

ABSTRACT
c-Myc (MYC) is an oncogenic transcription factor that is commonly overexpressed in a wide variety of human tumors. In breast cancer, MYC has recently been linked to the triple-negative subtype, a subtype that lacks any targeted therapy. Previously, we demonstrated that MYC behaves as a potent repressor of YAP and TAZ, 2 transcriptional coactivators that function as downstream transducers of the Hippo pathway. In this previous study, MYC repressed YAP/TAZ not only in primary breast epithelial cells but also in mouse models of triple-negative tumors. Here, we extend our previous bioinformatic and experimental analyses and demonstrate that MYC deregulation in primary breast epithelial cells leads to a robust repression of TEAD transcription factor activity, the transcription factor family mainly responsible for YAP/TAZ recruitment. Surprisingly, we find that MYC and TEAD activity is able to stratify different breast cancer subtypes in large panels of breast cancer patients. Thus, a deep understanding of the MYC-YAP/TAZ circuitry might yield new insights into the establishment and maintenance of specific breast cancer subtypes.
KEYWORDS: Breast Cancer, Hippo, Mammary Epithelial Cells, MYC, TAZ, TEAD, YAP
Introduction
The transcription factor c-Myc (MYC) belongs to the basic-helix-loop-helix leucine zipper (bHLHZ) family and has been implicated in multitudinous aspects of tumor development and tumor maintenance.1 MYC binds with the highest affinity to specific DNA sequences, called E-boxes (CACGTG), as a heterodimeric complex with its partner protein MAX, leading to transactivation of its target genes by recruitment of several cofactors, such as TRRAP and associated histone acetyltransferases.2,3
Given MYC's large number of binding sites and its many target genes, it has been very difficult to assign specific target genes to MYC's oncogenic role, even though it is clear that high MYC activity is predictive in terms of prognosis.4 Consequently, high MYC activity typically leads to a poor survival prognosis in a multitude of tumor entities.
Previously, we could identify a pathway that couples MYC-driven changes in mitochondrial dynamics to repression of YAP/TAZ target genes in primary breast epithelial cells and in mouse models of breast cancer.5 YAP and TAZ are transcriptional coactivators that act as the critical downstream transducers of the Hippo pathway. The canonical Hippo pathway cascade involves 2 types of kinases: Mammalian Ste20-like kinases (MST1/2) can get activated by a multitude of upstream signals and in turn activate the Large tumor suppressor (LATS1/2) kinases.6,7 LATS kinases in turn phosphorylate critical serine residues in YAP and TAZ leading to their cytoplasmic sequestration and proteasomal degradation.8,9
Besides the “canonical” MST-LATS-dependent pathway, YAP can get inactivated by other means, e.g. through mechanotransduction or 5′ AMP-activated kinases (AMPKs).10,11
In our studies, we could demonstrate that MYC induces Phospholipase D6 (PLD6) as a critical target gene being crucial for MYC's repressive effect on YAP/TAZ target gene expression. MYC-mediated transactivation of PLD6 (a mitochondrial phospholipase) leads to a change in mitochondrial dynamics since PLD6 promotes fusion of these organelles.12 In turn, altered dynamics of mitochondrial fusion/fission in conjunction with additional MYC-mediated changes in metabolism strain cellular energy resources, ultimately leading to activation of AMPK. AMPK then phosphorylates YAP at serine 94, a residue that is absolutely critical for YAP's interaction with Tea domain (TEAD) transcription factors. Thus, MYC, with the help of this pathway, is able to specifically disrupt the binding of YAP to TEAD, the transcription factor family mainly responsible for the recruitment of YAP. We initially discovered this pathway in the CD44high/CD24low population of immortalized breast epithelial cells (HMLE), a population demonstrating high activity of YAP/TAZ compared to the CD44low/CD24high HMLE population.5,13 However, we could further demonstrate that this pathway is also operational in mouse models of breast cancer since the inhibition of MYC activity led to a re-establishment of YAP/TAZ activity accompanied by a regain of differentiation in the respective tumors. Since bioinformatics analyses of large panels from breast cancer patients suggested that MYC is also able to interfere with YAP/TAZ function in human breast cancers, we set out to corroborate our recent findings and to investigate if MYC is indeed able to specifically interfere with TEAD-dependent functions of YAP/TAZ.
