

Abstract
Acetylated histone H3 lysine 56 (H3K56Ac) is one of the reversible histone post-translational modifications (PTMs) responsive to DNA damage. We previously described a biphasic decrease and increase of epigenetic mark H3K56Ac in response to ultraviolet radiation (UVR)-induced DNA damage. Here, we report a new function of UV damaged DNA-binding protein (DDB) in deacetylation of H3K56Ac through specific histone deacetylases (HDACs). We show that simultaneous depletion of HDAC1/2 compromises the deacetylation of H3K56Ac, while depletion of HDAC1 or HDAC2 alone has no effect on H3K56Ac. The H3K56Ac deacetylation does not require functional nucleotide excision repair (NER) factors XPA and XPC, but depends on the function of upstream factors DDB1 and DDB2. UVR enhances the association of DDB2 with HDAC1 and, enforced DDB2 expression leads to translocation of HDAC1 to UVR-damaged chromatin. HDAC1 and HDAC2 are recruited to UVR-induced DNA damage spots, which are visualized by anti-XPC immunofluorescence. Dual HDAC1/2 depletion decreases XPC ubiquitination, but does not affect the recruitment of DDB2 to DNA damage. By contrast, the local accumulation of γH2AX at UVR-induced DNA damage spots was compromised upon HDAC1 as well as dual HDAC1/2 depletions. Additionally, UVR-induced ATM activation decreased in H12899 cells expressing H3K56Ac-mimicing H3K56Q. These results revealed a novel role of DDB in H3K56Ac deacetylation during early step of NER and the existence of active functional cross-talk between DDB-mediated damage recognition and H3K56Ac deacetylation.
Keywords: Histone post-translational modifications, Nucleotide excision repair, Damaged DNA-binding protein 2, Histone deacetylases, Acetylated histone H3 lysine 56, Xeroderma pigmentosum group E
1. Introduction
Genomic integrity is constantly challenged by exposures from ubiquitously occurring endogenous and exogenous DNA damaging agents. To overcome this, living organisms have evolved elaborate surveillance systems, collectively termed DNA damage response (DDR) which is promptly deployed to ensure genomic integrity. The DDR uses sensor proteins to detect DNA damage, promote repair and initiate signaling checkpoint events that control cell cycle progression [1,2]. Inadequate DDR and failure of damage repair can result in cell death and contribute to human diseases, e.g., ataxia telangiectasia (A-T) [1], xeroderma pigmentosum (XP) syndrome [3], etc. The A-T syndrome is caused by a defect in the A-T mutated (ATM) which is responsible for managing the cell’s response to multiple forms of stress including double-strand breaks in DNA; XP syndrome results from mutation in any of the XP genes classified from A to G.
XP gene products are crucial components of nucleotide excision repair (NER) machinery, which removes diverse DNA lesions including UVR-induced 6-4-pyrimidine pyrimidone photoproducts (6-4PP), cyclobutane pyrimidine dimmers (CPD) and chemical-induced bulky lesions from genome [3]. NER consists of two sub-pathways: global genomic repair (GGR) which eliminates DNA damage from entire genome, and transcription-couple repair (TCR) which eliminates DNA damage located on transcribed strands of transcriptionally active genes [4]. In GGR, DNA lesions are first recognized by UV damaged DNA-binding proteins (UV-DDB or DDB), and handed over from DDB to XPC-RAD23B complexes [5–7]. DDB is composed of two components, DDB1 and DDB2, whose function is absent due to gene mutation in cells derived from XP-E patients [8–10]. Genetic deletion of DDB2 in mice mimics XP-E phenotype, impairing the repair of photolesions and causing hypersensitivity to UVR-induced skin cancers [11]. Biochemically, DDB2 is incorporated into a Cul4A-DDB1-ROC1 (CRL4), forming a CRL4DDB2 E3 ubiquitin ligase complex, where DDB2 functions as a substrate adapter [12]. Cul4A, DDB1 and DDB2 are rapidly recruited to UVR-induced lesions, with the kinetics consistent with the binding of a preassembled CRL4DDB2 complex [13,14]. The activity of CRL4DDB2 ubiquitin ligase is activated at damage sites upon irradiation and directed to chromatin or chromatin-bound proteins. Substrates of CRL4DDB2 ubiquitin ligase include histone 2A [15], histone 3 and 4 [16], XPC [17,18], and DDB2 itself [12,17,19]. Ubiquitin modified DDB2 is degraded by proteasome. Conversely, ubiquitination of XPC augments the DNA binding of XPC without altering its specificity [17]. Consequently, DNA damage is handed over from DDB to XPC [19]. Damage-bound XPC-RAD23B initiates the assembly of pre-incision complex composed of TFIIH, XPA, as well as endonucleases XPF and XPG, which make 5′ and 3′ incision, respectively, to remove a 24–32 bp oligonucleotide containing DNA lesion. The gap resulting from dual incision is filled by repair synthesis and the final nick is ligated to complete NER [20,21].
DNA repair, like other DNA-templated processes, is inevitably influenced by chromatin structures. The fundamental unit of chromatin is nucleosome [22], which consists of two copies each of histones H2A, H2B, H3 and H4, as well as ~146 bp of DNA wrapped nearly twice around histone octamer. Nucleosomes are interconnected by sections of linker DNA and a linker histone, H1. Higher order of chromatin structure is achieved by compacting nucleosomes into chromatin fiber. Eukaryotic genomes exist in different chromatin states in the nucleus: heterochromatin (also called ‘closed’ chromatin) and euchromatin (‘open’ chromatin) where active transcription occurs. Chromatin remodeling includes nucleosome repositioning [23], histone eviction/deposition [24] and histone variant incorporations [25]. Histone post-translational modifications (PTMs) is another mechanism that regulates the state of chromatin [26]. Histones are modified at distinct amino acid residues with different reversible PTMs, which include methylation, ubiquitination, sumoylation, phosphorylation and acetylation [27]. These PTMs can be reversed by specific enzymes. For example, acetylation of lysine residues is regulated by histone acetyltransferase (HATs) and histone deacetylases (HDACs) that catalyze addition and removal of acetyl moiety, respectively.
Histone PTMs, e.g., acetylation, have been known for transcription regulation [28]. It is becoming increasingly clear that histone PTMs also function in DDR [29,30]. The best characterized histone PTM is phosphorylation of serine 139 at the C-terminal tail of histone H2AX (also called γH2AX). The phosphorylation is mediated by ATM, ATM and RAD3 related (ATR) and DNA-dependent protein kinase (DNA-PK) in response to DNA damages including strand breaks and DNA photolesions [31,32]. Histone acetylation was first PTM linked to DDR [33]. Recently, acetylated histone H3 lysine 56 (H3K56Ac) and acetylated histone H3 lysine 9 (H3K9Ac) were identified as DNA damage responsive PTMs [34]. Specifically, H3K56Ac and H3K9Ac are rapidly diminished and subsequently restored in response to exposures to a variety of DNA damaging agents including UVR and ionizing radiation (IR). More recent studies reported that H3K56Ac is down-regulated by HDAC1 and HDAC2 at DNA double strand breaks to promote DNA double strand break repair by non-homologous end-joining [35]. The mechanism that regulates H3K56Ac after UVR-induced DNA damage is not yet known.
