

ABSTRACT
The repair of DNA double-strand breaks (DSBs) by homologous recombination (HR) is an essential process in maintenance of chromosomal stability. A key player of HR is the strand exchange factor RAD51 whose assembly at sites of DNA damage is tightly regulated. We detected an endogenous complex of RAD51 with the calcium-binding protein S100A11, which is localized at sites of DNA repair in HaCaT cells as well as in normal human epidermal keratinocytes (NHEK) synchronized in S phase. In biochemical assays, we revealed that S100A11 enhanced the RAD51 strand exchange activity. When cells expressing a S100A11 mutant lacking the ability to bind Ca2+, a prolonged persistence of RAD51 in repair sites and nuclear γH2AX foci was observed suggesting an incomplete DNA repair. The same phenotype became apparent when S100A11 was depleted by RNA interference. Furthermore, down-regulation of S100A11 resulted in both reduced sister chromatid exchange confirming the restriction of the recombination capacity of the cells, and in an increase of chromosomal aberrations reflecting the functional requirement of S100A11 for the maintenance of genomic stability. Our data indicate that S100A11 is involved in homologous recombination by regulating the appearance of RAD51 in DSB repair sites. This function requires the calcium-binding activity of S100A11.
KEYWORDS: DNA double-strand breaks, genomic aberrations, homogenous recombination, RAD51, S100A11
Introduction
DNA double-strand breaks (DSBs) severely threaten genomic integrity. DSBs are highly toxic and can lead to illegitimate DNA changes, such as deletions or translocations contributing to cellular dysfunction, cell deaths or various disorders including cancer.1-3 DSBs are caused by endogenous sources, like oxygen radicals or by collapsed DNA replication forks as well as by exogenous sources, such as carcinogenic compounds or ionizing radiation.4,5 To avoid their deleterious consequences, cells have developed strategies to recognize and repair DSBs. A main repair pathway for DSBs in S or G2 phase cells is homologous recombination (HR), a largely error-free process that engage sister chromatids as template for DNA repair.6 In higher eukaryotes, HR is initiated by enzymatic processing of the broken double-stranded DNA (dsDNA) ends to generate resected 3´-single-stranded DNA (ssDNA) overhangs by several nucleases and helicases. These ssDNA tails are stabilized by the ssDNA binding protein, RPA, which is subsequently replaced by RAD51 to form a helical nucleoprotein filament. RAD51 is an evolutionarily conserved key factor of HR that exchanges DNA strands between homologous partners.7 The formation of the nucleoprotein filament containing RAD51 and ssDNA is strictly regulated by a number of factors. Most of these factors bind autonomously to DNA, such as BRCA1, BRCA2, and the RAD51 paralogs (RAD51B/C/D, XRCC2/3) as well as the chromatin remodeling protein p400. But GEMIN2 works as a RAD51 mediator without any DNA binding capacity.8-13 Therefore, it must be elucidated in detail, how several mammalian RAD51 mediators cooperate for the formation of RAD51/ssDNA-containing nucleoprotein filaments. Once formed, the RAD51/ssDNA-filament searches for a homologous template, which becomes invaded to form a displacement loop (D-loop), which in turn performs a strand exchange to generate heteroduplex DNA (hDNA). D-loop formation is stimulated by several regulators such as RAD54, its paralog RAD54B, PALB2 or RAD51AP1.14-19 The invading strand primes DNA repair synthesis on the homologous DNA template. Thereafter, the newly synthesized strand dissociates from hDNA and re-anneals with the other end of the DSB to complete DSB by HR.6 It has been shown that the strand exchange activity of human RAD51 protein is stimulated by Ca2+.20,21 In the presence of Ca2+, the ATPase activity of RAD51 is modulated resulting in a stabilized active RAD51/ATP/ssDNA-nucleoprotein filament with a reduced ATP hydrolysis rate. Intriguingly, the activation of human RAD51 by modulation of the ATPase seems to be an evolutionary recent event since Ca2+ ions were not able to stimulate yeast Rad51.20
In a former study, we detected a functional interaction between S100A11 and RAD54B.22 S100A11 was shown to be involved in the dynamic relocalization of RAD54B into DSB repair foci. RAD54B is a homolog of human RAD54 contributing to HR by an association with RAD51.15,23-25 S100A11, on the other hand, is an EF hand-type Ca2+-binding protein that functions as a homodimer in cells. It belongs to the S100 protein family possessing functions concerning calcium homeostasis and interaction with cytoskeletal components. S100A11 has been implicated in several distinct cellular processes such as cell growth and motility, cell cycle progression, and cell differentiation.26 Moreover, S100A11 was detected to be up- or down-regulated in different tumor entities.27 S100A11 is sensitive to DNA damage as it redistributed from the cytoplasm into the nucleus and accumulated in nuclear foci after induction of DSBs.28 Therefore, we asked whether S100A11 contributes to the detection and repair of DSBs. In the present study, we identified a RAD51/S100A11 interaction and characterized the impact of S100A11 both on RAD51 activity and persistence at DSB repair sites, as well as its importance for genome maintenance after genotoxic insult.
Results
S100A11 colocalized with RAD51 at sites of DNA damage repair
We have shown, that S100A11 responded to DSBs by translocation from the cytoplasm into the nucleus.28 We also detected a functional interaction between S100A11 and RAD54B, a factor implicated in HR.22 Therefore, we wondered about the specific role of S100A11 in homologous recombination. HR occurs predominantly in S phase and with a lesser content in G2 phase of the cell cycle when sister chromatid sequences are available as template for DNA repair.29 The subcellular distribution of S100A11 in HaCaT cells, synchronized in S phase (∼80%), with and without bleomycin-induced DSBs was examined by confocal laser scanning microscopy. In parallel, we assessed the phosphorylation of histone H2AX, one of the first cellular responses of DNA damage.30 We detected focal localization of S100A11 in the nucleoplasm of HaCaT cells treated with bleomycin, whereas in untreated control cells, S100A11 was mainly localized in the cytoplasm (Fig. 1A). Hence, in response to DNA damage a significant fraction of S100A11 migrated into the nucleus, and a subfraction thereof localized to γH2AX foci. On average, 7% of all S100A11 foci per nucleus showed co-localization with γH2AX (Fig. 1C). Very similarly, a comparable translocation of S100A11 was observed in γ-ray induced “ionizing radiation induced foci” (IRIF) demonstrating that the S100A11 response was independent from the cause of DSBs (Figure S1A). RAD51 as well as S100A11 responded to DNA damage with nuclear translocation without a change of protein expression (Figure S1B).28,31,32 Interestingly, both S100A11 and RAD51 were frequently found at the same discrete sites throughout the nucleoplasm of human HaCaT cells containing DNA damage (Fig. 1B). On average 26% of all S100A11 foci colocalized with RAD51, clearly indicating that during the DNA repair process a subpopulation of HR sites was associated with S100A11 foci (Fig. 1C). The ratio of RAD51/S100A11 colocalization was comparable to the ratio of colocalization of RAD51 with well-known interacting partners such as RAD52, BRCA2-interacting protein and BRCA1.33,34 Two hours after induction of DSBs, on average 16% of all RAD51 foci showed a co-localization with γH2AX signals in S phase HaCaT cells (Figure S1C). We also detected a colocalization of RAD51/S100A11 with γH2AX foci corresponding to DNA damage sites (Figure S1D). We further confirmed the colocalization of S100A11 and RAD51 in human primary cells after induction of DNA damage by bleomycin using normal human epidermal keratinocytes (NHEK) of juvenile foreskins (Figure S1E). Moreover, a specific antibody against S100A11 coprecipitated RAD51 from a crude extracts of S phase HaCaT cells, and interaction was increased after treatment with bleomycin, whereas neither protein was pulled down with an unspecific antibody (Fig. 1D). In a reciprocal experiment, FLAG-S100A11 was pulled-down from bleomycin-treated U2OS cells expressing both RAD51-myc-6xHis and FLAG-S100A11 using Talon resin (Clontech) (Fig. 1E). Here, we detected a weak, but clearly visible signal compared to a control pull-down from cells transfected with the FLAG-S100A11 encoding plasmid only. These results suggested the existence of a RAD51/S100A11 interaction in cells that is augmented after DSB damage.
Figure 1.
