

Abstract
Huntington's disease (HD) is a hereditary neurodegenerative condition with no therapeutic intervention known to alter disease progression, but several trials are ongoing and biomarkers of disease progression are needed. Tau is an axonal protein, often altered in neurodegeneration, and recent studies pointed out its role on HD neuropathology. Our goal was to study whether cerebrospinal fluid (CSF) tau is a biomarker of disease progression in HD. After informed consent, healthy controls, pre‐symptomatic and symptomatic gene expansion carriers were recruited from two HD clinics. All participants underwent assessment with the Unified HD Rating Scale ’99 (UHDRS). CSF was obtained according to a standardized lumbar puncture protocol. CSF tau was quantified using enzyme‐linked immunosorbent assay. Comparisons between two groups were tested using ancova. Pearson's correlation coefficients were calculated for disease progression. Significance level was defined as p < 0.05. Seventy‐six participants were included in this cross‐sectional multicenter international pilot study. Age‐adjusted CSF tau was significantly elevated in gene expansion carriers compared with healthy controls (p = 0.002). UHDRS total functional capacity was significantly correlated with CSF tau (r = −0.29, p = 0.004) after adjustment for age, and UHDRS total motor score was significantly correlated with CSF tau after adjustment for age (r = 0.32, p = 0.002). Several UHDRS cognitive tasks were also significantly correlated with CST total tau after age‐adjustment. This study confirms that CSF tau concentrations in HD gene mutation carriers are increased compared with healthy controls and reports for the first time that CSF tau concentration is associated with phenotypic variability in HD. These conclusions strengthen the case for CSF tau as a biomarker in HD.
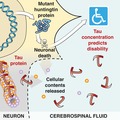
In the era of novel targeted approaches to Huntington's disease, reliable biomarkers are needed. We quantified Tau protein, a marker of neuronal death, in cerebrospinal fluid and found it was increased in patients with Huntington's disease and predicted motor, cognitive, and functional disability in patients. It is therefore likely to be a biomarker of disease progression, and possibly of therapeutic response.
Read the Editorial Highlight for this article on page 9.
Keywords: biomarkers, cerebrospinal fluid, Huntington disease, pilot projects, tau proteins
Abbreviations used
- α
alfa
- β
beta
- ANCOVA
analysis of covariance
- CAG
cytosine‐adenine‐guanine
- CSF
cerebrospinal fluid
- DCL
diagnostic confidence level
- HD
Huntington's disease
- p
p‐value
- r
Pearson's coefficient of correlation
- SD
standard deviation
- TFC
UHDRS total functional capacity
- TMS
UHDRS total motor score
- UCL
University College London
- UHDRS
Unified HD Rating Scale ’99
- UK
United Kingdom
Huntington's disease (HD) is a hereditary autosomal dominant neurodegenerative condition that affects movement control, behavior, and cognition (Bates et al. 2015). No therapeutic intervention has been found to alter disease progression but several drugs are currently being developed and tested (Bates et al. 2015). The progression of HD is slow, with half of the patients surviving at least 24 years after the motor diagnosis (Rodrigues et al. 2014). This makes studying the efficacy of disease‐modifying compounds challenging.
The development of sensitive biomarkers of disease progression that reflect neuropathology and treatment response is of vital importance to empower clinical trials.
Tau is a protein with microtubule‐stabilizing functions, which is mainly confined to axons, and when abnormally assembled or aggregated, is associated with multiple neurodegenerative conditions, Alzheimer's disease in particular (Spillantini and Goedert 2013). Indeed, it has been shown that tau concentration in cerebrospinal fluid (CSF) of patients with Alzheimer's disease is associated with disease progression (Blom et al. 2009). It has been shown that HD patients have higher CSF total tau concentrations compared with healthy controls (Constantinescu et al. 2011), but no associations with phenotype or independent validation were made.
Recently, two independent groups showed that human HD, while principally a ‘huntingtinopathy’, was also in effect a secondary tauopathy, with tau isoform imbalances, increased total tau, nuclear tau deposits, and tau co‐localization with mHTT (Fernandez‐Nogales et al. 2014; Vuono et al. 2015). Given all this, we set out to study whether CSF total tau is a biomarker of disease progression.