We previously reported that upon UVR-induced DNA damage, a fast initial deacetylation of H3K56Ac is followed by full renewal of H3K56Ac state [36]. H3K56Ac restoration requires histone chaperone Asf1 and appears to be a post-repair event of chromatin restoration. In this study, we investigated whether HDAC1 and HDAC2 participate in deacetylation of H3K56A in response to UVR exposure. We further explored whether deacetylation of H3K56A is linked to early steps of NER. Our data has uncovered a new function of DDB which is involved in specific deacetylation of H3K56A by HDAC1 and HDAC2.
2. Materials and methods
2.1. Cell lines
XP-A (GM04429), XP-C (GM02096), XP-E (GM01389, GM02415) cells were obtained from Coriell Institute for Medical Research (Camden, NJ 08103). HeLa-DDB2.com cells, expressing HA- and FLAG-tagged DDB2, were kindly provided by Dr. Yoshihiro Nakatani (Dana-Farber Cancer Institute, Boston, MA 02215). Li-Fraumeni syndrome (LFS) fibroblast 041 cell line (p53-Null, harboring a codon 184 frameshift, resulting in premature termination of translation of p53 protein) was provided by Dr. Michael Tainsky (M.D. Anderson Cancer Center, Houston, TX 77030). LFS-041-DDB2 cells stably expressing V5-His-tagged DDB2 were established in our laboratory. Human lung adenocarcinoma H1299 cells expressing Flag-tagged H3.1 (H1299–H3K56) or Flag-tagged H3K56Q (H3 lysine 56 to glutamine mutation, mimicking acetylation) were gifts from Dr. Zhenkun Lou at Department of Oncology, Mayo Clinic, MN 55905). XP-A, XP-C, LFS-041, HeLa and HeLa-DDB2.com cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). H1299 cells were grown in RPMI medium supplemented with 10% fetal bovine serum. XP-E cells were cultured in MEM supplemented with Earle’s salts and nonessential amino acids and 10% FBS. All cells were grown at 37 °C in a humidified atmosphere with 5% CO2.
HeLa HDAC1-KD and HeLa HDAC2-KD cell lines were generated upon transfection with HDAC1 and HDAC2 shRNA expressing constructs into HeLa cells using the FuGENE 6 transfection reagent. At 24 h post-transfection, puromycin was added to media at a final concentration of 2 μg/ml. The selection with puromycin was continued further for 1 week. The surviving cells were trypsinized, diluted and re-seeded so as to achieve colonies from single cells under puromycin selection. Clones exhibiting knockdown of HDAC1 or HDAC2 were recovered and expanded for further experiments.
2.2. Antibodies
Antibodies against H3K56Ac were obtained from Epitomics (Burlingame, CA 94010), HDAC1, HDAC2, γ-H2AX and phosphor-ATM (Ser 1981) antibodies were from Cell Signaling Technology (Danvers, MA 01923). Anti-XPC antibody (XPC-2) was generated by immunizing rabbits with a synthetic peptide corresponding to the C-terminus of human XPC protein. Anti-FLAG M2 agarose beads and anti-FLAG M2 antibody were purchased from Sigma–Aldrich (St. Louis, MO 63103). DDB1 and DDB2 antibodies were from Bethyl Laboratories (Montgomery, TX 77356).
2.3. Gene silencing
Small hairpin RNA (shRNA) targeting HDAC1 and HDAC2 were obtained from Sigma–Aldrich. DDB1 and DDB2 siRNA was from GE Dharmacon (Lafayette, CO 80026). The siRNA transfections were conducted using Lipofectamine™ 2000 transfection reagent from Invitrogen (Grand Island, NY 14072) according to the manufacturer’s instructions. In brief, ~0.3 × 106 HeLa cells were seeded in 60-mm tissue culture dishes and grown overnight. For transfection, Lipofectamine 2000 reagent was diluted in Opti-MEM medium and siRNA was added to it and incubated at room temperature for 20 min. The Lipofectamine–small interfering RNA (siRNA) complex was applied to the cells and incubated for 48 h before UV irradiation. The shRNA transfections were performed using FuGENE 6 transfection reagent (Roche Diagnostics, Indianapolis, IN 46250) according to the manufacturer’s protocol. Briefly, cells were grown to 50% confluency. ShRNA plasmid DNA was added to the diluted FuGENE 6 reagent in serum- and antibiotic-free medium and the FuGENE to DNA ratio was maintained at 6:1. After incubation at room temperature for 20 min, the FuGENE 6–DNA complex was added to cells. The cells were incubated for additional 24 h prior to UV irradiation of the cells.
2.4. Micropore irradiation and immunofluorescence
Cells were seeded on glass coverslips at a proper density and were grown to from a monolayer. Micropore UV irradiation was conducted by placing a 5-μm isopore polycarbonate filter (EMB Millipore, Billerica, MA 01821) followed by UV irradiation at indicated doses. The UV-irradiated cells were maintained for a desired time period for DNA repair to take place. Immunofluorescence staining was conducted according to the method established in our laboratory. Briefly, the cells were washed twice with cold PBS, permeabilized with 0.5% Triton X-100/PBS for about 8 min on ice as needed, and/or fixed with 2% paraformaldehyde in 0.5% Triton X-100 at 4 °C for 30 min. The fixed cells were rinsed twice with cold PBS and blocked with 20% normal goat serum in 0.1% Triton X-100/PBS, and stained with an appropriate primary antibody as well as fluorescein isothiocyanate (FITC) or Alexa Fluor 488 or Texas Red-conjugated secondary antibodies. The coverslips were mounted in Vectashield mounting medium with DAPI. The fluorescence images were obtained with a Nikon fluorescence microscope E80i and processed with SPOT software.
2.5. Cellular fractionation
Cellular fractionation was conducted as described by Anindya et al. [37], with modifications. Briefly, cells were lysed with 1 ml (~5× cell volume) of cytoplasmic lysis buffer (10 mM Tris–HCl [pH 7.9], 0.34 M sucrose 3 mM CaCl2, 2 mM magnesium acetate, 0.1 mM EDTA, 1 mM DDT, 0.5% NP-40 and a protease inhibitor cocktail). Nuclei were pelleted by centrifugation at 3500 × g for 15 min and washed with cytoplasmic lysis buffer without NP-40 and then lysed in 1 ml of nuclear lysis buffer (20 mM HEPES [pH 7.9], 3 mM EDTA, 10% glycerol, 1.5 mM MgCl2, 150 mM KOAc and protease inhibitors). The nucleoplasmic fractions were separated by centrifugation at 15,000 × g for 30 min and the pellet was resuspended in 0.2 ml of nuclease incubation buffer (150 mM HEPES [pH 7.9], 1.5 mM MgCl2, 150 mM KOAc and protease inhibitors) and incubated with 50 U benzonase (25 U/μl) for 30 min at room temperature. The nuclease-releasable or soluble chromatin fraction was collected by centrifugation at 20,000 × g for 30 min. Protein concentrations of each cellular fractions were determined by Dc Bio-Rad DC protein assay and the same amount of protein fractions in relative total protein yields of each fractions were used for Western blotting.