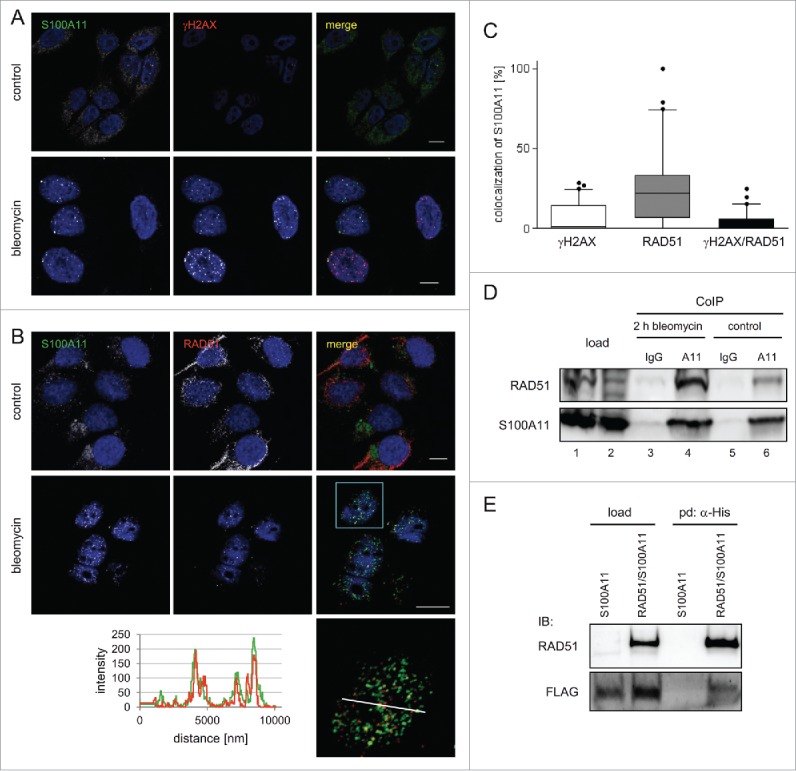
S100A11 interacts with RAD51 at sites of DSB repair. (A) HaCaT cells synchronized in S phase were treated with bleomycin (12.5 µg/ml) for 30 min and released in fresh medium for 90 min or were untreated (control) followed by immunostaining with antibodies against S100A11 (green) and γH2AX (red). Nuclear DNA was detected using DAPI (blue). Bar, 10 µm. (B) S phase HaCaT cells that were analyzed by laser scanning microscopy for S100A11 (green) and RAD51 (red) at sites of DNA damage 2 hour after bleomycin treatment. Bar, 10 µm. An area marked by a rectangle (light blue) in the overlay image (merge) is shown enlarged below the respective image. The intensities of the immunofluorescences in one cell derived from the RAD51 signal (red) and the S100A11 signal (green) are shown in a linescan (left side of the enlarged overlay). (C) Quantification of the colocalization of S100A11 with γH2AX (white Whisker box), RAD51 (gray Whisker box) and γH2AX/RAD51 (black Whisker box) in HaCaT cells after DSB induction. Colocalization events were determined in nuclei of cells treated as described in A. Thirty nuclei from 2 independent experiments were analyzed. Data are displayed as mean values (±SD). (D) Co-immunoprecipitation (CoIP) experiments between S100A11 and RAD51. A specific anti-S100A11 antibody precipitated RAD51 from whole cell extracts of S phase HaCaT cells treated with bleomycin (12.5 µg/ml) (lane 4) or untreated S phase cells (lane 6). In a control, an unspecific antibody used in CoIP experiments was unable to precipitate RAD51 from the same extracts (lanes 3 and 5). (E) The authenticity of the RAD51/S100A11 interaction was confirmed by pull-down with overexpressed proteins. U2OS cells were transfected with a plasmid encoding FLAG-S100A11 alone or together with a His-RAD51 encoding plasmid followed by synchronization in S phase and induction of DSBs by bleomycin treatment. Pull-down (pd) was carried out from cell extracts of the transfected cells for His-tagged proteins binding to Talon resins, and immunoblotting was performed using the antibodies as indicated. As loading control (load), 5% of the cell extract was used.
Intriguingly, S100A11 was only detected at sites of DSB repair in S phase cells when HR was active (Fig. 1A, B and Figure S1A,E). In bleomycin-damaged HaCaT cells synchronized at the G1/S boundary by aphidicolin treatment, S100A11 remained mainly in the cytoplasm, in strong contrast to the nuclear 53BP1 signal, a factor involved in DSB repair by NHEJ (Figure S1F).35
Furthermore, to elucidate potential factors involved in triggering of the S100A11 translocation after DNA damage we used pharmacological inhibitors. As it seems that the S100A11 translocation into the nucleus depended on the cell cycle phase in which the cell were damaged, we utilized the CDK inhibitor roscovitine and cells synchronized to the late S phase by release from a double thymidine block. We detected a significantly reduced amount of S phase HaCaT cells treated with both bleomycin and roscovitine showing nuclear S100A11 foci compared to control cells treated with only bleomycin (Figure S2). Treatment of the cells with 10 µM wortmannin, an ATM inhibitor, for 4 hours showed no impact on S100A11 distribution. This suggests that S100A11 translocation is regulated by S and/or G2 phase CDK activity.
S100A11 facilitated appropriate repair of DSBs by HR
To further explore the potential role of human S100A11 in DSB repair, we examined bleomycin-treated and S100A11-depleted cells by immunofluorescence and confocal laser scanning microscopy (Fig. 2 and Figure S3). The γH2AX signal was followed for 8 hours after induction of DSBs in S phase synchronized HaCaT cells both in terms of γH2AX positive cells and the γH2AX foci formation as an approximation of the number of DSBs and the kinetics of DNA repair.36 Our siRNAs efficiently depleted S100A11 (Fig. 2A). Before DSB induction, the average number of γH2AX foci per cell was comparable in controls treated with S100A11 and non-specific control (nsc) siRNA, respectively (Fig. 2B,C), even though a larger portion of the S100A11 knock-down cells showed γH2AX foci (Fig. 2B,D). Two hours after induction of DSBs, the number of γH2AX foci per cell increased and a comparable number of cells exhibiting γH2AX foci were detectable in S100A11- and mock-depleted cells (Fig. 2B-D), indicating comparable DNA damage signaling and initiation of DSB repair. As already described (Fig. 1A), a partial, focal colocalization of S100A11 with γH2AX at sites of DNA repair was consistently detectable in cells possessing S100A11. The knockdown of S100A11 abrogated the colocalization but had no influence on the formation of the γH2AX foci. Eight hours after damage induction, the average number of γH2AX foci in nsc siRNA mock-depleted HaCaT cells had returned to the level of untreated control cells indicating completion of repair (Fig. 2B-D). This was also reflected by the drop of γH2AX foci-positive cells to the control readings. In contrast, the number of S100A11 knockdown cells containing γH2AX signals as well as the number of foci per cell remained high reflecting a delay in the repair of DSBs.
Figure 2.

S100A11 is functionally required for complete DSB repair. (A) Down-regulation of S100A11 in HaCaT cells treated with specific siRNA against S100A11 (lane 2: S100A11 siRNA#6, lane 3: S100A11 siRNA#7) was confirmed by immunoblotting using a specific antibody against S100A11. As control, HaCaT cells transfected with nonspecific control (nsc) siRNA (lane1) were used. GAPDH served as loading control. (B) Experimental setup. HaCaT cells were transfected with the indicated siRNA and subsequently synchronized in S phase by double-thymidine block. DNA damage was induced by treatment with 12.5 µg/ml bleomycin for 30 min. Cells were harvested after different time points (2 and 8 h repair) for immunostaining against S100A11 and γH2AX. (C) γH2AX foci persist in damaged S100A11 knock-down cells. Control represents cells without bleomycin treatment. The number of γH2AX foci per cell is shown as Whisker graphs. Mean values represent 3 independent experiments with n > 30 cells analyzed for each condition. (D) Analysis of the percentage of individual cells possessing γH2AX foci. HaCaT cells were treated as described in B. Mean values represent analysis of n > 90 cells for each condition in 3 independent experiments. (E) RAD51 foci (white) persist upon DSB damage after S100A11 knock-down. HaCaT cells were treated as described in B and analyzed by immunostaining against RAD51 and S100A11. Nuclear DNA was detected using DAPI (blue). Bar, 10 µm. (F) Quantification of percentage of individual cell showing RAD51 foci. Mean values represent analysis of n > 85 cells for each condition in 3 independent experiments. * P < 0.05, *** P < 0.001.
The kinetics of RAD51 foci in both S100A11-depleted and undepleted cells displayed a temporal pattern that paralleled the appearance of γH2AX foci (Fig. 2B, E, F). The number of nsc siRNA treated cells with RAD51 foci decreased significantly between 2 and 8 hours of DNA repair, reaching background levels at the later time point. This was not the case after S100A11-depletion, where the number of RAD51-positive cells remained high (Fig. 2F). Hence, S100A11 did not affect the initiation of DNA damage repair by HR, but it strongly stimulated its completion.
S100A11 enhanced recombinase activities of RAD51
We purified recombinant RAD51 and S100A11 to near homogeneity in order to characterize their functional interaction in more detail (Figure S4). The authenticity of the proteins was confirmed by antibodies and mass spectrometry.
First, we assessed the effect of S100A11 concerning RAD51 within a strand exchange assay reflecting the HR process in vitro. Consistent with a previous study, the RAD51 strand exchange reaction was stimulated by Ca2+ compared to Mg2+ (Fig. 3A).20 Therefore, we performed strand exchange assays in the presence of Ca2+. The presence of the ssDNA-binding protein RPA was essential to stimulate the generation of both joint molecule intermediates and nicked-circular DNA product by RAD51. The addition of S100A11 to this reaction enhanced the formation of the joint molecule product (jm), as executed by RAD51, by more than 2-fold (Fig. 3B). From these data, we conclude that S100A11 promoted a RAD51-mediated strand exchange.
Figure 3.