Methods
Ethical approval
All human experiments were performed in accordance with the declaration of Helsinki and approved by the University College London (UCL)/UCL Hospitals Joint Research Ethics Committee (UK participants) and the University of British Columbia Clinical Research Ethics Board, as appropriate. All subjects gave informed written consent.
Participants
Subjects were recruited from two HD multidisciplinary clinics, one in London, United Kingdom and other in Vancouver, Canada. Healthy controls, pre‐symptomatic gene expansion carriers, and symptomatic gene expansion carriers were included. Individual with infectious, inflammatory, or other concomitant CNS disorders or significant comorbidities were excluded. Healthy controls were defined as individuals without family history of HD and without symptoms compatible with HD, or without a cytosine‐adenine‐guanine (CAG)‐expanded allele of the HD gene. Asymptomatic gene expansion carriers were defined as individuals with a CAG‐expanded allele of the HD gene and with a diagnostic confidence level inferior to 4. Symptomatic gene expansion carriers were defined as individuals with a CAG‐expanded allele of the HD gene and with a diagnostic confidence level of 4 (Reilmann et al. 2014).
Clinical assessment
All participants underwent a collection of demographic and clinical data as well as Unified HD Rating Scale ’99 (UHDRS) (Huntington's disease study group 1996), assessed by an experienced neurologist. Age, gender, CAG repeat length of gene expansion carriers, disease stage (Bates et al. 2014), total functional capacity (Shoulson and Fahn 1979) (TFC), and total motor score (Huntington's disease study group 1996) (TMS) were recorded. The UHDRS cognitive tasks (Symbol‐digit modality test, Stroop color matching task, Stroop word matching task, and Stroop interference task) were also recorded in the Vancouver cohort (Huntington's disease study group 1996). Disease burden was calculated according to CAG repeat number and age (Penney et al. 1997). Patients with motor abnormalities were defined as having early, moderate, or advanced disease using the TFC scale (13–7, early; 6–4, moderate; 3–0, advanced) assessed by experienced clinical raters (Bates et al. 2014).
CSF sample collection and storage
CSF was obtained by lumbar puncture performed between 9 : 00 and 11 : 00 am after fasting from midnight (water was permitted), examined by microscopy, and centrifuged to remove cells, and the acellular portion was frozen at −80°C. Further details were as previously published (Wild et al. 2015). Hemoglobin concentration using multiwavelength spectrophotometric readings was assessed to determine CSF contamination by blood.
CSF total tau quantification
CSF total tau was quantified using the INNOTEST enzyme‐linked immunosorbent assay according to the manufacturer's instructions (Fujirebio, Ghent, Belgium) in one round of experiments using one batch of reagents by board‐certified laboratory technicians who were blinded to clinical data.
Statistical analysis
Statistical analysis was performed with Stata version 14 software (StataCorp, College Station, Texas, USA). Potentially confounding demographic variables (age and gender) were examined in preliminary analyses. Total tau distribution was tested for normality using skewness and kurtosis, Shapiro–Wilk and Shapiro–Francia tests. Comparisons between two groups adjusted for covariates were tested using ancova. To study the association of total tau with disease progression, we calculated Pearson's and partial correlations coefficients. Bootstrapping with 1000 repetitions was applied to non‐normal variables. Significance level was defined as p < 0.05.