2.6. Immunoprecipitation
The immunoprecipitation was done at 4 °C overnight in RIPA buffer (50 mM Tris–HCl [pH 8.0], 150 mM NaCl, 1% NP40, 0.5% deoxycholate and protease inhibitors) using nuclease-releasable chromatin containing ~500–1000 μg protein and the anti-FLAG-M2 beads. The beads were washed 1 time with RIPA buffer and then with 3 times with TBS buffer (50 mM Tris–HCl [pH 7.4] 150 mM NaCl) and the bound proteins were eluted with FLAG peptide as described in the manufacturer’s protocol or released by boiling in SDS loading buffer.
3. Results
3.1. HDAC1 and HDAC2 redundantly function in deacetylation of H3K56Ac
We and others have reported a decrease in H3K56Ac level at low doses of UV exposure [34,36]. To probe whether HDACs are responsible for H3K56Ac deacetylation, we first treated HeLa cell with HDAC inhibiters sodium butyrate and trichostatin A immediately following cellular UV irradiation. As shown in Fig. 1A, increasing concentrations of both HDAC inhibitors significantly elevated the H3K56Ac levels, and essentially reversed the normally observed reduction of the UVR-induced cellular H3K56Ac levels. We then chose HDAC1 and HDAC2 of class I HDACs as candidate deacetylases and established stable HDAC1- or HDAC2-knockdown (KD) HeLa cell lines by transfection of shRNA expression vector into HeLa cells followed by clonal selection and expansion. The knockdown effects were confirmed by examination of HDAC1 and HDAC2 levels in corresponding cells (Fig. 1B). HeLa cells express higher levels of HDAC2 compared to HDAC1. The HDAC1 expression was seen in HeLa cells but was abolished in HDAC1-KD cells and it remained unchanged in HDAC2-KD cells. On the contrary, HDAC2 expression was seen in HeLa and HDAC1-KD cells but was significantly compromised in HDAC2-KD cells. We then examined H3K56Ac response to UVR-induced DNA damage at two different post-irradiation time-points of 8 and 24 h. These two time-points were chosen because of their distinct correspondence to H3K56Ac deacetylation and restoration [36]. In both HDAC1-KD and HDAC2-KD cells, UVR-induced H3K56Ac deacetylation was still seen at 8 h (Fig. 1C). Also, H3K56Ac was restored at 24 h to a level comparable to that in control cells without UV irradiation. These results indicated that the depletion of HDAC1 or HDAC2 alone do not significantly affect H3K56Ac deacetylation or restoration. To test if HDAC1 and HDAC2 work together in H3K56Ac deacetylation, we transiently transfected shHDAC1 expressing vector into HDAC2-KD HeLa cells to achieve simultaneous HDAC1 and HDAC2 knockdowns (Fig. 1D). Transfection of shHDAC1 #2, 3 and 4 constructs decreased HDAC1 levels to ~40%. Transfection with these constructs also reduced UVR-induced H3K56Ac deacetylation as compared to controls without transfection or shHDAC1 #1 transfection. These results indicated that HDAC1 and HDAC2 redundantly function in H3K56Ac deacetylation in early response to UVR-induced DNA damage.
Fig. 1.

HDAC1 and HDAC1 knockdown compromises UVR-induced reduction of H3K56Ac. (A) HDAC inhibition by sodium butyrate and trichostatin A reverses UVR-induced reduction of H3K56Ac. Hela cells were UV irradiated at 20 J/m2 and treated with sodium butyrate and trichostatin A at indicated concentration. The treated cells were harvested at 4 h post-UV, and the H3K56Ac levels were determined by Western blotting. (B) HDAC1 and HDAC2 levels were determined by Western blotting in stable HDAC1-KD and HDAC2-KD cells respectively expressing shRNA targeting HDAC1 or HDAC2. The actin blots were used as loading controls. (C) UVR-induced H3K56Ac reduction and restoration were not affected by HDAC1 or HDAC2 knockdown alone. HeLa HDAC1-KD and HDAC2-KD cells were exposed to 20 J/m2 of UV radiation. Whole cell extracts were prepared at indicated time points. H3K56Ac levels in equal amounts of protein extracts were determined by Western blotting using anti-H3K56Ac antibody. (D) HDAC1 depletion in HDAC2-KD cells affected UVR-induced H3K56Ac deacetylation at 8 h post-UV irradiation. HDAC2-KD cells were transiently transfected with HDAC1 shRNA expressing constructs #1–4 for 48 h. The transfected cells were UV irradiated at a dose of 20 J/m2 and the cells were harvested at 8 h post-UV. H3K56Ac and HDAC1 levels were determined by Western blotting and ImageJ software.
3.2. DDB1 and DDB2 are required for deacetylation of H3K56Ac
We have previously demonstrated that in HeLa cells as well as normal human fibroblasts (NHF) H3K56Ac deacetylation occurs as early as 0.25 h following UV irradiation [36]. So, we speculated that H3K56Ac deacetylation is primarily related to early steps, perhaps the damage recognition events of NER. Thus, we examined H3K56Ac deacetylation in NHF, XP-A and XP-C cells in response to UV irradiation (Fig. 2A). XP-A cells are fully deficient in NER, while XP-C cells are only deficient in GGR sub-pathway of NER. NHF showed a typical time-dependent H3K56Ac deacetylation. Similarly, in XP-C cells UV irradiation induced H3K56Ac deacetylation at 2, 4 and 8 h. In XP-A cells, UV irradiation was able to induce H3K56Ac deacetylation at 4 h, indicating that functional NER is not required for H3K56Ac deacetylation. We next examined H3K56Ac deacetylation in XP-E cells (Fig. 2B). Interestingly, in these cells, UV irradiation did not induce H3K56Ac deacetylation and the levels at 2, 4 and 8 h were comparable to the unirradiated control. Since, in yeast H3K56Ac is an established chromatin packaging mark, cellular cell cycle status may affect H3K56Ac deacetylation. To rule out the possibility that the defect in UVR-induced H3K56Ac deacetylation in XP-E is due to DNA replication in S-phase, we serum-starved the XP-E cultures and examined H3K56Ac in G1-arrested cells for extended post-irradiation time periods up to 24 h. Again, UV irradiation failed to cause H3K56Ac deacetylation at 4, 8 and 24 h. These results indicated that DDB2 function is required for H3K56Ac deacetylation. Examinations of HDAC1 and 2 in NHF indicated that HDAC1 and 2 are present in NHF and their levels were not significantly changed upon UV irradiation. Interestingly, HDAC1 level was very low in XP-E cells (Fig. 2C). We further dissected the effect of DDB function by examining the UVR-induced H3K56Ac deacetylation upon individual knockdown of DDB1 and DDB2 components (Fig. 2D). The efficiency of siRNA-mediated knockdown was confirmed by examination of DDB1 and DDB2 levels. Moreover, as expected, the UV irradiation lowered DDB2 levels in control and in DDB1 siRNA transfected cells, due to ubiquitin-mediated proteasomal degradation. UVR-induced H3K56Ac deacetylation was clearly impacted by the depletion of either DDB1 or DDB2. Taken together, these results indicated that UVR-induced H3K56Ac deacetylation is independent of functional NER, but requires the function of DDB but not XPC, suggesting that the initial damage recognition by DDB is intimately linked to H3K56Ac deacetylation. It is a noteworthy that HDAC1 level in XP-E cells, while very low, might not be the cause for defect of H3K56Ac deacetylation given that HDAC1 and HDAC2 redundantly function in H3K56Ac deacetylation.