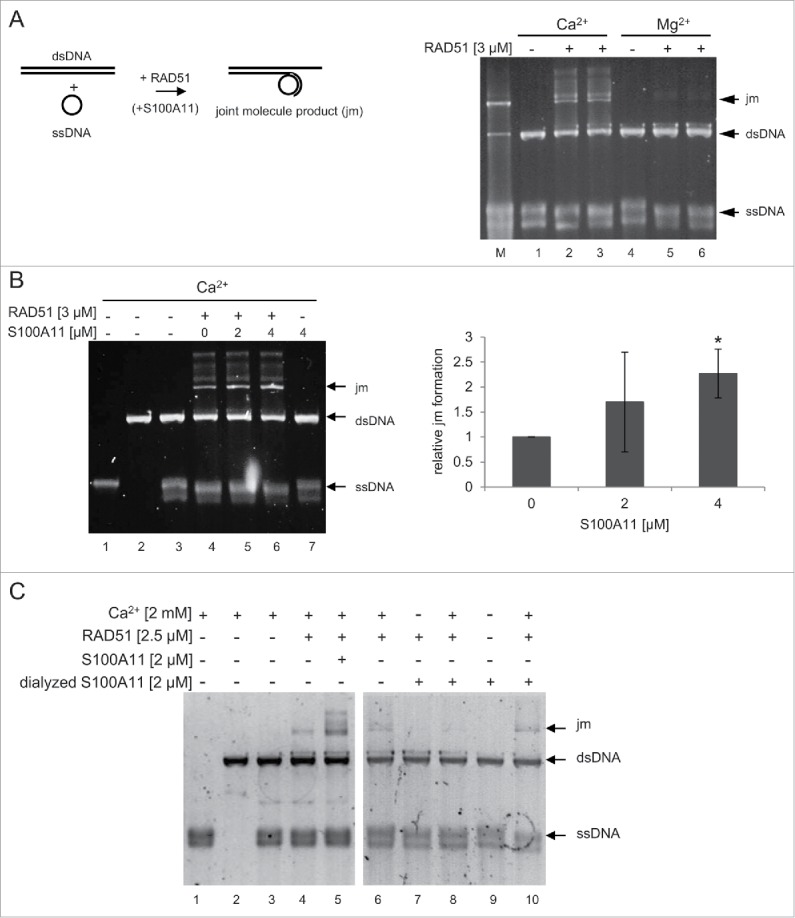
S100A11 stimulates the strand exchange activity of RAD51. (A) left panel Scheme of the strand exchange reaction between circular ssDNA and linearized dsDNA. right panel Strand exchange by human RAD51 requires Ca2+. RAD51, derived from 2 distinct purification procedures, (lanes 2–3 and 5–6: 3 µM) was incubated with 24 µM circular ΦX174 ssDNA in strand exchange buffer containing 2 mM of either magnesium or calcium acetate for 15 min at 37°C followed by incubation with 2.4 µM RPA for 5 min and addition of 24 µM linearized ΦX174 dsDNA to initiate strand exchange reaction for 2 h at 37°C. Lane M: constructed joint molecule DNA product derived from ssDNA/dsDNA annealing used as marker (B) left panel S100A11 enhances RAD51-mediated strand exchange. RAD51 (lanes 4–6: 3 µM) alone or with S100A11 (lane 5: 2 µM, lane 6: 4 µM) was incubated as described in (A) in strand exchange buffer containing calcium acetate (2 mM). As negative control, S100A11 (lane 7: 4 µM) was incubated alone. The joint molecule product (jm) was visualized by GelStar staining. right panel Quantification of S100A11-stimulated joint molecule formation by RAD51. Average values of 3 independent experiments are shown with standard derivation. (C) Dialysis of S100A11 abrogated the stimulating effect of S100A11 on RAD51 activity. RAD51 (lanes 4–8 and 10) together with undialyzed S100A11 (lane 5), S100A11 dialyzed in EGTA containing buffer (lanes 7–9), or S100A11 dialyzed in buffer without EGTA (lane 10), was incubated with 24 µM circular ΦX174 ssDNA in strand exchange buffer containing 2 mM of either magnesium or calcium acetate for 15 min at 37°C followed by incubation with 2.4 µM RPA for 5 min and addition of 24 µM linearized ΦX174 dsDNA to initiate strand exchange reaction for 2 h at 37°C. *P < 0.05.
Next, we assess the influence of the Ca2+-binding of S100A11 regarding the RAD51 activity as the binding of S100A11 to its interaction partners typically required the presence of the Ca2+ ion.37 Therefore, we dialyzed S100A11 in the presence of EGTA to detract Ca2+ ions from the purified protein before utilizing it in strand exchange assays (Fig. 3C). In contrast to “native” S100A11, the dialyzed S100A11 was not able to stimulate RAD51 in strand exchange reaction (Fig. 3C, compare lane 5 with lane 8). Hence, dialysis of S100A11 abrogated the stimulating effect of S100A11 on RAD51 activity.
Ca2+-binding deficient S100A11 mutant restricted DSB repair
It has been shown that the binding of Ca2+ by S100A11 triggers a structural change in the S100A11 homodimer inducing activated S100A11 protein capable to interact with its partners.38,39 Therefore, we constructed an S100A11 mutant protein deficient in binding of Ca2+ (S100A11ΔCa) and analyzed DNA repair after bleomycin treatment in S phase U2OS cells expressing S100A11wt or S100A11ΔCa (Fig. 4). The number of γH2AX foci 8 hours after induction of DNA damage, a period where DNA repair was usually completed in control cells (see Fig. 2), displayed a significant increase in the S100A11ΔCa mutant background compared to S100A11 wild-type expressing cells (Fig. 4A). Moreover, in cells expressing mutant S100A11ΔCa, γH2AX appeared as distinct focal signals probably reflecting uncompleted DNA repair. The corresponding analysis of S phase cells with RAD51 foci revealed similar results (Fig. 4B). Again, an increased number of cells with S100A11ΔCa showed RAD51 foci compared to cells expressing the wild-type form of S100A11 after 8 hours of DNA repair. Thus, S100A11ΔCa overexpression conferred a dominant-negative effect on DSB repair comparable to S100A11-depletion. As expected by eliminating of the Ca2+-binding capacity, the S100A11ΔCa mutant, in contrast to wild-type S100A11, did not seem to be able to interact with RAD51 as Talon resin failed to pull-down S100A11ΔCA from bleomycin-treated S phase U2OS cells expressing both RAD51-His and FLAG-S100A11ΔCa (Fig. 4D).
Figure 4.
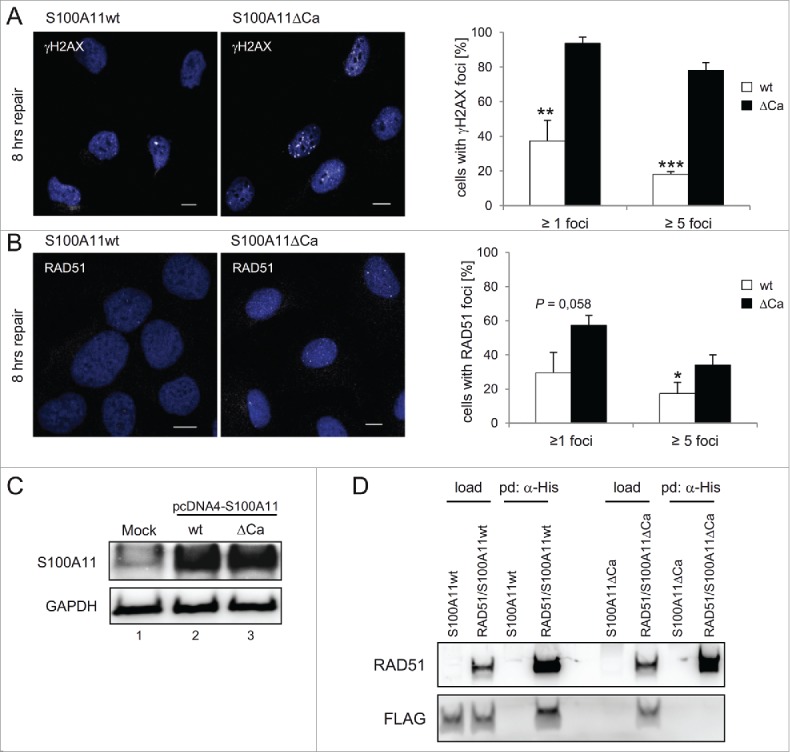
A S100A11 mutant without Ca2+-binding impairs DSB repair. (A) left panel Immunostaining of U2OS cells for γH2AX in cells expressing recombinant S100A11. Cells expressing S100A11ΔCa display significantly increased γH2AX levels 8 h after DSB induction. Cells transfected with a plasmid encoding S100A11wt or S100A11ΔCa, respectively, were synchronized in S phase, followed by treatment with 12.5 µg/ml bleomycin for 30 min. Cells were harvested 8 h after induction of DNA damage and analyzed by immunostaining against γH2AX (white). Nuclear DNA was detected using DAPI (blue). Bar, 10 µm. right panel Quantification of the percentage of cells showing γH2AX foci after S100A11wt (n = 88) or S100A11ΔCa (n = 99) transfection. The results of 3 independent experiments are presented. Data are shown as the mean ±SD. (B) Recombinant S100A11ΔCa expression increases RAD51 foci persistence. left panel U2OS cells were treated as above and analyzed by immunostaining against RAD51 (white). Nuclear DNA was detected using DAPI (blue). Bar, 10 µM. right panel Quantification of percentage of individual cells showing RAD51 foci. Mean values represent analysis of cells expressing S100A11wt (n = 94) or S100A11ΔCa (n = 99) of 3 independent experiments. (C) Expression of S100A11wt (lane 2) or S100A11ΔCa (lane 3) in pcDNA4-transfected U2OS cells was confirmed by immunoblotting against S100A11. An empty plasmid was used for mock-transfection of U2OS cells (lane 1). GAPDH served as loading control. (D) S100A11ΔCa mutant failed to interact with RAD51. U2OS cells were transfected with a plasmid encoding FLAG-S100A11 (wild-type or ΔCa mutant, respectively) alone or together with a His-RAD51 encoding plasmid followed by synchronization in S phase and induction of DSBs by bleomycin treatment. Pull-down (pd) was carried out from cell extracts of the transfected cells using Talon resins to precipitate His-tagged proteins together with interacting partners. For analysis of the pulled-down interacting proteins, immunoblotting was performed using the antibodies as indicated. As loading control (load), 5% of the cell extract was used. * P < 0.05, ** P < 0.01, *** P < 0.001.
S100A11 knockdown induced chromosomal aberrations and restricted cell viability
Here, we determined that S100A11 stimulated RAD51 strand exchange activity and was involved in HR as S100A11 downregulated cells possessed an impaired DSB repair. In extension to this, we elucidated the long-term consequences of an impaired S100A11 function on the cellular phenotype. To first assess the recombination capacity of S100A11-depleted cells, we analyzed sister chromatid exchange (SCE) formation. Here, S100A11 knock-down abolished SCEs derived from bleomycin-treated S phase HaCaT cells reflecting a restricted recombination (Fig. 5A), in line with the persistence of γH2AX and RAD51 foci after S100A11 depletion (Fig. 2), and thus confirming the results of the in vitro analysis that S100A11 enhanced recombination activity of RAD51 (Fig. 3). Since cellular HR defects should lead to gross chromosomal alterations, we monitored chromosomes from HaCaT metaphase cells after treatment with bleomycin (Fig. 5B–D). S100A11-depleted cells were more susceptible to the clastogenic impact of bleomycin whereas the level of chromosomal aberrations in mock-depleted controls was in line with previous studies with HaCaT cells.40 Chromosome breaks occurred more frequently in S100A11 down-regulated cells compared with control cells (Fig. 5B). Furthermore, S100A11-depleted cells exhibited also significantly more complex chromosomal aberrations (CCA) such as radial figures and double minutes than the controls (Fig. 5C). The increased number of chromosome breaks and the types of rearrangement in S100A11-depleted cells therefore reflected the deficient DNA repair by HR observed (Fig. 2). We also explored the effect of S100A11 depletion on the cell viability of DNA damaged HaCaT cells. The viability of S100A11-depleted HaCaT cells that were treated with bleomycin was significantly reduced (Fig. 5E). In contrast to this, mock-transfected HaCaT cells showed a comparable viability to untreated control cells (Fig. 5E). Hence, also these data confirm the important function of S100A11 in DNA repair.