Results
We conducted a cross‐sectional multicenter international pilot study of 76 participants (mean age 47.9; standard deviation [SD] 13.1; range 23–72 years; Table 1). Of these, 24 (31.6%) were healthy controls and 52 (68.4%) were gene expansion carriers – 13 (17.1%) pre‐manifest, 22 (29.0%) early HD, 6 (7.9%) moderate HD, and 11 (14.5%) advanced HD. The gene expansion group had a mean CAG repeat length of 43.8 (SD 3.9, range 36–63), a mean total functional score (TFC) of 8.6 (SD 4.5, range 0–13), and a Unified HD Rating Score (UHDRS) total motor score (TMS) of 34.6 (SD 29.6, range 0–96). Mean CSF total tau was correlated with age in healthy controls (r = 0.45, p = 0.006) and with disease burden score (a product of age and CAG repeat length) in gene expansion carriers (r = 0.23, p = 0.034). No difference was found in CSF total tau concentrations between genders (p = 0.436). Age‐adjusted CSF total tau was significantly elevated in gene expansion carriers compared with healthy controls (p = 0.002) (Fig. 1a). Total functional capacity was significantly correlated with CSF total tau (r = −0.29, p = 0.004) after adjustment for age, but not after adjustment for disease burden(r = −0.21, p = 0.146, Fig. 1b). UHDRS total motor score was significantly correlated with CSF total tau after adjustment for age (r = 0.32, p = 0.002) and also after adjustment for disease burden score (r = 0.30, p = 0.025, Fig. 1c). UHDRS symbol‐digit modality test, Stroop color matching task, and Stroop word matching task were significantly correlated with CSF total tau after adjustment for age (n = 24; r = −0.38, p = 0.020; r = −0.40, p = 1.335*10−4; r = −0.40, p = 2.426*10−4) and also after adjustment for disease burden score (r = −0.34, p = 0.008; r = 0.32, p = 0.002; r = −0.32, p = 0.039). Stroop interference task was not correlated with CSF total tau (n = 24; r = −0.21, p = 0.147). There was no association between CSF total tau and CSF hemoglobin concentration.
Table 1.
Characteristics of included participants
| Group | N n (%) | Age mean (SD) | Male n (%) | CAG mean (SD) | Burden score mean (SD) | TFC mean (SD) | TMS mean (SD) |
|---|---|---|---|---|---|---|---|
| Total | 76 (100) | 47.9 (13.1) | 33 (43.4) | 43.8 (3.9)* | 389.1 (151.8)* | 8.6 (4.5)* | 34.6 (29.6)* |
| Healthy controls | 24 (31.6) | 45.1 (14.3) | 8 (33.3) | N/A | N/A | N/A | N/A |
| Pre‐manifest HD | 13 (17.1) | 39.5 (12.5) | 6 (46.2) | 42.4 (2.5) | 255.0 (100.5) | 12.6 (0.7) | 2.4 (2.7) |
| Early HD | 22 (29.0) | 49.3 (10.4) | 8 (36.3) | 44.1 (2.7) | 402.6 (84.9) | 10.8 (1.5) | 28.3 (15.7) |
| Moderate HD | 6 (7.9) | 59.6 (6.2) | 5 (83.3) | 41.7 (2.7) | 365.8 (149.3) | 5.0 (0.9) | 51.8 (21.1) |
| Advanced HD | 11 (14.5) | 54.5 (11.7) | 6 (54.5) | 46.1 (6.2) | 533.4 (179.2) | 1.4 (1.4) | 75.7 (15.8) |
HD, Huntington's disease; N/A, non‐applicable; SD, standard deviation; TFC, UHDRS total functional capacity; TMS, UHDRS total motor score *, excluding healthy controls.
Figure 1.
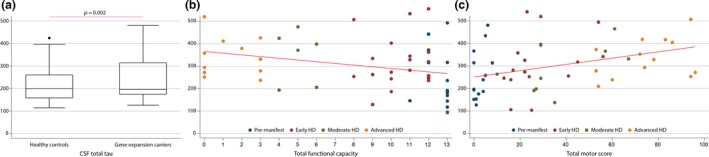
(a) Comparison between healthy controls versus gene expansion carriers; (b) Association between total tau and total functional capacity; (c) Association between total tau and Unified Huntington's Disease Rating Scale total motor score.
Discussion
This study confirms in two independent populations the previous finding that CSF total tau concentrations in HD gene mutation carriers are increased compared with healthy controls (Constantinescu et al. 2011), and the establishment of this difference persists after controlling for age – a possible confounder in the previous report.
To the best of our knowledge, ours is the first work where CSF total tau concentration has been shown to be associated with phenotypic variability in HD as measured by motor manifestations and cognitive dysfunction. These associations persist even after adjustment for disease burden score – indicating that CSF tau has independent power to predict clinical phenotype, beyond the known effects of age and CAG repeat length.