Fig. 2.
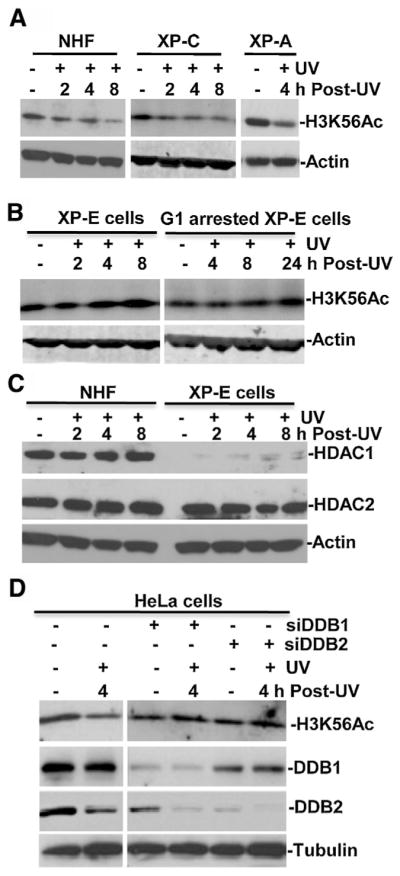
UVR-induced H3K56Ac deacetylation in early DDR requires DDB2 and DDB1 but not functional NER. (A) Normal human fibroblasts (NHF), XP-A, XP-C fibroblasts were UV irradiated at 20 J/m2, harvested at indicated time points post-UV, and the H3K56Ac levels were determined by Western blotting. The actin blots were used as loading controls. (B) The primary XP-E fibroblasts were arrested in G1 phase by serum starvation for 48 h. The XP-E cells and G1 arrested XP-E fibroblasts were UV irradiated, maintained and harvested at indicated post-UV time points. H3K56Ac levels in cell extracts were determined by Western blotting. (C) HDAC1 and HDAC2 were detected in NHF and XP-E cells with or without UV irradiation. (D) Knockdown of DDB1 and DDB2 affected H3K56Ac deacetylation. HeLa cells were transfected with 100 nM DDB1 or DDB2 siRNA using Lipofectamine transfection reagents. After transfection for 48 h, the cells were irradiated with 20 J/m2 of UV and harvested at 4 h time point. Levels of H3K56Ac, DDB1, DDB2 and tubulin in normal and transfected cells were determined by Western blotting using specific antibodies.
3.3. DDB2 enhances translocation of HDAC1 to UVR-damaged chromatin
We next explored the physical association of DDB2 to HDAC1 in vivo. Nuclease-releasable chromatin fractions from UV-irradiated FLAG-tagged DDB2 expressing HeLa-DDB2.com cells and control cells were subjected to immunoprecipitation by anti-FLAG gel. In immunoprecipitates, HDAC1 was detected in the immunoprecipitates from HeLa-DDB2.com cells upon UV irradiation but not from HeLa-DDB2.com or control HeLa cells without UV irradiation (Fig. 3A). These results confirmed our previous observations and indicated that DDB2 physically interacts with HDAC1 [38]. We further investigated whether DDB2 affects translocation of HDAC1 to chromatin. We used LFS-041 cell line which has low DDB2 protein level due to its p53-Null status [39] and established an enforced expression of DDB2 via stable transfection [40]. Because DDB2 protein level was low in LFS-041 cells, UV irradiation had no discernible influence on DDB2 translocation from nucleoplasmic to nuclease-releasable chromatin fractions. Similarly, UV irradiation had no appreciable effect on HDAC1 distribution (Fig. 3B). In LFS-041 cells with enforced DDB2 expression, UV irradiation triggered a DDB2 and HDAC1 translocation from nucleoplasmic fraction to chromatin fraction. These results indicated that UVR-induced a DDB2-dependent translocation of HDAC1 to damaged chromatin.
Fig. 3.

HDAC1 interacts with DDB2 and translocates to damaged chromatin. (A) DDB2 associates with HDAC1 in vivo. Parental HeLa cells and HA-FLAG-tagged DDB2 expressing HeLa-DDB2.com cells were UV irradiated at 20 J/m2, and nuclease-releasable chromatin fractions were made at 1 h post-UV. Immunoprecipitation was performed in RPIA buffer using nuclease-releasable chromatin from HeLa and HeLa DDB2.com and anti-FLAG antibody-conjugated gels. Asterisk mark (*) indicates immunoglobulin heavy chain (IgH). (B) LFS-041 fibroblasts and DDB2-expressing LFS-041 fibroblasts were UV irradiated at 50 J/m2 and harvested at 1 h post-UV. The chromatin fractions were prepared by cellular protein fractionation using protocol as described in Section 2. Equal protein amounts of nucleoplasmic fraction (nucleoplasmic) or nuclease-releasable chromatin fraction (soluble chromatin) in relative to total yields of each fraction were probed for DDB2 and HDAC1 by Western blotting.
3.4. HDAC1 regulates the damage handover from DDB to XPC at DNA damage sites
We next examined whether HDAC1 is recruited to UVR-induced DNA damage sites. HeLa cells were UV irradiated through a 5-μm pore filter, which generates localized DNA damage in sub-nuclear areas of irradiated cells. One hour after micropore irradiation, XPC, HDAC1 and HDAC2 were visualized by their cognate antibodies. HDAC1 and HDAC2 were seen to concentrate and form distinct foci in HeLa cell nuclei. More importantly, the HDAC1 and HDAC2 foci co-localized with repair factor XPC-marked foci, indicating HDAC1 and HDAC2 are recruited to DNA damage sites. We noticed that at some of the XPC foci, HDAC1 and HDAC2 accumulation was indistinct. This may be attributed to the engagement of limiting nuclear HDAC1 and HDAC2 in other cellular processes.