Figure 5.
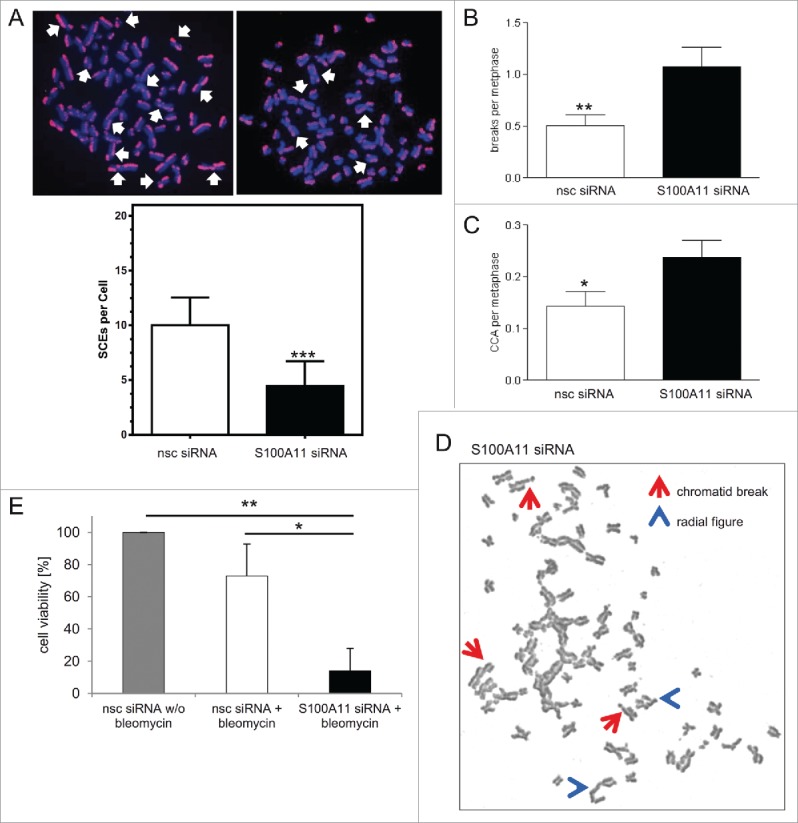
Downregulation of S100A11 results in restricted recombination capacity, chromosomal aberrations, and reduced cell viability. (A) S100A11 knock-down leads to a significant decrease in SCE. (top) Representative examples show multiple SCEs (left nsc siRNA, right S100A11 siRNA; white arrows) and (bottom) quantification of SCE per metaphase of 2 independent experiments. HaCaT cells with or without S100A11 siRNA were treated with bleomycin (12.5 µg/ml) for 30 min, allowed to recover for 2 h and transferred to medium containing 25 µM BrdU. After 4 h, 0.2 µg/ml Colcemid was added in fresh medium for 18 h to collect cells in metaphase. At least 40 metaphases per data point were analyzed. Error bars represent standard errors of the mean. (B-D) S100A11 knock-down leads to a significant increase of chromosomal abberations. HaCaT cells transfected with S100A11 siRNA or nsc siRNA were treated with 12.5 µg/ml bleomycin for 30 min. After further culture for 20 h, 0.1 µg/ml Colcemid was added for another 6 h to enrich cells in metaphase. The number of chromosome breaks (B) and complex chromosome aberrations (CCA) (C) are shown. Chromosome breaks and CCA were scored in n = 160 metaphase cells for each condition for 2 independent experiments. Error bars represent the standard errors of means. (D) Representative metaphase nuclei containing chromosomal aberrations as detected in S100A11-depleted HaCaT cells. Arrows (red) point to chromatid breaks; arrowheads (blue) point to radial figures. These aberrations are typical for DSBs occurred after DNA replication. (E) S100A11 knock-down leads to a significant loss of cell viability after bleomycin treatment. HaCaT cells transfected with specific S100A11 siRNA or control nsc siRNA for 72 h were treated with bleomycin (12.5 µg/ml) for 30 min. After this, medium was exchanged and the cells cultured for another 10 days. Then, the number of colonies formed was determined using Clono-Counter software in 3 independent experiments. HaCaT cells transfected with the control nsc siRNA without bleomycin treatment were used as control. * P < 0.05, ** P < 0.01, ***P < 0.001.
Discussion
We identified and characterized the functional interaction between RAD51, the key factor of HR, and S100A11, a protein that so far has been only found in vertebrates. S100A11 is differentially expressed in several tumors, where its expression pattern allows discrimination between distinct tumor entities.27,41 S100A11 is unique in the S100 family of Ca2+-binding proteins as it interacts with proteins involved in the repair of DSBs and is translocated to the nucleus in response to DNA damage.22,28 In contrast, another member of the S100 family, S100A6, translocates from the nucleus into the cytoplasm after induction of DNA damage.42
In the present study, we detected an endogenous RAD51/S100A11 complex that is formed in S phase HaCaT cells. Both proteins were found to localize at sites of DSB repair. There are 2 major pathways to repair double strand breaks: nonhomologous end-joining (NHEJ) and HR.3 NHEJ fixes DSBs in all cell cycle phases, whereas HR is solely involved in DSB repair in S and G2 phase cells, when template DNA in the form of sister chromatids is available. HR also contributes in multiple ways to the completion of DNA replication.43 In G1 phase cells, S100A11 predominantly resided in the cytoplasm even after DSB induction. In S phase cells S100A11 migrated into the nucleus where it colocalized with γH2AX at sites of DNA damage. It has been previously described that nuclear translocation of S100A11 is mediated by nucleolin and dependent on phosphorylation of residue threonine 10 by protein kinase Cα.44 Furthermore, an ATM-dependent phosphorylation of S100A11 inducing nuclear localization is suggested, whereas we did not observe an abrogation of nuclear localization employing the broad range PIKK inhibitor wortmannin in late S phase cells.45 Our data rather imply a requirement of S and/or G2 CDK activity for the translocation of S100A11 into the nucleus. In line with this, a cell cycle-dependent phosphorylation of the cyclin-dependent kinase consensus site serine 6 has been reported.46 This could represent a restriction of the S100A11 effect on HR to cell cycle phases were sister chromatids are available as HR templates, as reported for other HR factors such as BRCA1, RAP80, CtIP or EXO1.47-49
Only a minority of γH2AX foci colocalized with S100A11, which indicated that only a subpopulation of repair sites, possibly those undergoing HR, was associated with S100A11. Alternatively, S100A11 may only be transiently associated with the damage sites. Since S100A11 migration into foci was induced by bleomycin and γ-irradiation, we suggest a widespread involvement of S100A11 in DSB repair. As nuclear S100A11 foci occurred only in DNA damaged S phase cells, and were absent in cells at the G1/S transition, we deduced that S100A11 mainly was involved in DSB repair by HR rather than NHEJ.
This view was supported not only by the physical interaction between S100A11 and RAD51 demonstrated by immunoprecipitation, but also by biochemical assays using purified proteins. Our studies unveiled that S100A11 was able to enhance RAD51 activity corresponding to RAD51´s function in HR. We demonstrated a stimulating effect of S100A11 on the RAD51 strand exchange activity, which was comparable to the effect of several other RAD51 mediators.8,11,12,50,51 The stimulation of RAD51 reaction by S100A11 seemed to be dependent by Ca2+-binding of S100A11 as the removal of Ca2+ from S100A11 by dialysis resulted in an irreversible inactivation of the protein. Ca2+-binding of S100A11 is required to interact with partners which is agreeing to our result that an S100A11 mutant lacking Ca2+-binding (S100A11ΔCa), could not be pulled-down together with RAD51.37-39 Therefore, it seems that dialysis triggered an inhibitory conformation in the S100A11 protein resulting in the extinction of the RAD51 stimulation.
DSB repair in HaCaT S phase cells was completed 8 hours after induction of DNA damage, as judged by a disappearance of basically all γH2AX or RAD51 foci. This agreed well with published data indicating that DSB repair was near completion after 4 hours.52 In contrast to this, repair foci still persisted 8 hours after induction of DNA damage in cells depleted of S100A11. Since RAD51 localization to DSB repair foci was not impaired in S100A11-depleted cells, it is apparent that S100A11 affects DSB repair at a late step after RAD51 recruitment. Previously, we had already shown that S100A11 downregulation abrogated recognition of repair sites by RAD54B, another cofactor for RAD51-mediated strand exchange.22
Our in vitro assays demonstrated a stimulation of RAD51 by S100A11 and our cell biological studies suggested that in the absence of S100A11 cells could not appropriately complete DSB repair by HR. In contrast to its yeast ortholog, mammalian RAD51 is known to be Ca2+-dependent.20 In the light of the low intracellular Ca2+ concentrations in the cell, it is tempting to speculate that S100A11 is required for the intracellular delivery of Ca2+ to RAD51 during HR. In agreement with this view, S100A11 knockdown in control cells synchronized in S phase was sufficient to induce γH2AX formation suggesting a requirement of Ca2+-S100A11 for “background HR” caused by stalled or collapsed replication forks. The involvement of S100A11 in DSB repair was further elucidated using U2OS cells expressing an S100A11ΔCa mutant. With these cells we also confirmed the stimulatory effect of S100A11 on DSB repair, as U2OS cells expressing S100A11ΔCa still exhibited γH2AX as well as RAD51 foci representing unfinished DNA repair events even after 8 hours of DNA damage induction. Therefore, the depletion or inactivation of S100A11 in 2 cell lines caused a very similar phenotype as the disruption of other factors involved in DNA repair.12,53
Finally, an analysis of chromosome aberrations confirmed the suggested role of S100A11 in genome maintenance. We detected a significant increase of chromosomal aberrations such as chromosome breaks after S100A11 depletion unequivocally corroborating the diminished repair capacity of these cells. This is also underlined by the restricted capability for recombination of S100A11-depleted cells. Furthermore, an S100A11 knock-down provoked a serious drop in cell viability, also in support of an important role of S100A11 in DNA repair.
Based on the data presented here, we conclude that S100A11 assists the repair of DSBs by enhancing RAD51 activity and regulation of RAD51 persistence in DSB repair sites. Since HR and DSB repair are frequently targets of cancer treatment, S100A11 may become a future target of chemotherapeutic drugs.
Materials and methods
Cell culture and reagents
The human keratinocyte cell line HaCaT and U2OS osteosarcoma cells were cultured in DMEM supplemented with 10% fetal calf serum (FCS). Normal human epidermal keratinocytes (NHEK; kindly provided by P. Hemmerich, FLI Jena) were cultured in Keratinocyte Growth Medium 2 (ready-to-use; PromoCell, Heidelberg, Germany) supplemented with 10% FCS. Cells were grown to 80% confluence and were passaged at a split ratio of 1:5. Cells were harvested at 70–90% confluence and lysed in a buffer containing 100 mM sodium phosphate pH 7.5, 5 mM EDTA, 2 mM MgCl2, 0.1% CHAPS, 500 µM leupeptine, and 0.1 mM PMSF. After centrifugation (15 min; 15000 rpm) the supernatant was immediately processed further for immunoblotting. For immunoprecipitations, HaCaT cells synchronized in S phase by a double-thymidine block were treated with bleomycin (12,5 µg/ml) for 30 min. Afterwards, cells were released in fresh medium for further 90 min followed by lysis in RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 0.5% NP40, 0.25% SDS, 1 mM PMSF, 1 µg/ml leupeptine). For pull-down experiments using hexahistidine-tagged protein constructs, S phase U2OS cells expressing the desired proteins were treated with bleomycin (12,5 µg/ml) and lysed in buffer His (0.1 M Na-phosphate, pH 7.5, 2 mM MgCl2, CHAPS, 500 µM leupeptine 0.1 mM PMSF), and His-tagged proteins together with interacting partners were precipitated by Talon resin (Clontech, Mountain View, USA). For immunocytochemistry experiments, cells were synchronized in S phase by a double-thymidine block. Cells were treated with 2.5 mM thymidine for 16 h, released from the block for 9 hours by washing 3 times with PBS and blocked again by 2.5 mM thymidine for another 16 h. Afterwards, thymidine was washed off and cells were released in fresh medium at least for 1 h to enrich (∼80%) cells in S phase. For synchronization at the G1/S transition, cells were treated with 2 µg/ml aphidicolin for 24 h and enriched at G1/S (G1 cells >75%). Correct distribution of the cells in distinct cell cycle phases was confirmed by flow cytometric analysis. DBSs were induced by seeding cells on coverslips for 16 h and subsequent treatment with 12.5 µg/ml bleomycin for 30 min. After this, medium was exchanged and cells were harvested after different time points. When DNA damage was induced by γ-radiation, cells were irradiated with 5 Gy by exposure to a 137Cs source followed by a 1 h repair phase. For experiments using pharmacological inhibitors, S phase HaCaT cells were treated with wortmannin (10 µM) for 4.5 h or roscovitine (20 µM) for 2.5 h, respectively.
Plasmid constructions
Human RAD51 was PCR amplified from HaCaT cDNA using following oligonucleotides: 5′-GGAGAGGGTCCATGGCAATGGCAATGCAGATGCAGCTT-3′ (sense) and 5′-GTCTCCGGATCCGATTCAGTCTTTGGCATCTCCCACTCCATC-3′ (antisense). The PCR fragment was cloned between the NcoI and BamHI restriction site of a pET15b vector excluding the His-tag sequence of the vector. A pGEX-2T-S100A11 plasmid coding for wild-type S100A11 was kindly provided by N.H. Huh (Okayama University). An S100A11ΔCa construct with an eliminated Ca2+-binding capacity was created by site-directed exchanges of E38, D72, D76, and E79 to serine each (kindly provided by M. Sakaguchi, Okayama University).54 The S100A11wt gene as well as the S100A11ΔCa gene was cloned between the EcoRV and XbaI restriction sites of a pcDNA4-myc-6xHis vector. To generate RAD51-6xHis protein, human RAD51 gene was cloned between the BamHI and NotI restriction site of a pcDNA4-myc-6xHis vector. The S100A11wt gene or the S100A11ΔCa gene, respectively, was cloned between the EcoRV and BamHI restriction site of a p3xFLAG-CMV-10 vector. The correct insertion of all PCR products in the distinct vectors was confirmed in each case by DNA sequencing.
Protein expression and purification
The human RAD51 protein was overexpressed in the Escherichia coli strain BL21 (DE). About ten colonies of freshly transformed E. coli harboring the pET15b-RAD51 expression plasmid were used to inoculate each of 4 times 200 ml LB (supplemented with 100 µg/ml ampicillin). The cultures were left overnight at room temperature without shaking followed by growing with shaking (220 rpm) at 37°C to an OD600 of approx. 0.7 at the next morning. These four starter cultures were then filled up each to 1 l of LB medium supplemented with 100 µg/ml ampicillin and growth continued at 37°C up to OD600 0.9 – 1. Then, protein expression was induced by the addition of 0.5 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG). Thereafter, growth temperature was decreased to 30°C and cells were further cultured for 4 h. Then cells were harvested by centrifugation (10,000 × g for 12 min at 4°C), resuspended in buffer TA (30 mM Tris, pH 7.4, 100 mM KCl, 1 mM DTT, 10% glycerol (v/v), 0.5 mM EDTA) and centrifuged again. The cell pellet was processed immediately or stored at −80°C after snap-freezing for a maximum of 7 days. For purification of hRAD51, cell pellets derived from 4 liters of culture were resuspended and combined using prechilled buffer TA (supplemented with 0.1 mM PMSF and 1 µg/ml leupeptine). The cells were disrupted by 2 freezing/thawing cycles using liquid nitrogen followed by 3 times of sonication (Branson Sonifier) for 30 sec at 20% amplitude. The cell extract was clarified by centrifugation at 25,000 × g for 35 min at 4°C. Afterwards, the supernatant containing soluble hRAD51 protein was subjected for ammonium sulfate precipitation. Protein from 50 ml of the cell lysate was precipitated by 14 g of finely ground ammonium sulfate powder under stirring on ice for 40 min. The precipitate was collected at 25,000 × g for 35 min at 4°C and the pellet was subsequently resuspended in 50 ml of buffer TA. The solution was loaded onto a Q-Sepharose column HP 16/10 (GE Healthcare) pre-equilibrated with buffer TA. The column was washed with 5 column volumes of buffer TA. Proteins were eluted by a linear 0 – 600 mM KCl gradient in buffer TA using an Äkta Avant 25 device (GE Healthcare). Fractions containing RAD51 were pooled and dialyzed in buffer NaPi (100 mM sodium phosphate, pH 7.4, 100 mM KCl, 1 mM DTT, 10% glycerol (v/v)) for 7 h at 4°C. The dialyzed fraction was mixed with phosphocellulose resin pre-equilibrated with buffer NaPi in a batch-wise manner for 1 h at 4°C. The resin was washed 3 times with 10 batch volumes of buffer NaPi followed by fractionated elution with elution buffer (250 mM sodium phosphate, pH 7.3, 100 mM KCl, 1 mM DTT, 10 mM Na2S2O5, 10% glycerol (v/v)). Individual fractions containing RAD51 were dialyzed in storage buffer R (25 mM Tris, pH 7.5, 1 mM DTT, 200 mM KCl, 10% glycerol (v/v)) for 7 h at 4°C. Finally, individual fractions were concentrated using a Vivaspin 20 concentrator (GE Healthcare; molecular weight cut-off of 3,000 Da) at 4,000 × g at 4°C.
Human S100A11 protein was overexpressed in the E. coli strain BL21 (DE) and purified as described below. All purification steps were carried out at ≤4°C. One colony of freshly transformed E. coli harboring the pGEX-2T-S100A11 expression plasmid was used to inoculate 200 ml LB medium (supplemented with 100 µg/ml ampicillin). The culture was grown overnight with shaking (220 rpm) at 37°C. Afterwards, the culture was filled up with 1,200 ml LB medium supplemented with 100 µg/ml ampicillin and incubated at 37°C to OD600 0.6 – 0.8. Then, protein expression was induced for 18 h at 21°C by the addition of 0.23 mM IPTG. Cells were harvested by centrifugation (4,000 × g at 4°C for 15 min) and the cell pellet was processed immediately or stored at −80°C. For purification of the S100A11 protein, a cell pellet was resupended in 40 ml of prechilled lysis buffer (20 mM Tris, pH 7.5, 20% sucrose, 0.2 M NaCl, 10 mM MgCl2, 0.1 mM PMSF, 1 µg/ml leupeptine). The cells were disrupted by 2 freezing/thawing cycles using liquid nitrogen followed by sonication (3 times for 30 sec at 20% amplitude). For removal of DNA, 3 ml polyethyleneimine (PEI 600; 10% PEI-HCl, pH 7.8) was slowly added to the lysate under stirring over 20 min. Cell debris was separated from the protein crude extract by centrifugation (10,000 × g at for 15 min). Thereafter, the protein extract was mixed with glutathione agarose beads that were pre-equilibrated with lysis buffer (without protease inhibitors) in a batch-wise manner for 3 h at 4°C. After pelletizing (1,000 × g for 5 min), the beads were washed 4 times with 10 batch volumes lysis buffer. Then, S100A11 was separated from the glutathione-S-transferase (GST)-tag bound to the glutathione agarose beads by the addition of 120 units thrombin (diluted in 2.5 ml PBS) for 7 hours at room temperature. After a centrifugation step (4,000 × g for 15 min), the supernatant was incubated again with pre-equilibrated glutathione agarose beads for 8 h to eliminate possible GST contaminations from the protein extract. Afterwards, the solution was cleared by centrifugation (4,000 × g for 5 min) and the resulting protein extract was concentrated to 1 ml volume using a Vivaspin 20 concentrator at 4,000 × g. The protein extract containing S100A11 was loaded onto a Superdex 75 HighLoad 16/60 column (GE Healthcare) pre-equilibrated with storage buffer S (50 mM HEPES, pH 7.5, 100 mM NaCl, 10% glycerol) and a gel filtration chromatography was performed at 6°C using an Äkta Avant 25 device (GE Healthcare). The S100A11 containing fractions were pooled and incubated again with glutathione agarose beads pre-equilibrated with storage buffer S for 1 h. Finally, after clearance by centrifugation, the sample containing human S100 protein was concentrated using a Vivaspin 20 concentrator at 4,000 × g.
To detract Ca2+ ions from S100A11, the purified S100A11 protein was dialyzed in buffer D (50 mM HEPES, pH 7.5, 100 mM NaCl, 10% glycerol) with EGTA (2 mM) for 3 h and re-dialyzed in buffer D without EGTA for 3 h.
The purity of the proteins was examined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and the identity of the purified RAD51 protein was confirmed by both Western blot analysis and mass spectrometry. The identity of the S100A11 variants was confirmed by Western blot analysis. Purified proteins were stored at −80°C. Human RPA protein was purified as described earlier.55
DNA substrates for in vitro reactions
Single-stranded ΦX174 and ΦX174 replicative form I DNA were purchased from New England Biolabs (Ipswich, USA). Linearized double-stranded ΦX174 (replicative form III) DNA was prepared by XhoI digestion generating a 5´-overhang. In this study, the DNA concentrations for the ssDNA and dsDNA are expressed as moles of nucleotides and moles of base pairs, respectively.
Strand exchange assay
The ΦX174 circular ssDNA (24 µM nucleotides) was first incubated with RAD51 protein and S100A11 protein in 10 µl of strand exchange buffer (25 mM Tris-HCl, pH 7.5, 2.5 mM ATP, 1 mM DTT, 50 µg/ml BSA, 20 mM phosphocreatine, 0.1 µg/ml creatine kinase) with either 2 mM magnesium acetate or 2 mM calcium acetate for 15 min at 37°C. Then, 2.4 µM RPA was added and the reaction incubated for 5 min followed by the addition of linearized ΦX174 dsDNA (24 µM base pairs) to initiate strand exchange reaction for 2 hours at 37°C. The strand exchange products were deproteinized by addition of 0.7% SDS and 4.3 mg/ml proteinase K for 20 min at 37°C. Strand exchange products were separated by 1% agarose gel electrophoresis in 1 × TBE buffer at 25 V for 15 h at 4°C and visualized by GelStar (Lonza) staining for 1 h at 4°C followed by image recording using an Image Station (Syngene). Gel bands were densitometrically quantified using LabImage-1D (Kapelan Bio-Imaging, Leipzig, Germany).
RNA interference
Small interfering RNA (siRNA) duplex oligonucleotides were based on the human cDNAs encoding S100A11. siRNAs as well as a non-silencing control siRNA (nsc siRNA) were obtained from QIAGEN GmbH (Hilden, Germany). The siRNA applied to the S100A11 target sequence was 5′-CAGAACTAGCTGCCTTCACAA-3′ (S100A11si#6) or 5′-CAGCTAGATTTCTCAAGAATTT-3′ (S100A11si#7), respectively. The negative controls were 5′-UUCUCCGAACGUGUCACGUdTdT-3′ (sense) and 5′-ACGUGACACGUUCGGAGAAdTdT-3′ (antisense). Cells (1 × 105) were plated on 6-well plates 18 h prior to transfection and were 30% confluent when siRNA was added. The amount of siRNA duplexes applied was 1.5 µg/well. Transfection was performed using the amphiphilic delivery system SAINT-RED (Synvolux Therapeutics B.V., Groningen, The Netherlands) as described.22
Antibodies
Anti-S100A11 rabbit polyclonal antibody (BC001410; Protein Tech Group) at 1:1,000, anti-S100A11 sheep polyclonal antibody (PAS9364; Randox Laboratories Inc.) at 1:2,000, anti-RAD51 rabbit polyclonal antibody (sc-8349; Santa Cruz) at 1:200, anti-RAD51 goat polyclonal antibody (sc-7410; Santa Cruz) at 1:200, anti-GAPDH monoclonal antibody (5174; Cell Signaling) at 1.1,000, normal mouse IgG (500-M00; Pepro Tech Inc.) at 1:1,000, and anti-FLAG mouse monoclonal antibody (F1804; Sigma) at 1:500 were used in coimmunoprecipitation experiments and immunoblotting. Anti-S100A11 chicken polyclonal antibody (BP4007; Acris Antibodies) at 1:50, anti-RAD51 rabbit polyclonal antibody (ABE257; Millipore) at 1:250, anti-6xHis mouse monoclonal antibody (ab18184; Abcam) at 1:100, anti-53BP1 rabbit polyclonal antibody (sc-22760; Santa Cruz) at 1:50, anti-γH2AX (Ser139) (clone JBW301; 05-636; Millipore) at 1:750, and anti-γH2AX rabbit serum (kindly provided by P. Hemmerich, FLI Jena) at 1:100 were used in 2- or 3-color immunofluorescence staining as primary antibodies which were detected with species-specific secondary antibodies linked to fluorescein, Cy3, or Cy5 (Dianova) all at 1:200.
Coimmunoprecipitation and pull-down experiments
Coimmunoprecipitation assays were carried out as described.22 Briefly, specific anti-S100A11 antibody or, as negative control, normal mouse IgG were bound on protein A-agarose beads. Cell extracts (100 µl) from S phase HaCaT cells, treated with bleomycin (12.5 µg/ml) for 30 min or untreated, was incubated with antibody loaded beads for 1 h at 4°C. The resins were washed 3 times with RIPA buffer. Bound proteins were subjected to 12% SDS–PAGE and detected by immunoblotting. For pull-down experiments, U2OS cells transfected with a plasmid encoding FLAG-tagged S100A11 alone or together with a plasmid encoding His-tagged RAD51 were synchronized in S phase. Cells were treated with bleomycin (12.5 µg/ml) for 30 min and released in fresh medium for another 90 min. Cell extracts of these cells was applied to Talon resin (Clontech) (equilibrated in lysis buffer His, 10 mg/ml BSA) and allowed to bind at room temperature for 1 h on an end-to-end mixer. Unbound proteins were washed away using lysis buffer His and the Talon resin with the bound protein complexes was subsequently subjected to 12% SDS-PAGE and immunoblotting.
Immunocytochemistry and confocal microscopy
Cells grown on coverslips were fixed with 4% para-formaldehyde for 10 min at room temperature. Immunofluorescence was performed as previously described in ref. 22 Samples were scanned with a Zeiss LSM 510 laser scanning confocal device attached to an Axioplan 2 microscope using a 63x Plan-Apochromat oil objective (Carl Zeiss, Jena, Germany). Fluorescein, Cy3, or Cy5 dyes were excited by laser light at 488, 552, or 633 nm, respectively. DNA was detected using DAPI (4,6-diamidino-2-phenylindole) and 406 nm laser excitation. To avoid bleed-through effects, each dye was scanned independently using the multitracking function of the LSM 510 unit. Single optical sections were selected either from manual scans, or taken from stack projections. Images were merged using the LSM 510 (Carl Zeiss, Jena, Germany) software and stored as TIFF files. Figures were assembled from the TIFF files using Adobe Photoshop software.
Colony forming assay
To assess the survival rate of S100A11-depleted HaCaT cells after bleomycin treatment, 1 × 104 HaCaT cells were seeded into 6-well dishes overnight followed by treatment with S100A11 siRNA or, as control, with nsc siRNA. After 72 hours, the cells were treated with 12.5 µg/ml bleomycin for 30 min. After this, medium was exchanged and the cells cultured for another 10 days. Colonies were than washed twice with PBS, fixed with prechilled methanol for 15 min, washed once with PBS, stained with Giemsa dye for 15 min, rinsed with Aqua bidest, and finally air-dried. The number of colonies formed was then determined using Clono-Counter software.56
Chromosomal breaks analysis
HaCaT cells (7.5 × 104) were grown for 24 h before the cells were treated with S100A11 siRNA or with the control nsc siRNA for 48 h. To induce DSBs, cells were incubated with 12,5 µg/ml bleomycin for 30 min. After washing out with PBS (twice) the cells were cultured in fresh medium for another 20 h. To collect mitotic cells, 0.1 µg/ml Colcemid was added for 6 h. The cells were harvested and smoothly resuspended in 10 ml of freshly prepared prewarmed hypotonic solution (75 mM KCl) at 37°C for 35 min. The resuspended cells were swung shortly with 1 ml freshly prepared prechilled fixation solution (absolute methanol/glacial acetic acid, 3:1 (v/v)) and centrifuged (450 × g, 5 min). The pellet was resuspended in 10 ml of the prechilled fixation solution and centrifuged. This procedure was repeated twice and the metaphases were incubated in 1.5 ml fixation solution at −20°C for at least 3 h. Glass slides were prechilled in Milli-Q water for 24 h before 40 µl of the metaphase suspension was dripped on the slides localized in a humidity chamber preheated at 40°C. After dripping, slides were dried shortly (30 s) at 42°C on a heating plate. After continued drying at room temperature for 24 h, chromosomes were stained by Giemsa-GTG (G-bands by trypsin using Giemsa) as follows: Slides were treated with trypsin for 12 s in a Coplin jar, washed twice with PBS Gurr buffer (pH 6.88) and then stained with Giemsa solution for 10 min. Chromosomes were recorded by Metafer platform (MetaSystems Hard & Software GmbH, Altlussheim, Germany) and assessed using IKAROS software (MetaSystems). Chromosomal breaks as well as complex chromosome aberrations (CCA) were scored in > 150 metaphases for each condition in 2 independent experiments.
Sister chromatid exchange assay
Analysis of sister chromatid exchange (SCE) was performed as previously described.57 Briefly, exponentially growing HaCaT cells were grown for 52 h in the presence of S100A11 siRNA or control siRNA. Cells were then treated with bleomycin (12.5 µg/ml) for 30 min, allowed to recover for 2 h and transferred to medium containing 25 µM BrdU. After 4 h, 0.2 µg/ml Colcemid was added in fresh medium for 18 h to collect cells in metaphase. Metaphase spreads were prepared as described above. Slides were then incubated for 1 h with rat anti-BrdU (AbD Serotec, ICR1) at a dilution of 1:1000. After being washed 3 times with cold PBS, metaphases were incubated for 1.5 h with anti-rat Alexa Fluor 555 (Molecular Probes) at a dilution of 1:1000. DAPI was used for counterstaining and immunofluorescence was observed with the Zeiss AxioObserver.Z1 microscope as described above. At least 40 metaphases of 2 independent experiments per data point were analyzed and expressed as means, unpaired t-test was used for statistical analysis.
Statistical analysis
Each experiment was repeated at least 3 times unless stated otherwise. Data are presented as the mean ±SD. Statistical comparisons were performed using 2-sided Student's t test, Spearman´s rank correlation coefficient, or Mann-Whitney U test (for data sets diverging from normal distribution). A P value of < 0.05 was considered statistically significant. All statistical tests were 2-sided.
Supplementary Material
Abbreviations
- CCA
complex chromosomal aberrations
- DSBs
double-strand breaks
- HR
homologous recombination
- NHEJ
nonhomology end-joining
- NHEK
normal human epidermal keratinocytes
- SCE
sister chromatid exchange
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Drs Peter Hemmerich, Masakiyo Sakaguchi and Nam-ho Huh for materials, Christoph Biskup for support, Shamci Monajembashi for assistance at the γ-irradiation source and Peter Hemmerich as well as Heinz Peter Nasheuer for critical reading of the manuscript. Excellent technical assistance by Annerose Gleiche, Annett Wiesenburg, and Anita Willitzer is acknowledged.
Funding
This work was supported by the Wilhelm Sander-Stiftung (2011.004.1 to C.M.).
References
- [1].Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature 2001; 411:366-74; PMID:11357144; http://dx.doi.org/ 10.1038/35077232 [DOI] [PubMed] [Google Scholar]
- [2].Jackson SP, Bartek J. The DNA-damage response in human biology and diseases. Nature 2009; 461:1071-8; PMID:19847258; http://dx.doi.org/ 10.1038/nature08467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chapman JR, Taylor MRG, Boulton SJ. Playing the end game: DNA double-strand repair pathway choice. Mol Cell 2012; 47:497-510; PMID:22920291; http://dx.doi.org/ 10.1016/j.molcel.2012.07.029 [DOI] [PubMed] [Google Scholar]
- [4].Pfeiffer P, Goedecke W, Obe G. Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations. Mutagenesis 2000; 5:289-302; http://dx.doi.org/ 10.1093/mutage/15.4.289 [DOI] [PubMed] [Google Scholar]
- [5].Lehmann AR, Fuchs RP. Gaps and forks in DNA replication: rediscovering old models. DNA Repair 2006; 5:1495-8; PMID:16956796; http://dx.doi.org/ 10.1016/j.dnarep.2006.07.002 [DOI] [PubMed] [Google Scholar]
- [6].San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 2008; 77:229-57; PMID:18275380; http://dx.doi.org/ 10.1146/annurev.biochem.77.061306.125255 [DOI] [PubMed] [Google Scholar]
- [7].Sung P. Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science 1994; 265:1241-43; PMID:8066464; http://dx.doi.org/ 10.1126/science.8066464 [DOI] [PubMed] [Google Scholar]
- [8].Sigurdson S, Van Komen S, Bussen W, Schild D, Albala JS, Sung P. Mediator function of the human Rad51B-Rad51C complex in Rad51/RPA-catalyzed DNA strand exchange. Genes Dev 2001; 15:3308-18; PMID:11751636; http://dx.doi.org/ 10.1101/gad.935501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Thacker J, Zdzienicka MZ. The mammalian XRCC genes: their role in DNA repair and genetic stability. DNA Repair 2003; 2:655-72; PMID:12767346; http://dx.doi.org/ 10.1016/S1568-7864(03)00062-4 [DOI] [PubMed] [Google Scholar]
- [10].Nagaraju G, Scully R. Minding the gap: the underground functions of BRCA1 and BRCA2 at stalled replication forks. DNA Repair 2007; 6:1018-31; PMID:17379580; http://dx.doi.org/ 10.1016/j.dnarep.2007.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jensen RB, Carreira A, Kowalczykowski SC. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010; 467:678-83; PMID:20729832; http://dx.doi.org/ 10.1038/nature09399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Takizawa Y, Qing Y, Takaku M, Ishida M, Morozuma Y, Tsujita T, Kogame T, Hirota K, Takahashi M, Shibata T, et al.. GEMIN2 promotes accumulation of RAD51 at double-strand breaks in homologous recombination. Nucleic Acids Res 2010; 38:5059-74; PMID:20403813; http://dx.doi.org/ 10.1093/nar/gkq271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Courilleau C, Chailleux C, Jaurneau A, Grimal F, Briois S, Boutet-Robinet E, Boudsocq F, Trouche D, Canitrot Y. The chromatin remodeler p400 ATPase facilitates Rad51-mediated repair of DNA double-strand breaks. J Cell Biol 2012; 199:1067-81; PMID:23266955; http://dx.doi.org/ 10.1083/jcb.201205059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Alexeev A, Mazin AV, Kowalczykowski SC. Rad54 protein possesses chromatin-remodeling activity stimulated by the Rad51-ssDNA nucleoprotein filament. Nat Struct Biol 2003; 10:182-6; PMID:12577053; http://dx.doi.org/ 10.1038/nsb901 [DOI] [PubMed] [Google Scholar]
- [15].Wesoly J, Agarwal S, Sigurdsson S, Bussen W, Van Komen S, Qin J, van Steeg H, van Benthem J, Wassenaar E, Baarends WM, et al.. Differential contributions of mammalian Rad54 paralogs to recombination, DNA damage repair, and meiosis. Mol Cell Biol 2006; 26:976-89; PMID:16428451; http://dx.doi.org/ 10.1128/MCB.26.3.976-989.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Modesti M, Budzowska M, Baldeyron C, Demmers JA, Ghirlando R, Kanaar R. RAD51AP1 is a structure-specific DNA binding protein that stimulates joint molecule formation during RAD51-mediated homologous recombination. Mol Cell 2007; 28:468-81; PMID:17996710; http://dx.doi.org/ 10.1016/j.molcel.2007.08.025 [DOI] [PubMed] [Google Scholar]
- [17].Dray E, Etchin J, Wiese C, Saro D, Williams GJ, Hammel M, Yu X, Galkin VE, Liu D, Tsai MS, et al.. Enhancement of RAD51 recombinase activity by the tumor suppressor PALB2. Nat Struct Mol Biol 2010; 17:1255-9; PMID:20871616; http://dx.doi.org/ 10.1038/nsmb.1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dunlop MH, Dray E, Zhao W, San Filippo J, Tsai MS, Leung SG, Schild D, Wiese C, Sung P. Mechanistic insights into RAD51-associated protein 1 (RAD51AP1) action in homologous DNA repair. J Biol Chem 2012; 287:12343-7; PMID:22375013; http://dx.doi.org/ 10.1074/jbc.C112.352161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wright WD, Heyer WD. Rad54 functions as a heteroduplex DNA pump modulated by its DNA substrates and Rad51 during D-loop formation. Mol Cell 2014; 53:420-32; PMID:24486020; http://dx.doi.org/ 10.1016/j.molcel.2013.12.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bugreev DV, Mazin AV. Ca2+ activates human homologous recombination protein Rad51 by modulating its ATPase activity. Proc Natl Acad Sci USA 2004; 101:9988-93; PMID:15226506; http://dx.doi.org/ 10.1073/pnas.0402105101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mazina OM, Mazin AV. Human Rad54 protein stimulates DNA strand exchange activity of hRad51 protein in the presence of Ca2+. J Biol Chem 2004; 279:52042-51; PMID:15466868; http://dx.doi.org/ 10.1074/jbc.M410244200 [DOI] [PubMed] [Google Scholar]
- [22].Murzik U, Hemmerich P, Weidtkamp-Peters S, Ulbricht T, Bussen W, Hentschel J, von Eggeling F, Melle C. Rad54B targeting to DNA double-strand repair sites requires complex formation with S100A11. Mol Biol Cell 2008; 19:2926-35; PMID:18463164; http://dx.doi.org/ 10.1091/mbc.E07-11-1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tanaka K, Hiramoto T, Fukuda T, Miyagawa K. A novel human Rad54 homologue, Rad54B, associates with Rad51. J Biol Chem 2000; 275:26316-21; PMID:10851248; http://dx.doi.org/ 10.1074/jbc.M910306199 [DOI] [PubMed] [Google Scholar]
- [24].Miyagawa K, Tsuruga T, Kinomura A, Usui K, Katsura M, Tashiro S, Mishima H, Tanaka K. A role for RAD54B in homologous recombination in human cells. EMBO J 2002; 21:175-80; PMID:11782437; http://dx.doi.org/ 10.1093/emboj/21.1.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Otterlei M, Bruheim P, Ahn B, Bussen W, Karmakar P, Baynton K, Bohr VA. Werner syndrome protein participates in a complex with RAD51, RAD54, RAD54B and ATR in response to ICL-induced replication arrest. J Cell Sci 2006; 119:5137-46; PMID:17118963; http://dx.doi.org/ 10.1242/jcs.03291 [DOI] [PubMed] [Google Scholar]