Results
Previously, we performed RNA-Sequencing in HMLE cells infected with a doxycycline-inducible MYC allele (Fig. 1A). First, the cells were sorted according to their CD44/CD24 surface profile into either the sphere-forming CD44high/CD24low population or the CD44low/CD24high population. Afterwards, MYC was induced for 8 hours by doxycycline addition, and the gene expression profiles were subsequently analyzed by RNA-Sequencing. To gain insights into the pathways that might get altered in response to MYC induction, we performed GSEA analyses using our previously published RNA-Sequencing data set. GSEA analyses test if there is an uneven distribution of a given gene set in gene expression profiles. The so-called C2 or C6 collection of gene sets are commonly used for this kind of analyses, and they consist of gene sets that were significantly up- or down-regulated in previous gene expression analyses after a specific manipulation (e.g., overexpression of YAP).
Figure 1.

MYC restrains TEAD-dependent YAP functions. A: Workflow for the RNA-Sequencing experiment performed in FACS-sorted HMLE cells expressing a doxycycline-inducible HA-MYC allele (pInducer21-HA-MYC). B: Gene set enrichment analysis for the C3 gene set (motifs) in the CD44high/CD24low HMLE population 8 hours after MYC induction. MYC and MYCMAX represent potential binding sites for the MYC/MAX heterodimer, whereas TEF1 represents predicted binding sites for TEAD1 (also called TEF1). NES = normalized enrichment score; FDR = false discovery rate. C: CD44high/CD24low HMLE cells were infected with the indicated YAP 5SA mutants (YAP 5SA or the TEAD-binding compromised YAP 5SA S94A mutant) and analyzed by immunoblots. VINCULIN served as a loading control. D: qRT-PCR analyses of the direct YAP/TAZ target genes ANKRD1 and CTGF, as well as additional genes (BDNF, ARHGAP29 and RAB30) from the WGGAATGY_V$TEF1_Q6 C3 gene set. E: qChIP analysis in MCF7 cells for TEAD binding to the promoters of the given genes. IgG served as a negative control. Ctrl region = U2 promoter region.
Here, we concentrated for our analyses on the C3 gene set, a gene set that is solely derived from bioinformatic analyses (e.g., presence of evolutionarily conserved transcription factor binding sites in the promoter of a given gene). Thus, unlike the commonly used C2 or C6 gene sets, they are not experimentally derived and are consequently independent of a specific experimental setup or cellular context. Consistent with MYC's high affinity for E-boxes, the gene sets that were most strongly induced upon MYC induction represented binding sites for the MYC/MAX heterodimer (Fig. 1B). Importantly, MYC strongly repressed those genes bearing a binding site for TEF1 (=TEAD1) in their promoter, supporting our previous findings that MYC is able to disrupt the interaction between YAP/TAZ and TEAD transcription factors.5 To substantiate these bioinformatics analyses, we tested if those genes are indeed induced in a TEAD-YAP/TAZ-dependent manner. To this end, we infected CD44high/CD24low HMLE cells either with a constitutively active YAP 5SA mutant, lacking all 5 LATS phosphorylation sites, or a YAP 5SA mutant with an additional S94A mutation (Fig. 1C). The S94A mutation in YAP leads to a specific disruption of YAP's interaction with TEAD transcription factors and removes the particular serine residue that gets phosphorylated in response to MYC induction.5,14 Consequently, this mutant allowed us to exclude the involvement of other transcription factors in the YAP-mediated regulation of these genes.14,15 Here, YAP 5SA was able to potently induce expression of known TEAD-YAP/TAZ target genes (ANKRD1 and CTGF) and additional putative TEAD-YAP/TAZ target genes included in the 2 TEF1 C3 gene sets (Fig. 1B–D). In contrast to this, the YAP 5SA S94A mutant was strongly compromised in induction of these genes (Fig. 1C, D). Consistent with the specific inability of YAP 5SA S94A to induce the expression of these genes, we could confirm binding of TEAD1 to the promoters of several induced genes (CTGF, EPS8L2, ARHGAP29 and RAB30) by chromatin-immunoprecipitation (ChIP) analyses in MCF7 cells, validating these genes as bona fide TEAD target genes (Fig. 1E).
