decomposition pathways can be suppressed, leading to higher catalytic turnover numbers.10
In principle, an encapsulation event could also change the catalytic pathway of a reaction, and as such can be used as a switch for a catalytic transformation. Switchable catalysis is an interesting upcoming field of research as it provides new tools to control the reaction process with external stimuli such as light, pH, or metal coordination, a concept that is important to control reactions in nature.11 In a recent study, encapsulation of a photoredox catalyst was shown to be a feasible stimulus to steer reactivity.12 In a previous paper we reported the encapsulation of an N‐heterocyclic carbene (NHC) mononuclear gold(I) complex inside a self‐assembled hexameric resorcin[4]arene cage 1 6 and showed that the encapsulated catalyst gives a different product distribution than the free complex.13 This supramolecular complex is formed in apolar, water‐saturated solvents (Scheme 1).14, 15 In the current contribution, we report how we change the active gold complex from dinuclear to mononuclear by reversible encapsulation and demonstrate that this can be used for both switching on/off a reaction, as well as for controlling its selectivity during the course of a reaction. Changing the reactivity or selectivity of a gold catalyst has been shown before by changing the ligands16 or Brønsted acid/base effects17 and even by guest binding by a rotaxane,18 but reversibly changing the active species of a gold‐catalyzed reaction as a result of encapsulation has, to the best of our knowledge, not been shown before.
Scheme 1.
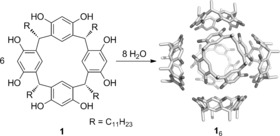
Formation of the self‐assembled hexameric resorcin[4]arene cage.
In this study we use a dinuclear hydroxyl‐bridged gold complex [{Au(NHC)}2(μ‐OH)][X] instead of a mononuclear gold complex.19 This complex reacts through different mechanisms than mononuclear gold complexes20 and is a privileged catalyst for dual activation reactions.21 It, however, is too large to fit inside the cage 1 6, 22 and should be split into mononuclear complexes upon encapsulation. The NHC‐Au‐X fragment displays the traditional Lewis acidic character of mononuclear cationic gold catalysts, activating unsaturated substrates such as alkynes through π‐coordination, making them more electrophilic and susceptible towards nucleophilic attack.23 Meanwhile, the NHC‐Au‐OH species behaves as a Brønsted base and is known to σ‐activate substrates such as terminal alkynes or phenols.24 Together the two species, NHC‐Au‐X and NHC‐Au‐OH, can dually activate substrates through both π‐ and σ‐activation (Scheme 2). This dual‐activation mode was originally proposed by Toste and Houk25 and is well established and employed nowadays. In addition, this dual activation has elegantly been explored by Hashmi26 and Zhang27 for the formation of gold acetylides, which can react as a nucleophile and attack the π‐activated bond of the substrate, and as such represents a common strategy in gold‐mediated synthesis.
Scheme 2.
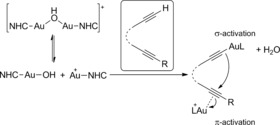
Dinuclear complexes [{Au(NHC)}2(μ‐OH)][X] can dually activate substrates.
We envisioned that encapsulation of the two fragments of the dinuclear gold catalyst [{Au(NHC)}2(μ‐OH)][X] in separate cages would alter its typical reactivity as they undergo transformations in a site isolated fashion. We anticipated that by complex encapsulation we could reversibly switch between the dinuclear and mononuclear catalyst and, therefore, have a tool to shift from dual gold catalysis to a mononuclear reaction mechanism. This forms the basis for an on/off switchable system, but can also alter the product distribution during a gold‐catalyzed transformation.
Indeed, when complex [{Au(IPr)}2(μ‐OH)][BF4] 2 (Scheme 3) and the capsule were mixed together, encapsulation of a mononuclear gold complex was confirmed by 1H NMR and 1H 2D DOSY NMR (see the Supporting Information). To demonstrate the principle of switching catalyst activity by selective encapsulation, the dual gold‐catalyzed hydrophenoxylation reaction was studied.21, 28 This reaction requires σ‐activation of a phenol by the NHC‐Au‐OH moiety and π‐activation of an alkyne by the cationic NHC‐Au‐X fragment (Scheme 4). Indeed, it was observed that the standard reaction between phenol (4) and diphenylacetylene (5) readily takes place when using 2 as catalyst in the absence of the cage (full conversion within 60 min). However, in the presence of the cage, the dinuclear complex 2 is broken and encapsulated as mononuclear species. As the dual reaction pathway is no longer available, no conversion to vinyl ether 6 is obtained, even after 24 h.29 We wondered if the reaction could be switched on again by adding a competing guest that would bind more strongly to the cage than the gold catalyst. For this purpose we selected tetraalkylammonium salts, as they are known to bind very strongly inside the hexamer.15 Gratifyingly, upon addition of Et4N+BF4 − (7) to expel the gold catalyst from the cage, the catalytic activity was restored and product 6 was obtained in 89 % yield after one hour.