Tau is a microtubule‐associated protein, which under certain pathologic conditions, aggregates abnormally. Still, it is controversial whether these accumulations are cytotoxic and cause cellular dysfunction, contributing to neuronal cell death, or are just an epiphenomenon (Bretteville and Planel 2008). Moreover, while tau expression is particularly high in neurons with thin unmyelinated axons, this is not specific to a particular region or neuronal subpopulation and its elevation in CSF is considered a non‐specific marker of neuronal death (Zetterberg et al. 2013).
Our findings strengthen the case for CSF total tau concentration as a biomarker in HD, reflecting neuronal death and/or secondary tau pathology (Fig. 2). They suggest the need for longitudinal analysis of CSF total tau to try to establish its ability to predict disease progression, phenotypic variability and, eventually, therapeutic response. Further, this work suggests the need for further investigation of microtubule‐associated protein dynamics in HD.
Figure 2.
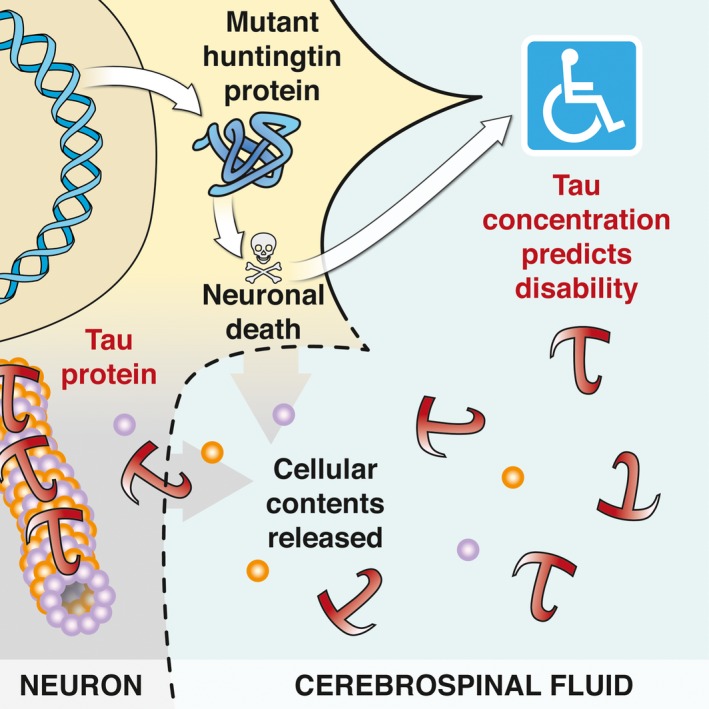
Tau is a neuronal cell death biomarker and is correlated with the disability caused by mHTT protein.
Supporting information
Appendix S1. Study methods.
Acknowledgments and conflict of interest disclosure
We thank all participants who volunteered to contribute their cerebrospinal fluid and for their enthusiasm for assisting in research to help the global HD community. This study was funded by CHDI Foundation Inc, GSK, Medical Research Council UK, Swedish Research Council, European Research Council, the Knut and Alice Wallenberg Foundation and the Wolfson Foundation. This work was supported in part by the National Institute for Health Research University College London Hospitals Biomedical Research Centre and the UCL Leonard Wolfson Experimental Neurology Centre.
SJT receives Grant funding for her research from the Medical Research Council UK, the Wellcome Trust, CHDI Foundation, the BBSRC, Rosetrees Trust, and the UCL/UCLH Biomedical Research Centre. In the past year, SJT has been on advisory boards or had consultancies with Ixico Technologies, Shire Human Genetic Therapies, and TEVA Pharmaceuticals. All honoraria paid for these consultancies and advisory boards were paid to UCL. EJW has undertaken consultancy work for Ionis, Roche and Shire Pharmaceuticals, from which all fees were paid to his employer UCL to support his research. BRL has received research funding from Teva Pharmaceuticals, Lifemax, the CHDI Foundation, and the Huntington Society of Canada and has been a paid scientific advisor or consultant to Raptor, LifeMax, Roche, Pfizer, Teva, and Ionis Pharmaceuticals. The other authors have no conflict of interest to declare.