In early steps of NER, DNA damage is first recognized by DDB and then handed over to XPC. Consequently, XPC is ubiquitinated by CRL4DDB2 E3 ubiquitin ligase complex. To investigate if HDAC1 and HDAC2 depletion affect damage handover, we examined XPC ubiquitination as a specific readout of this cellular event. Upon cellular UV exposures, XPC is rapidly ubiquitinated by CRL4DDB2 E3 ubiquitin ligase complex, generating slower migrating protein bands which can be detected by anti-XPC western blotting [17,18,41]. In HDAC1-KD and HDAC2-KD cells, UV irradiation was able to induce XPC ubiquitination at 1 h and the XPC ubiquitination disappeared at 4 h (Fig. 4B and C). Conversely, HDAC1 and HDAC2 double depletion in both cells compromised XPC ubiquitination at 1 h. These results indicated that HDAC1 and HDAC2, similar to their regulation of deacetylation of H3K56Ac, also work redundantly in regulation of XPC ubiquitination. Thus, HDAC1 and HDAC2 act as upstream regulators of damage recognition by XPC.
Fig. 4.

HDAC1 and HDAC2 are recruited to DNA damage sites and modulate XPC ubiquitination upon UV irradiation. (A) HDAC1 and HDAC2 colocalize with XPC at DNA damage spots. HeLa cells were grown on coverslips and UV irradiated at 100 J/m2 through a 5-μm micropore filter placed on the cell monolayers. The cells were fixed with 2% paraformaldehyde at 1 h post-UV and immunofluorescence staining was performed with primary anti-XPC, anti-HDAC1 specific antibody and Texas Red-or fluorescein isothiocyanate (FITC)-conjugated secondary antibodies. Arrows indicate UVR-induced HDCA1, 2 or XPC immunofluorescence spots. (B) and (C) HDAC1 and HDAC2 knockdowns decrease XPC ubiquitination upon UV irradiation. Stable HDAC2-KD and HDAC1-KD cells were transiently transfected with shHDAC1 (B) or shHDAC2 (C) constructs for 48 h, respectively, the transfected cells were irradiated with UV at 20 J/m2 and harvested at 1 or 4 h post-UV. Cell extracts containing equal amounts were used for probing XPC and ubiquitin-modified XPC forms by Western blotting. (Ub)n represents multiple (n) ubiquitin moieties. Lamin B blots were used as loading control. Asterisk mark (*) represents a nonspecific protein band from cross-reacting of XPC antibody.
3.5. HDAC1 and HDAC2 depletion decreases accumulation of γH2AX at DNA damage sites
To determine if HDAC1 and HDAC2 depletion affects damage recognition by DDB2, we examined the recruitment of DDB2 to DNA damage sites upon HDAC1 or HDAC1/2 double depletion. Local DNA damage was generated by micropore irradiation, and, the photolesions were visualized by CPD-specific antibody (Fig. 5A, upper panel). In most CDP spots in HDAC1-KD cells, DDB2 foci co-localized with CPD foci, indicating single HDAC1 depletion did not affect DDB2 recruitment to DNA damage sites. Interestingly, HDAC1 and HDAC2 double depletion did not significantly affect the DDB2 recruitment to damage sites either (Fig. 5A, lower panel). Thus, we concluded that HDAC1 and HDAC2 do not play a direct role in DDB-mediated damage recognition.
Fig. 5.

(A) HDAC1/2 knockdown does not affect DDB2 recruitment to DNA damage sites. (A) Stable HeLa HDAC1-KD cells or HDAC1-KD cells transiently transfected with shHDAC2 DNA constructs were UV irradiated at 100 J/m2 through a 5-μm micropore filter and maintained for 1 h. The cells were then fixed with 2% paraformaldehyde, and, DDB2 and CPD were visualized by immunofluorescence using anti-DDB2 or anti-CPD specific antibodies. Arrows indicated typical UVR-induced, antibody-decorated immunofluorescence spots of DDB2 and CPD. (B) HDAC1 and HDAC2 knockdown decreases γH2AX accumulation at DNA damage after UV irradiation. Stable HeLa HDAC1-KD cells or HDAC1-KD cells transiently transfected with shHDAC2 DNA constructs were UV irradiated at 20 J/m2 through a 5-μm micropore filter. The cells were fixed with 2% paraformaldehyde 1 h after UV irradiation, and, γH2AX foci (pointed by arrows) were visualized at DNA damage spots by immunofluorescence. (C) Quantitative data are from the analysis of γH2AX foci in HeLa, HeLa HDAC1-KD cells and hHDAC2-tranfected HDAC1-KD cells. Mean ± SE were calculated from multiple microscopic fields of three independent experiments. Double asterisk mark (**) indicates p ≤ 0.01. (D) H1299 cells expressing H3K56 or H3K56Q were irradiated at 20 J/m2 and harvested at indicated time points. ATM, phospho-ATM and γH2AX were determined by specific antibodies. Actin blots were used as loading control.
We next asked whether HDAC1 and HDAC2 depletion affect the accumulation of γH2AX at DNA damage sites. Local γH2AX accumulation was determined at 1 h after micropore UV irradiation. As shown in Fig. 5B and C, micropore UV irradiation efficiently induced γH2AX accumulation in HeLa cells with about 60% of cells positive for γH2AX foci. By contrast, only ~20% of cells were positive for γH2AX foci in HDAC1-KD cells. Interestingly, HDAC1/2 double depletion did not cause further decrease in local γH2AX accumulation, suggesting that HDAC1 is a major player in regulation of local γH2AX accumulation. The γH2AX is a well known DNA damage marker with direct correlation to the presence of DNA strand breaks in cells. In case of UVR-induced DNA damage, H2AX phosphorylation is triggered by NER intermediate (strand break) or Exo1-processed intermediate resulting from dual incision via NER of photolesion [32,42]. Thus, the results suggested that HDAC1 and HDAC2 participate in DDR following UVR-induced DNA damage.
We finally tested whether H3 acetylation status affects ATM activation using H1299 cells expressing H3K56Q, which mimics H3K56Ac. As shown in Fig. 5D, without UVR, ATM levels were same in H3K56 or H3K56Q-expressing H1299 cells. Following UVR, ATM level decreased slightly at 24 h time-points in both cells. Interestingly, the UVR-induced ATM phosphorylation in H3K56-expressing H1299 cells was apparent at 4 h time-point and the phosphorylation declined in 8–24 h periods. More importantly, phospho-ATM levels were lower in H3K56Q-expressing H1299 cells at 4 and 8 h as compared with that in H3K56-expressing H1299 at the same time-points. Conversely, γH2AX level was only slightly lower in the H3K56Q expressing cells, indicating H3K56Q expression does not have significant effect on global level of UVR-induced γH2AX. Given the recruitment of HDAC1 and HDAC2 to local DNA damage sites, we surmised that H3K56Ac deacetylation has a preferential on-site effect on local γH2AX accumulation. Taken together, these results suggested that H3K56Ac deacetylation facilitates ATM phosphorylation/activation and local γH2AX accumulation during early events of DDR.