Scheme 3.
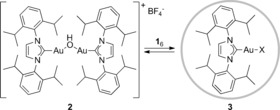
Encapsulation of [{Au(IPr)}2(μ‐OH)][BF4] in capsule 1 6.
Scheme 4.
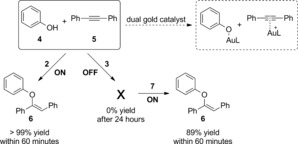
Switching the gold‐catalyzed hydrophenoxylation off and on. Reaction conditions: solvent=toluene, T=80 °C, [4]=550 mm, [5]=500 mm, [2]=2.5 mm (0.025 mol %), [1]=50 mm.
Next we explored the cage effect on changing the product distribution of a reaction. For this purpose we explored the conversion of 4‐phenyl‐1‐butyne (8). Depending on the available reaction pathways, the transformation can yield up to four products (Table 1). In the absence of the cage, 2 can activate two substrates in a σ‐ and π‐activation mode and, therefore, dimerization of 8 takes place through a dual‐activation mechanism, yielding the branched and linear products, 11 and 12 respectively (Table 1, entry 1; Scheme 5).26b, 30 As anticipated, this dual‐activation reaction pathway is completely blocked upon encapsulation of the catalyst and the production of 11 and 12 stopped. Instead, when in the cage, the catalyst produced the intramolecularly cyclized product 10, which is known to form via a mononuclear pathway inside the cage (Table 1, entry 2).13 In the presence of the cage and the competing guest 7, product 10 was not formed and dimerization products 11 and 12 were obtained (Table 1, entry 3), indicating that under these conditions the catalysis proceeds via a dual‐gold mechanism and thus takes place outside the cage.
Table 1.
Gold catalyzed conversion of 4‐phenyl‐1‐butyne.[a]
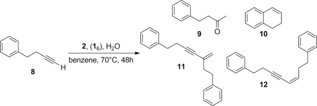
| ||||||
|---|---|---|---|---|---|---|
| Entry | 1 6 | 7 | Yield 9 [%] | Yield 10 [%] | Yield 11 [%] | Yield 12 [%] |
| 1 | − | − | 7 | 0 | 38 | 8 |
| 2 | + | − | 3 | 16 | 0 | 0 |
| 3 | + | + | 47 | 0 | 28 | 4 |
| 4 | − | + | 6 | 0 | 47 | 8 |
[a] Reaction conditions: [8]=66 mm, [2]=1.7 mm (2.5 mol %), [H2O]=44 mm, [1]=33 mm, [7]=33 mm. Yields are averaged from two measurements.
Scheme 5.
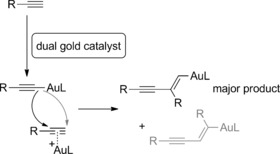
Dimerization of terminal alkynes through σ‐ and π‐activation.
Due to the presence of water molecules to facilitate the self‐assembly of the capsule, the well‐documented hydration product 9 was formed in all cases through π‐activation of the triple bond,31, 32 but only in minor amounts. We observed an increased rate of formation of this product in the presence of both cage and competing guest 7, which is interesting but difficult to explain. The addition of only the competing guest 7, in the absence of cage but with the same amount of water present, does not lead to increased formation of product 9 (Table 1, entry 4).