- [26].Sakaguchi M, Huh NH. S100A11, a dual growth regulator of epidermal keratinocytes. Amino Acids 2011; 41:797-807; PMID:20872027; http://dx.doi.org/ 10.1007/s00726-010-0747-4 [DOI] [PubMed] [Google Scholar]
- [27].Salama I, Malone PS, Mihaimeed F, Jones JL. A review of the S100 proteins in cancer. Eur J Surg Oncol 2008; 34:357-64; PMID:17566693; http://dx.doi.org/ 10.1016/j.ejso.2007.04.009 [DOI] [PubMed] [Google Scholar]
- [28].Gorsler T, Murzik U, Ulbricht T, Hentschel J, Hemmerich P, Melle C. DNA damage-induced translocation of S100A11 into the nucleus regulates cell proliferation. BMC Cell Biol 2010; 11:100; PMID:21167017; http://dx.doi.org/ 10.1186/1471-2121-11-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mao Z, Bozzella M, Seluanov A, Gorbunova V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008; 18:2902-6; http://dx.doi.org/ 10.4161/cc.7.18.6679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rogakou EP, Pilch DR, Orr OH, Ivanova VS, Bonner WM. DNA double-strand breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998; 273:5858-68; PMID:9488723; http://dx.doi.org/ 10.1074/jbc.273.10.5858 [DOI] [PubMed] [Google Scholar]
- [31].Chen F, Nastasi A, Shen Z, Brenneman M, Crissman H, Chen DJ. Cell cycle-dependent protein expression of mammalian homologs of yeast DNA double-strand break repair genes Rad51 and Rad52. Mutat Res 1997; 384:205-11; PMID:9330616; http://dx.doi.org/ 10.1016/S0921-8777(97)00020-7 [DOI] [PubMed] [Google Scholar]
- [32].Gildemeister OS, Sage JM, Knight KL. Cellular redistribution of Rad51 in response to DNA damage: novel role for Rad51C. J Biol Chem 2009; 284:31945-52; PMID:19783859; http://dx.doi.org/ 10.1074/jbc.M109.024646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wray J, Lui J, Nickoloff JA, Shen Z. Distinct RAD51 associations with RAD52 and BRCCIP in response to DNA damage and replication stress. Cancer Res 2008; 68:2699-707; PMID:18413737; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-6505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Huh MS, Ivanochko D, Haschem LE, Curtin M, Delorme M, Goodall E, Yan K, Picketts DJ. Stalled replication forks within heterochromatin require ATRX for protection. Cell Death Dis 2016; 7:e2220; http://dx.doi.org/ 10.1038/cddis.2016.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Xiong X, Du Z, Wang Y, Feng Z, Fan P, Yan C, Willers H, Zhang J. 53BP1 promotes microhomology-mediated end-joining in G1-phase cells. Nucleic Acids Res 2015; 43:1659-70; PMID:25586219; http://dx.doi.org/ 10.1093/nar/gku1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 1999; 146:905-16; PMID:10477747; http://dx.doi.org/ 10.1083/jcb.146.5.905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Santamaria-Kisiel L, Rintala-Dempsey AC, Shaw GS. Calcium-dependent and –independent interactions of the S100 protein family. Biochem J 2006; 396:201-14; PMID:16683912; http://dx.doi.org/ 10.1042/BJ20060195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rety S, Osterloh D, Arie JP, Tabaries S, Seeman J, Russo-Marie F, Gerke V, Lewit-Bentley A. Structural basis of the Ca(2+)-dependent association between S100C (S100A11) and its target, the N-terminal part of annexin I. Structure 2000; 8:175-84; PMID:10673436; http://dx.doi.org/ 10.1016/S0969-2126(00)00093-9 [DOI] [PubMed] [Google Scholar]
- [39].Dempsey AC, Walsh MP, Shaw GS. Unmasking the annexin I interaction from the structure of Apo-S100A11. Structure 2003; 11:887-97; PMID:12842051; http://dx.doi.org/ 10.1016/S0969-2126(03)00126-6 [DOI] [PubMed] [Google Scholar]
- [40].Boukamp P, Popp S, Krunic D. Telomere-dependent chromosomal instability. J Investig Dermatol Symp Proc 2005; 10:89-94; PMID:16358816; http://dx.doi.org/ 10.1111/j.1087-0024.2005.200401.x [DOI] [PubMed] [Google Scholar]
- [41].Melle C, Ernst G, Schimmel B, Bleul A, von Eggeling F. Colon-derived liver metastasis, colorectal carcinoma, and hepatocellular carcinoma can be discriminated by Ca2+-binding proteins S100A6 and S100A11. PLoS ONE 2008; 3:e3767; PMID:19048101; http://dx.doi.org/ 10.1371/journal.pone.0003767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Orre LM, Pernemalm M, Lenggvist J, Lewensohn R, Lehtiö J. Up-regulation, modification, and translocation of S100A6 induced by exposure to ionizing radiation revealed by proteomics profiling. Mol Cell Proteomics 2007; 6:2122-31; PMID:17785350; http://dx.doi.org/ 10.1074/mcp.M700202-MCP200 [DOI] [PubMed] [Google Scholar]
- [43].Aze A, Zhou JC, Costa A, Costanzo V. DNA replication and homologous recombination factors: acting together to maintain genome stability. Chromosoma 2013; 122:401-13; PMID:23584157; http://dx.doi.org/ 10.1007/s00412-013-0411-3 [DOI] [PubMed] [Google Scholar]
- [44].Sakaguchi M, Miyazaki M, Sonegawa H, Kashiwagi M, Ohba M, Kuroki T, Namba M, Huh NH. PKCa mediates TGFb-induced growth inhibition of human keratinocytes via phosphorylation of S100C/A11. J Cell Biol 2004; 164:979-84; http://dx.doi.org/ 10.1083/jcb.200312041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kozlov SV, Waardenberg AJ, Engholm-Keller K, Arthur JW, Graham ME, Lavin MF. Reactive oxygen species (ROS)-activated ATM-dependent phosphorylation of cytoplasmic substrates identified by large scale phosphoproteomics screen. Mol Cell Proteomics 2016; 15:1032-47; PMID:26699800; http://dx.doi.org/ 10.1074/mcp.M115.055723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dephoure N, Zhou C, Villen J, Beausoleil CE, Elledge SJ, Gygi SP. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci USA 2008; 105:10762-7; PMID:18669648; http://dx.doi.org/ 10.1073/pnas.0805139105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Johnson N, Cai D, Kennedy RD, Pathania S, Arora M, Li YC, D'Andrea AD, Parvin JD, Shapiro GI. Cdk1 participates in BRCA1-dependent S phase checkpoint control in response to DNA damage. Mol Cell 2009; 35:327-39; PMID:19683496; http://dx.doi.org/ 10.1016/j.molcel.2009.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cho HJ, Oh YJ, Han SH, Chung HJ, Kim CH, Lee NS, Kim WJ, Choi JM, Kim H. Cdk1 protein-mediated phosphorylation of receptor-associated protein 80 (RAP80) serine677 modulates DNA damage-induced G2/M checkpoint and cell survival. J Biol Chem 2013; 288:3768-76; PMID:23264621; http://dx.doi.org/ 10.1074/jbc.M112.401299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tomimatsu N, Mukherjee B, Catherine Hardebeck M, Ilcheva M, Vanessa Camacho C, Louise Harris J, Porteus M, Llorente B, Khanna KK, Burma S. Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nat Commun 2014; 5:3561; PMID:24705021; http://dx.doi.org/ 10.1038/ncomms4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Seong C, Colavito S, Kwon Y, Sung P, Krejci L. Regulation of RAD51 recombinase presynaptic filament assembly via interactions with the RAD52 mediator and the Src2 anti-recombinase. J Biol Chem 2009; 284:24363-71; PMID:19605344; http://dx.doi.org/ 10.1074/jbc.M109.032953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Buisson R, Dion-Cote AM, Coulombe Y, Launay H, Cai H, Stasiak AZ, Stasiak A, Xia B, Masson J. Y. Cooperation of breast cancer proteins PALP2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol 2010; 17:1247-54; PMID:20871615; http://dx.doi.org/ 10.1038/nsmb.1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 2004; 118:699-713; PMID:15369670; http://dx.doi.org/ 10.1016/j.cell.2004.08.015 [DOI] [PubMed] [Google Scholar]
- [53].Bolderson E, Tomimatsu N, Richard DJ, Boucher D, Kumar R, Pandita TK, Burma S, Khanna KK. Phosphorylation of Exo1 modulates homologous recombination repair of DNA double-strand breaks. Nucleic Acids Res 2010; 38:1821-31; PMID:20019063; http://dx.doi.org/ 10.1093/nar/gkp1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Jaiswal JK, Lauritzen SP, Scheffer L, Sakaguchi M, Bunkenborg J, Simon SM, Kallunki T, Jäättelä M, Nylandsted J. S100A11 is required of efficient plasma membrane repair and survival of invasive cancer cells. Nat Commun 2014; 5:3795; PMID:24806074; http://dx.doi.org/ 10.1038/ncomms4795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Henricksen LA, Umbrich CB, Wold MS. Recombinant replication protein A: expression, complex formation, and functional characterization. J Biol Chem 1994; 269:11121-32; PMID:8157639 [PubMed] [Google Scholar]
- [56].Niyazi M, Niyazi I, Belka C. Counting colonies of clonogenic assays by using densitometric software. Radiat Oncol 2007; 2:4; PMID:17212832; http://dx.doi.org/ 10.1186/1748-717X-2-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Conrad S, Künzel J, Löbrich M. Sister chromatid exchanges occur in G2-irradiated cells. Cell Cycle 2011; 10:222-8; PMID:21224723; http://dx.doi.org/ 10.4161/cc.10.2.14639 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.