Thus, we concluded that MYC's ability to interfere with YAP's function depends on its ability to specifically disrupt the interaction between YAP and TEAD transcription factors.
Consistent with MYC's inhibitory effect on YAP/TAZ activity, we could demonstrate previously that high MYC activity shows a strong correlation with low YAP activity in gene expression profiles of human breast cancer patients. Given our findings that MYC specifically interferes with TEAD-dependent YAP functions, we reasoned that we should be able to detect an anti-correlation between MYC activity and the expression of genes within the TEAD C3 gene set (WGGAATGY_V$TEF1_Q6) that gets potently repressed after MYC induction in CD44high/CD24low HMLE cells (Fig. 1B). To this end, we analyzed the expression of MYC target genes and the expression of genes within this particular TEAD C3 gene set and performed an association score analysis for these gene sets as described previously.5 In this kind of analysis, a given gene set is used in a GSEA-like manner per patient. Here, a ranked list (according to fold change) of all expressed genes is used to test for an uneven distribution of a specific gene set in the respective patient. We tested 2 of the biggest published breast cancer patient panels, METABRIC (1302 patients) and TCGA (1215 patients), for a potential relationship between MYC and TEAD activity. In both data sets, we could observe a highly significant anti-correlation between MYC activity and TEAD activity, respectively (p = 2.65×10−49 for METABRIC, p = 1.0×10−74 for TCGA) (Fig. 2A, B). Furthermore, MYC activity correlated with high-grade tumors (p = 2.11×10−30), whereas TEAD activity correlated with low-grade tumors (p = 1.14×10−6) (Fig. 2A). Thus, we extended the analysis and tested if MYC or TEAD activity, respectively, might stratify breast cancer patients into specific breast cancer subtypes (Fig. 2C). Consistent with previous analyses, this analysis demonstrated an association of high MYC/low TEAD activity with basal tumors.5 Strikingly, this analysis revealed that MYC and TEAD activity is also unevenly distributed among additional breast cancer subtypes: high MYC/low TEAD activity was associated with a high prevalence of Her2-amplified tumors and tumors of the Luminal B subtype, whereas low MYC/high TEAD activity was associated with a high prevalence of the “Normal” subtype.
Figure 2.
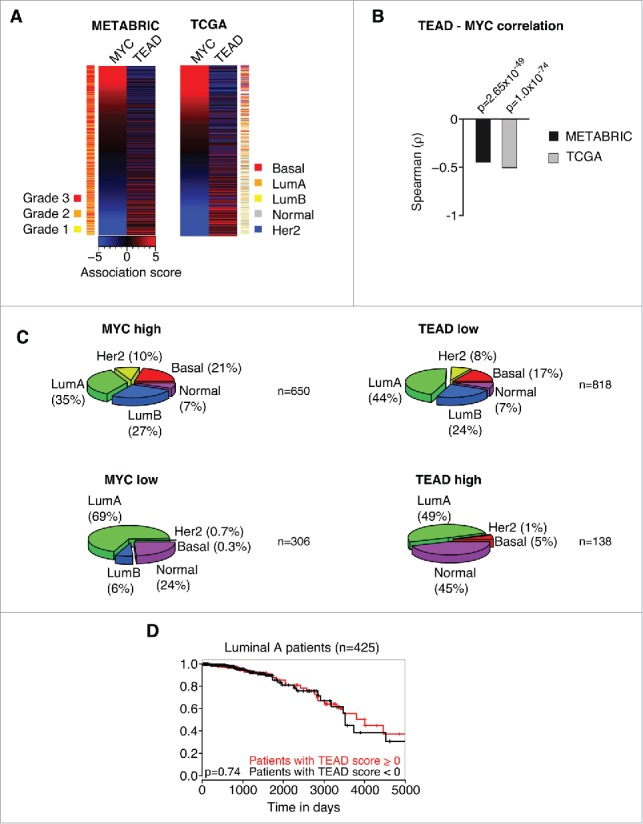
MYC and TEAD activity stratify human breast cancer patients. A: Association score analysis for MYC target genes and TEAD target genes (C3 WGGAATGY_V$TEF1_Q6 gene set) in the METABRIC or TCGA breast cancer data sets, respectively. The association score for the expression of MYC and TEAD target genes was calculated for every single patient and subsequently sorted according to the MYC association score. A negative association score describes the repression of the respective gene set, whereas a positive association score describes an induction of the respective gene set in the respective patient compared to the median. A color code is given to annotate the grade (METABRIC) or subtype (TCGA) per patient. LumA = Luminal A; LumB = Luminal B; Her2 = Her2-amplified. B: Spearman rank correlation and associated significance test for the correlation between MYC and TEAD activity as determined in A. C: Patients from the TCGA data set were stratified according to their association score (AS) for MYC and TEAD, respectively. The prevalence of breast cancer subtypes within the given groups is depicted as a piechart. MYC