Next we investigated whether it would be possible to switch the product selectivity during the course of the reaction (Figure 1). The reaction was initiated using only the dinuclear gold catalyst 2. Under these reaction conditions, the branched dimer 11 was the main product formed, indicating that the dual activation pathway is dominating. After 3 h, an excess of cage 16 was added, thereby removing the dinuclear gold complex from the reaction mixture by encapsulation of the cationic mononuclear gold fragment. The formation of 11 immediately stopped, while production of compound 10 started. After another 3 h, compound 7 was added as a competing guest for 16, releasing the catalyst from the cage. As compound 10 can only be formed while the cationic gold species is encapsulated in 16, its production stopped, whereas formation of 11 started again. The formation of product 12 followed the same trend as 11, but it was less clear as this compound was formed in much smaller amounts. As ketone 9 can be formed both inside and outside the cage (Table 1), the formation of this product could not be switched; the faster formation of this product after addition of both cage and Et4N+BF4 − (7) corresponds with the observations in Table 1.
Figure 1.
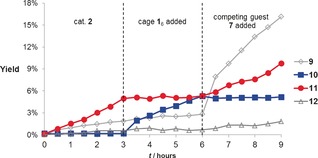
Formation of products from the reaction of substrate 8 in the presence of stepwise addition of cage and competing guest. Reaction conditions: [8]=66 mm, [2]=1.7 mm (2.5 mol %), [H2O]=44 mm, [1]=33 mm, [7]=33 mm. Yields are averaged from two measurements.
In conclusion, we have demonstrated that it is possible to control the pathway by which gold complexes convert substrate molecules by complex encapsulation events. In the presence of a self‐assembled hexameric resorcin[4]arene cage the dinuclear complex [{Au(NHC)}2(μ‐OH)][X] breaks into mononuclear units that are encapsulated as the dinuclear gold complex 2 is too large to fit inside the cage. By doing so, the dual activation reactivity typical for complex 2 is switched into (re)activity that is typical for mononuclear complexes. By using this strategy on a reaction that can only proceed through a dual‐activation pathway, we have demonstrated on/off switching of a gold‐catalyzed reaction. Moreover, this approach also provides tools to change the product distribution during the course of a reaction, which is demonstrated in the activation of 4‐phenyl‐1‐butyne. In summary, this strategy of switching the active species of a gold‐catalyzed reaction by means of host‐guest interactions provides a new approach to controlling catalytic transformations, even during the course of the reaction.
Experimental Section
Hydrophenoxylation experiments: The catalyst 2 (0.025 mol %) was added to a solution of 4 (550 mm) and 5 (500 mm) and in selected experiments cage 1 6 (50 mm) and/or competing guest 7 (50 mm) in [D8]toluene. The mixture was heated to 80 °C and yields were monitored using GC and 1H NMR of the crude mixture.
Catalytic experiments with substrate 8: The catalyst 2 (2.5 mol %), H2O (44 mm), substrate 8 (66 mm), and, in selected experiments, cage 1 6 (33 mm) and/or competing guest 7 (33 mm) were mixed in [D6]benzene and heated to 70 °C for 48 h. Yields were monitored using GC and 1H NMR of the crude mixture and were determined as the average of two experiments.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Support for this work was generously provided by King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under Award No. OSR‐2015‐CCF‐1974‐03 (to SPN) and by ERC grant nr. ERC‐2013‐AdG 339786 NAT‐CAT (to JNHR).