Read the Editorial Highlight for this article on page 9.
EJW was the guarantor of this project. EJW, HZ, BL, and SJT conceptualized the project. EJW, PM, and NR were responsible to organize and execute the project. The statistical analysis was conducted by FBR. The first manuscript was written by FBR and LB. All authors reviewed and authorized the final version of the manuscript.
References
- Bates G, Tabrizi S. and Jones L. (2014) Huntington's Disease, Oxford University Press, Oxford. [Google Scholar]
- Bates G. P., Dorsey R., Gusella J. F. et al (2015) Huntington disease. Nat. Rev. Dis. Primers, 1, 15005. doi: 10.1038/nrdp.2015.5. [DOI] [PubMed] [Google Scholar]
- Blom E. S., Giedraitis V., Zetterberg H. et al (2009) Rapid progression from mild cognitive impairment to Alzheimer's disease in subjects with elevated levels of tau in cerebrospinal fluid and the APOE epsilon4/epsilon4 genotype. Dement. Geriatr. Cogn. Disord. 27, 458–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretteville A. and Planel E. (2008) Tau aggregates: toxic, inert, or protective species? J. Alzheimer's Dis. 14, 431–436. [DOI] [PubMed] [Google Scholar]
- Constantinescu R., Romer M., Zetterberg H., Rosengren L. and Kieburtz K. (2011) Increased levels of total tau protein in the cerebrospinal fluid in Huntington's disease. Parkinsonism Relat. Disord. 17, 714–715. [DOI] [PubMed] [Google Scholar]
- Fernandez‐Nogales M., Cabrera J. R., Santos‐Galindo M., Hoozemans J. J., Ferrer I., Rozemuller A. J., Hernandez F., Avila J. and Lucas J. J. (2014) Huntington's disease is a four‐repeat tauopathy with tau nuclear rods. Nat. Med. 20, 881–885. [DOI] [PubMed] [Google Scholar]
- Huntington's disease study group (1996) Unified Huntington's Disease Rating Scale: reliability and consistency. Huntington Study Group. Movement Disord. 11, 136–142. [DOI] [PubMed] [Google Scholar]
- Penney J. B., Jr , Vonsattel J. P., MacDonald M. E., Gusella J. F. and Myers R. H. (1997) CAG repeat number governs the development rate of pathology in Huntington's disease. Ann. Neurol. 41, 689–692. [DOI] [PubMed] [Google Scholar]
- Reilmann R., Leavitt B. R. and Ross C. A. (2014) Diagnostic criteria for Huntington's disease based on natural history. Movement Disord. 29, 1335–1341. [DOI] [PubMed] [Google Scholar]
- Rodrigues F., Abreu D., Damásio J., Gonçalves N., Correia‐Guedes L., Coelho M. and Ferreira J. (2014) I21 causes of death in a European Huntington's disease cohort (REGISTRY). J. Neurol. Neurosurg. Psychiatry 85, A64–A65. [Google Scholar]
- Shoulson I. and Fahn S. (1979) Huntington disease: clinical care and evaluation. Neurology 29, 1–3. [DOI] [PubMed] [Google Scholar]
- Spillantini M. G. and Goedert M. (2013) Tau pathology and neurodegeneration. Lancet Neurol. 12, 609–622. [DOI] [PubMed] [Google Scholar]
- Vuono R., Winder‐Rhodes S., de Silva R., Cisbani G., Drouin‐Ouellet J., Spillantini M. G., Cicchetti F. and Barker R. A. (2015) The role of tau in the pathological process and clinical expression of Huntington's disease. Brain 138, 1907–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild E. J., Boggio R., Langbehn D. et al (2015) Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington's disease patients. J. Clin. Investig. 125, 1979–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterberg H., Smith D. H. and Blennow K. (2013) Biomarkers of mild traumatic brain injury in cerebrospinal fluid and blood. Nat. Rev. Neurol. 9, 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Study methods.