high: AS > −2; MYC low: AS ≤ −2; TEAD high: AS > 3; TEAD low: AS ≤ 3. LumA = Luminal A; LumB = Luminal B; Her2 = Her2-amplified. n denotes the number of patients per piechart. Please note that the breast cancer subtype information was not available for every patient in the TCGA data set. D: Kaplan-Meier plot for patients with a Luminal A subtype. The patients were stratified according to their TEAD activity (TEAD score > 0 ; TEAD score < 0, respectively) and analyzed for their disease-specific survival. The p-value was determined by a chi-square test.
In summary, the data suggest that MYC is also able to restrain TEAD activity in human tumors and that this relationship is able to stratify breast cancer subtypes. Importantly, we could not observe any significant differences in survival of the patients when applying this stratification, based on TEAD activity, within the Luminal A subtype (Fig. 2D). We analyzed this subtype since it is evenly distributed among TEAD low and TEAD high patients (44% and 49%, respectively) yielding a sufficiently high number of patients for statistical analyses. Thus, TEAD activity is not an independent predictor of survival, implying that the MYC-TEAD/YAP pathway might rather be involved in the evolution of specific breast cancer subtypes.
Discussion
We previously demonstrated that MYC behaves as a potent repressor of YAP/TAZ activity in primary breast epithelial cells and mouse models of breast cancer.5 Additionally, we observed that the MYC-YAP/TAZ antagonism is preserved in gene expression profiles of breast cancer patients and that high MYC/low YAP/TAZ activity leads to a poor survival for the patient. This suggests that it might indeed be beneficial for the bulk tumor to maintain this antagonism; a hypothesis supported by the fact that antagonizing MYC activity in a MMTV-Wnt1 breast cancer mouse model leads to more differentiated tumors coinciding with re-establishment of YAP/TAZ activity.
Here, we provide a detailed analysis of MYC's effect on TEAD-dependent YAP activity in primary breast epithelial cells and panels of breast cancer gene expression profiles.
MYC specifically interferes with TEAD-dependent YAP functions, consistent with its stimulating effect on YAP S94 phosphorylation, a serine residue that is crucial for YAP's interaction with TEAD transcription factors. As one would expect from these findings, we could observe an anti-correlation between MYC activity and TEAD activity in 2 large gene expression data sets of breast cancer patients. Importantly, we used gene sets for TEAD that are completely unbiased, since they were solely generated based on the presence of evolutionarily conserved TEAD binding sites in the promoters of the respective genes.
Surprisingly, the activity of these gene sets was sufficient to stratify human breast cancer patients into different breast cancer subtypes, e.g. Her2-amplified tumors showed a very strong preference for high MYC/low TEAD activity. In the future, it will be interesting to determine whether MYC's effect on TEAD activity contributes to the establishment of certain subtypes, or if this correlation simply reflects a specific cell of origin, even though both scenarios are not necessarily mutually exclusive and evidence for both exist. YAP and TAZ are involved in lineage commitment of primary breast epithelial cells.16 Deregulated MYC activity might consequently shift the balance of differentiation in an early (pre-)tumorigenic cell of origin which is later reflected in gene expression profiles of the respective tumor.