A. C. H. Jans, A. Gómez-Suárez, S. P. Nolan, J. N. H. Reek, Chem. Eur. J. 2016, 22, 14836.
References
- 1.For selected reviews on catalysis in confined spaces, see:
- 1a. Yoshizawa M., Klosterman J. K., Fujita M., Angew. Chem. Int. Ed. 2009, 48, 3418–3438; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 3470–3490; [Google Scholar]
- 1b. Wiester M. J., Ulmann P. A., Mirkin C. A., Angew. Chem. Int. Ed. 2011, 50, 114–137; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 118–142; [Google Scholar]
- 1c. Raynal M., Ballester P., Vidal-Ferran A., van Leeuwen P. W. N. M., Chem. Soc. Rev. 2014, 43, 1734–1787; [DOI] [PubMed] [Google Scholar]
- 1d. Zarra S., Wood D. M., Roberts D. A., Nitschke J. R., Chem. Soc. Rev. 2015, 44, 419–432; [DOI] [PubMed] [Google Scholar]
- 1e. Leenders S. H. A. M., Gramage-Doria R., de Bruin B., Reek J. N. H., Chem. Soc. Rev. 2015, 44, 433–448; [DOI] [PubMed] [Google Scholar]
- 1f. Brown C. J., Toste F. D., Bergman R. G., Raymond K. N., Chem. Rev. 2015, 115, 3012–3035; [DOI] [PubMed] [Google Scholar]
- 1g. Bistri O., Reinaud O., Org. Biomol. Chem. 2015, 13, 2849–2865. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Slagt V. F., Reek J. N. H., Kamer P. C. J., Van Leeuwen P. W. N. M., Angew. Chem. Int. Ed. 2001, 40, 4271–4274; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 4401–4404; [Google Scholar]
- 2b. Slagt V. F., Kamer P. C. J., van Leeuwen P. W. N. M., Reek J. N. H., J. Am. Chem. Soc. 2004, 126, 1526–1536; [DOI] [PubMed] [Google Scholar]
- 2c. Kuil M., Soltner T., van Leeuwen P. W. N. M., Reek J. N. H., J. Am. Chem. Soc. 2006, 128, 11344–11345; [DOI] [PubMed] [Google Scholar]
- 2d. Bocokić V., Kalkan A., Lutz M., Spek A. L., Gryko D. T., Reek J. N. H., Nat. Commun. 2013, 4, 2670. [DOI] [PubMed] [Google Scholar]
- 3. Fiedler D., Leung D. H., Bergman R. G., Raymond K. N., Acc. Chem. Res. 2005, 38, 349–358. [DOI] [PubMed] [Google Scholar]
- 4. Leung D. H., Bergman R. G., Raymond K. N., J. Am. Chem. Soc. 2007, 129, 2746–2747. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Wang Z. J., Brown C. J., Bergman R. G., Raymond K. N., Toste F. D., J. Am. Chem. Soc. 2011, 133, 7358–7360; [DOI] [PubMed] [Google Scholar]
- 5b. Kaphan D. M., Levin M. D., Bergman R. G., Raymond K. N., Toste F. D., Science 2015, 350, 1235–1238. [DOI] [PubMed] [Google Scholar]
- 6. Hart-Cooper W. M., Clary K. N., Toste F. D., Bergman R. G., Raymond K. N., J. Am. Chem. Soc. 2012, 134, 17873–17876. [DOI] [PubMed] [Google Scholar]
- 7. Hetterscheid D. G. H., Chikkali S. H., de Bruin B., Reek J. N. H., ChemCatChem 2013, 5, 2785–2793. [Google Scholar]
- 8. Walczuk E. B., Kamer P. C. J., van Leeuwen P. W. N. M., Angew. Chem. Int. Ed. 2003, 42, 4665–4669; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 4813–4817. [Google Scholar]
- 9. Hong S. H., Day M. W., Grubbs R. H., J. Am. Chem. Soc. 2004, 126, 7414–7415. [DOI] [PubMed] [Google Scholar]
- 10. Otte M., Kuijpers P. F., Troeppner O., Ivanović-Burmazović I., Reek J. N. H., de Bruin B., Chem. Eur. J. 2013, 19, 10170–10178. [DOI] [PubMed] [Google Scholar]
- 11.For overviews of switchable catalysis, see:
- 11a. Lüning U., Angew. Chem. Int. Ed. 2012, 51, 8163–8165; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 8285–8287; [Google Scholar]
- 11b. Imahori T., Kurihara S., Chem. Lett. 2014, 43, 1524–1531; [Google Scholar]
- 11c. Blanco V., Leigh D. A., Marcos V., Chem. Soc. Rev. 2015, 44, 5341–5370; [DOI] [PubMed] [Google Scholar]
- 11d. McConnell A. J., Wood C. S., Neelakandan P. P., Nitschke J. R., Chem. Rev. 2015, 115, 7729–7793. [DOI] [PubMed] [Google Scholar]
- 12. Bianchini G., Scarso A., La Sorella G., Strukul G., Chem. Commun. 2012, 48, 12082–12084. [DOI] [PubMed] [Google Scholar]
- 13. Cavarzan A., Scarso A., Sgarbossa P., Strukul G., Reek J. N. H., J. Am. Chem. Soc. 2011, 133, 2848–2851. [DOI] [PubMed] [Google Scholar]
- 14. Macgillivray L. R., Atwood J. L., Nature 1997, 389, 469–472. [Google Scholar]
- 15. Shivanyuk A., J. Rebek, Jr. , Proc. Natl. Acad. Sci. 2001, 98, 7662–7665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gorin D. J., Sherry B. D., Toste F. D., Chem. Rev. 2008, 108, 3351–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Handa S., Subramanium S. S., Ruch A. A., Tanski J. M., Slaughter L. M., Org. Biomol. Chem. 2015, 13, 3936. [DOI] [PubMed] [Google Scholar]
- 18. Galli M., Lewis J. E. M., Goldup S. M., Angew. Chem. Int. Ed. 2015, 54, 13545—13549; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13749–13753. [Google Scholar]
- 19.