4. Discussion
Our previous work described that the H3K56Ac in cells responds in a biphasic manner following introduction of photolesion in cells [36], and the response is akin to that resulting from induction of DNA double strand breaks [34]. In this study, we have provided evidence that the photolesion recognition by DDB is linked to H3K56Ac deacetylation in which HDAC1 and HDAC2 function redundantly. Our results are analogous to the report that H3K56Ac is down-regulated by HDAC1 and HDAC2 at DNA double strand breaks [35]. The results are also consistent with HDAC-regulated DDR of UV-mimetic 4-nitroquinoline 1-oxide (4NQO) [43] and the situation where DDB2 plays a role in activation of ATM and ATR kinases in response to UVR-induced DNA damage [44]. In the former situation, yeast rpd3 hda1 (HDAC1/2 homolog) double mutants showed hypersensitivity to 4NQO and hydroxyurea, whereas in the latter, ATM and ATR phosphorylation were negatively affected in XP-E cells and DDB2-depleted NHF cells.
Our findings link H3K56Ac deacetylation to DDB-mediated photolesion recognition which constitutes a very early event of NER. It can be speculated that H3K56Ac deacetylation is related to an ‘open’ state of chromatin, while H3K56Ac refurbishment reflects the chromatin structure restoration following successful completion of repair. The scenario fits with the ‘access-repair-restore’ model of NER [45]. However, it should be recognized that functional significance of H3K56Ac in chromatin regulation is largely unclear. In a genome-wide study in human cells, H3K56Ac was found enriched on promoter regions of both active and inactive genes [46]. On the other hand, DNA damage was found to result in significant reduction of H3K56Ac on promoters of active genes that are not repressed upon DNA damage [34]. So, the H3K56Ac level may not directly correlate to active or repressed transcription. In case of DDR, H3K56 acetylation drives chromatin assembly after DNA repair and signals completion of repair in yeast [47]. Recently, it was reported that the acute sensitivity of H3K56Ac-deficient cells to methyl methane sulfonate (MMS) and camptothecin (CPT), which cause DNA damage during replication, is due to the failure to complete recombination repair or DNA replication [48]. In our previous work, H3K56R mutation which cannot be acetylated did not have a discernible effect on GGR, indicating H3K56 acetylation does not significantly affect GGR [36]. Here, we have demonstrated that H3K56Ac deacetylation requires DDB but not XPC and XPA functions, indicating that deacetylation would be only associated with photolesion recognition event mediated by DDB. At present, understanding of H3K56Ac function remains a challenge. Nevertheless, the novel function of DDB in H3K56Ac deacetylation provides an impetus for further dissection of the role of DDB2 in chromatin regulation at the sites of DNA damage.
It is known that DDB2 function is not essential for NER in vitro but is required for CPD repair and significantly promotes 6-4PP repair in vivo [9,39]. This observation has led to the idea that DDB-mediated photolesion recognition may link chromatin remodeling to DNA repair [49]. To support this notion, INO80 chromatin remodeling complex was shown to promote the removal of UV lesions. INO80 appears to associate with DDB1 [50]. In the same vein, it was recently reported that DDB2 elicits large-scale chromatin unfolding [51]. In chromatin containing UVR-induced photolesions, a marked ATP-dependent reduction in density of core histones was found to require DDB2 function. Our data suggests that HDAC1 and HDAC2 are additional components of DDB2-mediated regulation of chromatin structure. We demonstrated that both HDAC1 and HDAC2 are involved in UVR-induced H3K56Ac deacetylation and, these HDACs are all recruited to UVR-induced lesion sites. Moreover, DDB2 was found to associate with HDAC1 in vivo and enforced DDB2 expression results in translocation of HDAC1 to damaged chromatin. Given that DDB2 elicits large-scale chromatin unfolding in the vicinity of UVR-induced DNA damage, the deacetylation of H3K56Ac by HDAC1/2 may be a part of, or an indicator of, the chromatin unfolding event. Additionally, we found that HDAC1 level was very low in XP-E cells. Although it might not be the cause for the defect of H3K56Ac deacetylation in XP-E cells, it would be interesting to learn whether low level of HDAC1 influences histone acetylations, e.g., H3K9Ac, H3K14ac or H4K16ac. In a sense, DDB-mediated photolesion recognition may not only be linked to chromatin remodeling, but also to histone PTM regulation including histone acetylation, deacetylation and ubiquitination. These events act coordinately to create a local chromatin environment permissive to the initiation of NER by XPC.
In this study, HDAC1/2 depletion was found to decrease the accumulation of γH2AX at DNA damage sites. At present, it is unclear how DDB2-mediated H3K56Ac deacetylation contributes to such changes in γH2AX. The finding, however, is consistent with the observations that HDACs regulate DDR in yeast [43]. In particular, HDAC inhibition/ablation counteracts Mec (yeast orthologue of human ATR) activation and causes nuclease Exo1 and Sae2 degradation. In our study, HDAC1 was found to interact with DDB2 and translocate to UVR damaged chromatin in a DDB2-dependent manner. It could be expected that HDAC1 and HDAC2 are recruited to damage sites and exert their effect locally, e.g., through direct interaction with ATM [52]. Nevertheless, we cannot rule out the possibility that HDAC1 or HDAC2 depletion also has off-site effects. For example, it was recently reported that siRNA-mediated HDAC1/2 depletion globally down-regulates a number of DRR components including Rad50, p53, BRCA1 and ATM through transcription [53].
HDAC1 and HDAC2 are overexpressed in many cancers [54]. HDAC inhibitors sensitize cancer cell to DNA damage based therapies and are being combined with radiation therapies in clinical trials [55,56]. The findings in this study suggest that HDAC inhibitors may be used in combination with DNA-damaging drugs, e.g. cisplatin, whose DNA lesions are targets of NER.
Acknowledgments
Authors thank Dr. Yoshihiro Nakatani (Dana-Farber Cancer Institute, Boston, MA 02215) for providing HeLa-DDB2.com cells, Dr. Michael Tainsky (M.D. Anderson Cancer Center, Houston, TX 77030) for providing Li-Fraumeni syndrome (LFS) fibroblast 041 cell lines. Authors are also grateful to Dr. Zhenkun Lou (Department of oncology, Mayo Clinic, MN, 55905) for Flag-tagged H3.1 expressing H1299-cell lines. Aruna Battu was partly supported by a Pelotonia fellowship of The Ohio State University. This work was supported by Public Health service Grants (ES2388, ES12991) from National Institute of Health
Footnotes
Conflict of interest
The authors declare that there are no conflicts of interest.
References
- 1.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28(5):739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 3.De Boer J, Hoeijmakers JH. Nucleotide excision repair and human syndromes. Carcinogenesis. 2000;21(3):453–460. doi: 10.1093/carcin/21.3.453. [DOI] [PubMed] [Google Scholar]
- 4.Hanawalt PC. Subpathways of nucleotide excision repair and their regulation. Oncogene. 2002;21(58):8949–8956. doi: 10.1038/sj.onc.1206096. [DOI] [PubMed] [Google Scholar]
- 5.Wakasugi M, Kawashima A, Morioka H, Linn S, Sancar A, Mori T, Nikaido O, Matsunaga T. DDB accumulates at DNA damage sites immediately after UV irradiation and directly stimulates nucleotide excision repair. J Biol Chem. 2002;277(3):1637–1640. doi: 10.1074/jbc.C100610200. [DOI] [PubMed] [Google Scholar]
- 6.Fitch ME, Nakajima S, Yasui A, Ford JM. In vivo recruitment of XPC to UV-induced cyclobutane pyrimidine dimers by the DDB2 gene product. J Biol Chem. 2003;278(47):46906–46910. doi: 10.1074/jbc.M307254200. [DOI] [PubMed] [Google Scholar]
- 7.Sugasawa K, Okamoto T, Shimizu Y, Masutani C, Iwai S, Hanaoka F. A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 2001;15(5):507–521. doi: 10.1101/gad.866301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu G, Chang E. Xeroderma pigmentosum group E cells lack a nuclear factor that binds to damaged DNA. Science. 1988;242(4878):564–567. doi: 10.1126/science.3175673. [DOI] [PubMed] [Google Scholar]
- 9.Tang JY, Hwang BJ, Ford JM, Hanawalt PC, Chu G. Xeroderma pigmentosum p48 gene enhances global genomic repair and suppresses UV-induced mutagenesis. Mol Cell. 2000;5(4):737–744. doi: 10.1016/s1097-2765(00)80252-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nichols AF, Ong P, Linn S. Mutations specific to the xeroderma pigmentosum group E Ddb-phenotype. J Biol Chem. 1996;271(40):24317–24320. doi: 10.1074/jbc.271.40.24317. [DOI] [PubMed] [Google Scholar]
- 11.Alekseev S, Kool H, Rebel H, Fousteri M, Moser J, Backendorf C, De Gruijl FR, Vrieling H, Mullenders LH. Enhanced DDB2 expression protects mice from carcinogenic effects of chronic UV-B irradiation. Cancer Res. 2005;65(22):10298–10306. doi: 10.1158/0008-5472.CAN-05-2295. [DOI] [PubMed] [Google Scholar]
- 12.Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113(3):357–367. doi: 10.1016/s0092-8674(03)00316-7. [DOI] [PubMed] [Google Scholar]
- 13.Luijsterburg MS, Goedhart J, Moser J, Kool H, Geverts B, Houtsmuller AB, Mullenders LH, Vermeulen W, van Driel R. Dynamic in vivo interaction of DDB2 E3 ubiquitin ligase with UV-damaged DNA is independent of damage-recognition protein XPC. J Cell Sci. 2007;120(Pt 15):2706–2716. doi: 10.1242/jcs.008367. [DOI] [PubMed] [Google Scholar]
- 14.Alekseev S, Luijsterburg MS, Pines A, Geverts B, Mari PO, Giglia-Mari G, Lans H, Houtsmuller AB, Mullenders LH, Hoeijmakers JH, Vermeulen W. Cellular concentrations of DDB2 regulate dynamic binding of DDB1 at UV-induced DNA damage. Mol Cell Biol. 2008;28(24):7402–7413. doi: 10.1128/MCB.01108-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kapetanaki MG, Guerrero-Santoro J, Bisi DC, Hsieh CL, Rapic-Otrin V, Levine AS. The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc Natl Acad Sci USA. 2006;103(8):2588–2593. doi: 10.1073/pnas.0511160103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H, Zhai L, Xu J, Joo HY, Jackson S, Erdjument-Bromage H, Tempst P, Xiong Y, Zhang Y. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol Cell. 2006;22(3):383–394. doi: 10.1016/j.molcel.2006.03.035. [DOI] [PubMed] [Google Scholar]
- 17.Sugasawa K, Okuda Y, Saijo M, Nishi R, Matsuda N, Chu G, Mori T, Iwai S, Tanaka K, Tanaka K, Hanaoka F. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell. 2005;121(3):387–400. doi: 10.1016/j.cell.2005.02.035. [DOI] [PubMed] [Google Scholar]
- 18.Wang QE, Zhu Q, Wani G, El-Mahdy MA, Li J, Wani AA. DNA repair factor XPC is modified by SUMO-1 and ubiquitin following UV irradiation. Nucl Acids Res. 2005;33:4023–4034. doi: 10.1093/nar/gki684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El-Mahdy MA, Zhu Q, Wang QE, Wani G, Praetorius-Ibba M, Wani AA. Cullin 4A-mediated proteolysis of DDB2 protein at DNA damage sites regulates in vivo lesion recognition by XPC. J Biol Chem. 2006;281(19):13404–13411. doi: 10.1074/jbc.M511834200. [DOI] [PubMed] [Google Scholar]
- 20.De Laat WL, Jaspers NG, Hoeijmakers JH. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999;13(7):768–785. doi: 10.1101/gad.13.7.768. [DOI] [PubMed] [Google Scholar]
- 21.Sancar A. DNA excision repair. Annu Rev Biochem. 1996;65:43–81. doi: 10.1146/annurev.bi.65.070196.000355. [DOI] [PubMed] [Google Scholar]
- 22.Kornberg RD. Structure of chromatin. Annu Rev Biochem. 1977;46:931–954. doi: 10.1146/annurev.bi.46.070177.004435. [DOI] [PubMed] [Google Scholar]
- 23.Bowman GD. Mechanisms of ATP-dependent nucleosome sliding. Curr Opin Struct Biol. 2010;20(1):73–81. doi: 10.1016/j.sbi.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Das C, Tyler JK, Churchill ME. The histone shuffle: histone chaperones in an energetic dance. Trends Biochem Sci. 2010;35(9):476–489. doi: 10.1016/j.tibs.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Altaf M, Auger A, Covic M, Cote J. Connection between histone H2A variants and chromatin remodeling complexes. Biochem Cell Biol. 2009;87(1):35–50. doi: 10.1139/O08-140. [DOI] [PubMed] [Google Scholar]
- 26.Lee JS, Smith E, Shilatifard A. The language of histone crosstalk. Cell. 2010;142(5):682–685. doi: 10.1016/j.cell.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 28.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447(7143):407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 29.Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair. Cell. 2007;128(4):721–733. doi: 10.1016/j.cell.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 30.Escargueil AE, Soares DG, Salvador M, Larsen AK, Henriques JA. What histone code for DNA repair? Mutat Res. 2008;658(3):259–270. doi: 10.1016/j.mrrev.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 31.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273(10):5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 32.Hanasoge S, Ljungman M. H2AX phosphorylation after UV irradiation is triggered by DNA repair intermediates and is mediated by the ATR kinase. Carcinogenesis. 2007;28(11):2298–2304. doi: 10.1093/carcin/bgm157. [DOI] [PubMed] [Google Scholar]
- 33.Ramanathan B, Smerdon MJ. Changes in nuclear protein acetylation in UV-damaged human cells. Carcinogenesis. 1986;7(7):1087–1094. doi: 10.1093/carcin/7.7.1087. [DOI] [PubMed] [Google Scholar]
- 34.Tjeertes JV, Miller KM, Jackson SP. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 2009;28(13):1878–1889. doi: 10.1038/emboj.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S, Jackson SP. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010;17(9):1144–1151. doi: 10.1038/nsmb.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Battu A, Ray A, Wani AA. ASF1A and ATM regulate H3K56-mediated cell-cycle checkpoint recovery in response to UV irradiation. Nucl Acids Res. 2011;39(18):7931–7945. doi: 10.1093/nar/gkr523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anindya R, Aygun O, Svejstrup JQ. Damage-induced ubiquitylation of human RNA polymerase II by the ubiquitin ligase Nedd4, but not Cockayne syndrome proteins or BRCA1. Mol Cell. 2007;28(3):386–397. doi: 10.1016/j.molcel.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 38.Zhao R, Han C, Eisenhauer E, Kroger J, Zhao W, Yu J, Selvendiran K, Liu X, Wani AA, Wang QE. DNA damage-binding complex recruits HDAC1 to repress Bcl-2 transcription in human ovarian cancer cells. Mol Cancer Res. 2014;12(3):370–380. doi: 10.1158/1541-7786.MCR-13-0281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hwang BJ, Ford JM, Hanawalt PC, Chu G. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc Natl Acad Sci USA. 1999;96(2):424–428. doi: 10.1073/pnas.96.2.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barakat BM, Wang QE, Han C, Milum K, Yin DT, Zhao Q, Wani G, Arafa E, El-Mahdy MA, Wani AA. Overexpression of DDB2 enhances the sensitivity of human ovarian cancer cells to cisplatin by augmenting cellular apoptosis. Int J Cancer. 2010;127(4):977–988. doi: 10.1002/ijc.25112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He J, Zhu Q, Wani G, Sharma N, Han C, Qian J, Pentz K, Wang QE, Wani AA. Ubiquitin-specific protease 7 regulates nucleotide excision repair through deubiquitinating XPC protein and preventing XPC protein from undergoing ultraviolet light-induced and VCP/p97 protein-regulated proteolysis. J Biol Chem. 2014;289(39):27278–27289. doi: 10.1074/jbc.M114.589812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sertic S, Pizzi S, Cloney R, Lehmann AR, Marini F, Plevani P, Muzi-Falconi M. Human exonuclease 1 connects nucleotide excision repair (NER) processing with checkpoint activation in response to UV irradiation. Proc Natl Acad Sci USA. 2011;108(33):13647–13652. doi: 10.1073/pnas.1108547108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R, Botrugno OA, Parazzoli D, Oldani A, Minucci S, Foiani M. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011;471(7336):74–79. doi: 10.1038/nature09803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ray A, Milum K, Battu A, Wani G, Wani AA. NER initiation factors, DDB2 and XPC, regulate UV radiation response by recruiting ATR and ATM kinases to DNA damage sites. DNA Repair (Amst) 2013;12(4):273–283. doi: 10.1016/j.dnarep.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Green CM, Almouzni G. When repair meets chromatin. First in series on chromatin dynamics. EMBO Rep. 2002;3(1):28–33. doi: 10.1093/embo-reports/kvf005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie W, Song C, Young NL, Sperling AS, Xu F, Sridharan R, Conway AE, Garcia BA, Plath K, Clark AT, Grunstein M. Histone h3 lysine 56 acetylation is linked to the core transcriptional network in human embryonic stem cells. Mol Cell. 2009;33(4):417–427. doi: 10.1016/j.molcel.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen CC, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, Tyler JK. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell. 2008;134(2):231–243. doi: 10.1016/j.cell.2008.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wurtele H, Kaiser GS, Bacal J, St-Hilaire E, Lee EH, Tsao S, Dorn J, Maddox P, Lisby M, Pasero P, Verreault A. Histone H3 lysine 56 acetylation and the response to DNA replication fork damage. Mol Cell Biol. 2012;32(1):154–172. doi: 10.1128/MCB.05415-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones KL, Zhang L, Seldeen KL, Gong F. Detection of bulky DNA lesions: DDB2 at the interface of chromatin and DNA repair in eukaryotes. IUBMB Life. 2010;62(11):803–811. doi: 10.1002/iub.391. [DOI] [PubMed] [Google Scholar]
- 50.Jiang Y, Wang X, Bao S, Guo R, Johnson DG, Shen X, Li L. INO80 chromatin remodeling complex promotes the removal of UV lesions by the nucleotide excision repair pathway. Proc Natl Acad Sci USA. 2010;107(40):17274–17279. doi: 10.1073/pnas.1008388107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luijsterburg MS, Lindh M, Acs K, Vrouwe MG, Pines A, van Attikum H, Mullenders LH, Dantuma NP. DDB2 promotes chromatin decondensation at UV-induced DNA damage. J Cell Biol. 2012;197(2):267–281. doi: 10.1083/jcb.201106074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim GD, Choi YH, Dimtchev A, Jeong SJ, Dritschilo A, Jung M. Sensing of ionizing radiation-induced DNA damage by ATM through interaction with histone deacetylase. J Biol Chem. 1999;274(44):31127–31130. doi: 10.1074/jbc.274.44.31127. [DOI] [PubMed] [Google Scholar]
- 53.Thurn KT, Thomas S, Raha P, Qureshi I, Munster PN. Histone deacetylase regulation of ATM-mediated DNA damage signaling. Mol Cancer Ther. 2013;12(10):2078–2087. doi: 10.1158/1535-7163.MCT-12-1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26(37):5420–5432. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- 55.Smith KT, Workman JL. Histone deacetylase inhibitors: anticancer compounds. Int J Biochem Cell Biol. 2009;41(1):21–25. doi: 10.1016/j.biocel.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 56.Camphausen K, Tofilon PJ. Inhibition of histone deacetylation: a strategy for tumor radiosensitization. J Clin Oncol. 2007;25(26):4051–4056. doi: 10.1200/JCO.2007.11.6202. [DOI] [PubMed] [Google Scholar]