Apart from that, it has also been shown that MYC is able to generate a heterogeneous set of tumor types in mouse models of MYC-driven tumorigenesis.17 This suggests that additional genetic/epigenetic modifiers (such as Her2-amplification) might be able to act in concert with MYC to generate specific subtypes in the course of MYC-driven tumorigenesis. If and how the TEAD/YAP network is involved in this tumor evolution, will be an important issue that needs to be addressed in the future.
However, the most important question in our view is: why do we detect such a strong anti-correlation between MYC activity and YAP/TAZ activity in human breast cancer tumors? There is no doubt that YAP behaves as a potent oncogene in several tissues such as liver and skin.18-20 Nevertheless, in our analyses YAP and TEAD activity are always correlated with low-grade tumors. One explanation for this seeming discrepancy is the fact that gene expression profiles from breast cancer patients measure the gene expression profiles of the bulk tumor. Thus, it may sometimes be hard to bring the results from this kind of analyses in line with genetic experiments, e.g., where YAP is needed for the oncogenic capacity of polyama middle T antigen (PyMT)-driven breast cancer development.21 It is conceivable that YAP/TAZ might indeed have an oncogenic role, also in breast cancer. Since YAP and TAZ have been correlated with cancer stem cell-related traits, YAP and TAZ most likely exert their oncogenic function in a small population of cells such as cancer stem cells.13 Due to their low frequency within the tumor, they would not significantly contribute to the gene expression profiles of bulk tumors. Still, the bulk tumor seems to select for low YAP/TAZ activity, suggesting that this reflects a benefit for the tumor. Since, up to now, no frequent upstream mutations have been detected that would lock a tumor cell in either of the 2 states (high MYC/low YAP/TAZ vs. low MYC/high YAP/TAZ), it might be possible that a certain degree of plasticity in tumors exists. This kind of plasticity was demonstrated previously for the oncogenic transcription factor Twist1.22 Here, Twist1 activation very efficiently drove the establishment of primary squamous cell carcinomas but needed to be switched off for efficient colonization of metastases at distant sites.
Thus, it will be interesting to see whether such a dynamic relationship between MYC and YAP/TAZ plays a role in the development of breast cancer tumors.
Materials and methods
Tissue culture and lentiviral transduction
HMLE cells were kept in MEGM medium (Lonza) and infected as described previously.5 HMLE cells were infected with a doxycycline-inducible lentiviral plasmid bearing a HA-MYC allele (pInducer21-HA-MYC) that also contains a constitutive co-expression cassette for GFP. After FACS sorting for GFP-positive cells, HMLE pInducer-21-HA-MYC cells were induced for 8 hours with 1 µg/ml doxycycline to induce HA-MYC expression. The YAP 5SA S94A mutant was generated by site-directed mutagenesis and cloned into the same backbone as the YAP 5SA mutant (LeGO-iG2-Puro).
Gene set enrichment analyses
We used our previously published RNA-Sequencing data set (GSE66250) to perform gene set enrichment analyses (GSEA) using the Broad GSEA analysis tool (gsea2-2.2.2).
Antibodies
HMLE cells were lysed in RIPA buffer and equal amounts of protein were used for SDS-PAGE and subsequent immunoblotting. The following antibodies were used: YAP (Cell Signaling, 14074), CTGF (Santa Cruz, sc-14939), Vinculin (Sigma, V9131), TEAD1 (BD Biosciences, 610923).
Chromatin-immunoprecipitation (ChIP)
ChIP assays were performed from MCF7 cells as described previously.23
qRT-PCR
Total RNA was extracted using peqGOLD TriFast Reagent (Peqlab). First-strand cDNA synthesis was performed with M-MLV Reverse Transcriptase (Invitrogen) and random hexamer primers (Roche) according to standard procedures. Gene expression was analyzed on a StepOne plus platform (Thermo Scientific) in technical triplicates using ABsolute SYBR Green Mix (Thermo Scientific). The expression values were normalized to b2M as housekeeping gene using the ddCt method. The following primers were used for qRT-PCR:
ARHGAP29 Fw: CCTTATGGGAGATGTAGGCAATG
ARHGAP29 Rev: AGCTCGATAGAGTCAGTGTTCT
BDNF Fw: TAACGGCGGCAGACAAAAAGA
BDNF Rev: TGCACTTGGTCTCGTAGAAGTAT
EPS8L2 Fw: ACTCAGCGAGCCAGGTTTC
EPS8L2 Rev: CAGTTGAGGATTTGCGTCTCC The primers for CTGF and ANKRD1 have been described previously.5
Analysis of breast cancer data sets
All analyses were performed in the R (v 3.2.3) environment. Gene expression data and corresponding clinical data for TCGA and METABRIC were downloaded from the respective webpages (https://tcga-data.nci.nih.gov/tcga/ or https://www.ebi.ac.uk/, respectively). The enrichment of specific gene signatures per patient was performed using a GSEA-like algorithm as described previously using a Kolgomorov-Smirnov test for normality.5,24
The C3 motif gene set was downloaded from the Molecular Signatures Database (MSigDB, http://software.broadinstitute.org/gsea/msigdb) and used for subsequent analyses of breast cancer patients and gene set enrichment analysis (GSEA).
Statistical analysis
To correlate the association scores of MYC or TEAD with each other or with grade, respectively, a Spearman rank correlation test was performed in R.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed and the authors declare no competing financial interests.
Acknowledgments
HMLE cells were a kind gift from Robert Weinberg (Whitehead Institute for Biomedical Research, Cambridge, MA). We thank all members of the von Eyss and the Eilers lab for helpful discussions.
Funding
The Fritz Lipmann Institute (FLI) is a member of the Leibniz Association and is financially supported by the Federal Government of Germany and the State of Thuringia. Dana Elster was supported by a LGSA (Leibniz Graduate School on Aging) fellowship. This work was supported by grants (M.E.) from the Deutsche Krebshilfe (109696), the Thyssen Foundation and the Deutsche Forschungsgemeinschaft (Ei 222/12-1).
References
- [1].Kress TR, Sab∫ A, Amati B. MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer 2015; 15:593-607; PMID:26383138; http://dx.doi.org/ 10.1038/nrc3984 [DOI] [PubMed] [Google Scholar]
- [2].McMahon SB, Van Buskirk HA, Dugan KA, Copeland TD, Cole MD. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell 1998; 94:363-74; PMID:9708738; http://dx.doi.org/ 10.1016/S0092-8674(00)81479-8 [DOI] [PubMed] [Google Scholar]
- [3].Jaenicke LA, Eyss von B, Carstensen A, Wolf E, Xu W, Greifenberg AK, Geyer M, Eilers M, Popov N. Ubiquitin-Dependent turnover of MYC antagonizes MYC/PAF1C complex accumulation to drive transcriptional elongation. Mol Cell 2015; 61(1):54-67; PMID:26687678 [DOI] [PubMed] [Google Scholar]
- [4].Walz S, Lorenzin F, Morton J, Wiese KE, Eyss B, Herold S, Rycak L, Dumay-Odelot H, Karim S, Bartkuhn M, et al.. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014; 511:483-7; PMID:25043018; http://dx.doi.org/ 10.1038/nature13473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Eyss von B, Jaenicke LA, Kortlever RM, Royla N, Wiese KE, Letschert S, McDuffus L-A, Sauer M, Rosenwald A, Evan GI, et al.. A MYC-Driven Change in Mitochondrial Dynamics Limits YAP/TAZ Function in Mammary Epithelial Cells and Breast Cancer. Cancer Cell 2015; 28:743-57; PMID:26678338; http://dx.doi.org/ 10.1016/j.ccell.2015.10.013 [DOI] [PubMed] [Google Scholar]
- [6].Yu F-X, Zhao B, Guan K-L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015; 163:811-28; PMID:26544935; http://dx.doi.org/ 10.1016/j.cell.2015.10.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ni L, Zheng Y, Hara M, Pan D, Luo X. Structural basis for Mob1-dependent activation of the core Mst-Lats kinase cascade in Hippo signaling. Genes Dev 2015; 29:1416-31; PMID:26108669; http://dx.doi.org/ 10.1101/gad.264929.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhao B, Li L, Tumaneng K, Wang C-Y, Guan K-L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev 2010; 24:72-85; PMID:20048001; http://dx.doi.org/ 10.1101/gad.1843810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al.. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 2007; 21:2747-61; PMID:17974916; http://dx.doi.org/ 10.1101/gad.1602907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al.. Role of YAP/TAZ in mechanotransduction. Nature 2011; 474:179-83; PMID:21654799; http://dx.doi.org/ 10.1038/nature10137 [DOI] [PubMed] [Google Scholar]
- [11].Mo J-S, Meng Z, Kim YC, Park HW, Hansen CG, Kim S, Lim D-S, Guan K-L. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat Cell Biol 2015; 17:500-10; PMID:25751140; http://dx.doi.org/ 10.1038/ncb3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Choi S-Y, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol 2006; 8:1255-62; PMID:17028579; http://dx.doi.org/ 10.1038/ncb1487 [DOI] [PubMed] [Google Scholar]
- [13].Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR, Poletti A, et al.. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011; 147:759-72; PMID:22078877; http://dx.doi.org/ 10.1016/j.cell.2011.09.048 [DOI] [PubMed] [Google Scholar]
- [14].Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang C-Y, Chinnaiyan AM, et al.. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 2008; 22:1962-71; PMID:18579750; http://dx.doi.org/ 10.1101/gad.1664408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li Z, Zhao B, Wang P, Chen F, Dong Z, Yang H, Guan K-L, Xu Y. Structural insights into the YAP and TEAD complex. Genes Dev 2010; 24:235-40; PMID:20123905; http://dx.doi.org/ 10.1101/gad.1865810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Skibinski A, Breindel JL, Prat A, Galván P, Smith E, Rolfs A, Gupta PB, Labaer J, Kuperwasser C. The Hippo transducer TAZ interacts with the SWI/SNF complex to regulate breast epithelial lineage commitment. Cell Rep 2014; 6:1059-72; PMID:24613358; http://dx.doi.org/ 10.1016/j.celrep.2014.02.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Leung JY, Andrechek ER, Cardiff RD, Nevins JR. Heterogeneity in MYC-induced mammary tumors contributes to escape from oncogene dependence. Oncogene 2012; 31:2545-54; PMID:21996730; http://dx.doi.org/ 10.1038/onc.2011.433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007; 130:1120-33; PMID:17889654; http://dx.doi.org/ 10.1016/j.cell.2007.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan S-T, Luk JM, Wigler M, Hannon GJ, et al.. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006; 125:1253-67; PMID:16814713; http://dx.doi.org/ 10.1016/j.cell.2006.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, Kreger BT, Vasioukhin V, Avruch J, Brummelkamp TR, et al.. Yap1 acts downstream of α-catenin to control epidermal proliferation. Cell 2011; 144:782-95; PMID:21376238; http://dx.doi.org/ 10.1016/j.cell.2011.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chen Q, Zhang N, Gray RS, Li H, Ewald AJ, Zahnow CA, Pan D. A temporal requirement for Hippo signaling in mammary gland differentiation, growth, and tumorigenesis. Genes Dev 2014; 28:432-7; PMID:24589775; http://dx.doi.org/ 10.1101/gad.233676.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012; 22:725-36; PMID:23201165; http://dx.doi.org/ 10.1016/j.ccr.2012.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Eyss von B, Maaskola J, Memczak S, Möllmann K, Schuetz A, Loddenkemper C, Tanh M-D, Otto A, Muegge K, Heinemann U, et al.. The SNF2-like helicase HELLS mediates E2F3-dependent transcription and cellular transformation. EMBO J 2012; 31:972-85; PMID:22157815; http://dx.doi.org/ 10.1038/emboj.2011.451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mizuno H, Spike BT, Wahl GM, Levine AJ. Inactivation of p53 in breast cancers correlates with stem cell transcriptional signatures. Proc Natl Acad Sci USA 2010; 107:22745-50; PMID:21149740; http://dx.doi.org/ 10.1073/pnas.1017001108 [DOI] [PMC free article] [PubMed] [Google Scholar]