- 19a. Ramón R. S., Gaillard S., Poater A., Cavallo L., Slawin A. M. Z., Nolan S. P., Chem. Eur. J. 2011, 17, 1238–1246; [DOI] [PubMed] [Google Scholar]
- 19b. Gómez-Suárez A., Oonishi Y., Meiries S., Nolan S. P., Organometallics 2013, 32, 1106–1111. [Google Scholar]
- 20. Nolan S. P., Acc. Chem. Res. 2011, 44, 91–100. [DOI] [PubMed] [Google Scholar]
- 21. Oonishi Y., Gómez-Suárez A., Martin A. R., Nolan S. P., Angew. Chem. Int. Ed. 2013, 52, 9767–9771; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 9949–9953. [Google Scholar]
- 22. Adriaenssens L., Escribano-Cuesta A., Homs A., Echavarren A. M., Ballester P., Eur. J. Org. Chem. 2013, 1494–1500. [Google Scholar]
- 23. Hashmi A. S. K., Gold Bull. 2003, 36, 3–9. [Google Scholar]
- 24. Fortman G. C., Poater A., Levell J. W., Gaillard S., Slawin A. M. Z., Samuel I. D. W., Cavallo L., Nolan S. P., Dalton Trans. 2010, 39, 10382–10390. [DOI] [PubMed] [Google Scholar]
- 25. Cheong P. H. Y., Morganelli P., Luzung M. R., Houk K. N., Toste F. D., J. Am. Chem. Soc. 2008, 130, 4517–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.
- 26a. Braun I., Asiri A. M., Hashmi A. S. K., ACS Catal. 2013, 3, 1902–1907; [Google Scholar]
- 26b. Hashmi A. S. K., Lauterbach T., Nösel P., Vilhelmsen M. H., Rudolph M., Rominger F., Chem. Eur. J. 2013, 19, 1058–1065; [DOI] [PubMed] [Google Scholar]
- 26c. Hashmi A. S. K., Acc. Chem. Res. 2014, 47, 864–876. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Ye L., Wang Y., Aue D. H., Zhang L., J. Am. Chem. Soc. 2012, 134, 31–34; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27b. Wang Y., Yepremyan A., Ghorai S., Todd R., Aue D. H., Zhang L., Angew. Chem. Int. Ed. 2013, 52, 7795–7799; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7949–7953. [Google Scholar]
- 28.
- 28a. Gómez-Suárez A., Oonishi Y., Martin A. R., Vummaleti S. V. C., Nelson D. J., Cordes D. B., Slawin A. M. Z., Cavallo L., Nolan S. P., Poater A., Chem. Eur. J. 2016, 22, 1125–1132; [DOI] [PubMed] [Google Scholar]
- 28b. Gómez-Suárez A., Oonishi Y., Martin A. R., Nolan S. P., Beilstein J. Org. Chem. 2016, 12, 172–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The fact that the reaction between diphenylacetylene and phenol does not take place in the presence of the cage could also be caused by the unability of the substrates and catalyst to coencapsulate inside the cage. However, the reaction between diphenylacetylene and thiophenol, which can be catalyzed by a mononuclear, π-activating catalyst due to the stronger nucleophile, did yield the coupling product. This shows that coencapsulation of the two substrates and the catalyst is possible and the hydrophenoxylation is switched off due to preventing the dinuclear pathway. For references on hydrothiolation:
- 29a. Zhang X., Wang K., RSC Adv. 2015, 5, 34439–34446; [Google Scholar]
- 29b. Tamai T., Fujiwara K., Higashimae S., Nomoto A., Ogawa A., Org. Lett. 2016, 18, 2114–2117. [DOI] [PubMed] [Google Scholar]
- 30. Sun S., Kroll J., Luo Y., Zhang L., Synlett 2012, 54–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marion N., Ramón R. S., Nolan S. P., J. Am. Chem. Soc. 2009, 131, 448–449. [DOI] [PubMed] [Google Scholar]
- 32. Leyva A., Corma A., J. Org. Chem. 2009, 74, 2067–2074. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary