

Abstract

The structure-based design of 1, 2, 3, 4-tetrahydroisoquinoline derivatives as selective DDR1 inhibitors is reported. One of the representative compounds, 6j, binds to DDR1 with a Kd value of 4.7 nM and suppresses its kinase activity with an IC50 value of 9.4 nM, but it is significantly less potent for a panel of 400 nonmutated kinases. 6j also demonstrated reasonable pharmacokinetic properties and a promising oral therapeutic effect in a bleomycin-induced mouse pulmonary fibrosis model.
Introduction
Discoidin domain receptors (i.e., DDR1 and DDR2) are transmembrane receptor tyrosine kinases (RTKs) that specifically recognize fibrillar collagens as extracellular ligands.1−3 DDR1 and DDR2 are highly involved in fundamental cellular processes, including cell proliferation, migration, adhesion, and matrix remodeling.4−11 The dysregulation of DDR1 has been linked to a variety of human cancers and inflammatory conditions such as fibrotic disorders and atherosclerosis.4−11 Collective evidence indicates a critical link between DDR1 and pulmonary fibrosis, a lethal disease with few therapeutic options.6,8,11,12 For instance, a DDR1 deletion has been reported to alleviate bleomycin (BLM)-induced lung inflammation and pulmonary fibrosis by blocking P38 mitogen-activated protein kinase (p38 MAPK) activation.12
We and others have identified several classes of DDR1 inhibitors with different selectivity profiles and have demonstrated their therapeutic potential for various human cancers (Figure 1).13−18,28 However, these small molecules still have relatively poor target specificity and there are limited reports on the efficacy of the pharmacologic inhibition of DDR1 in models of pulmonary fibrosis.8 In this article, we report the structure-based design of tetrahydroisoquinoline derivatives as new highly selective DDR1 inhibitors with promising therapeutic effects in a BLM-induced pulmonary fibrosis mouse model.
Figure 1.

Selective DDR1/DDR2 kinase inhibitors.
Results and Discussion
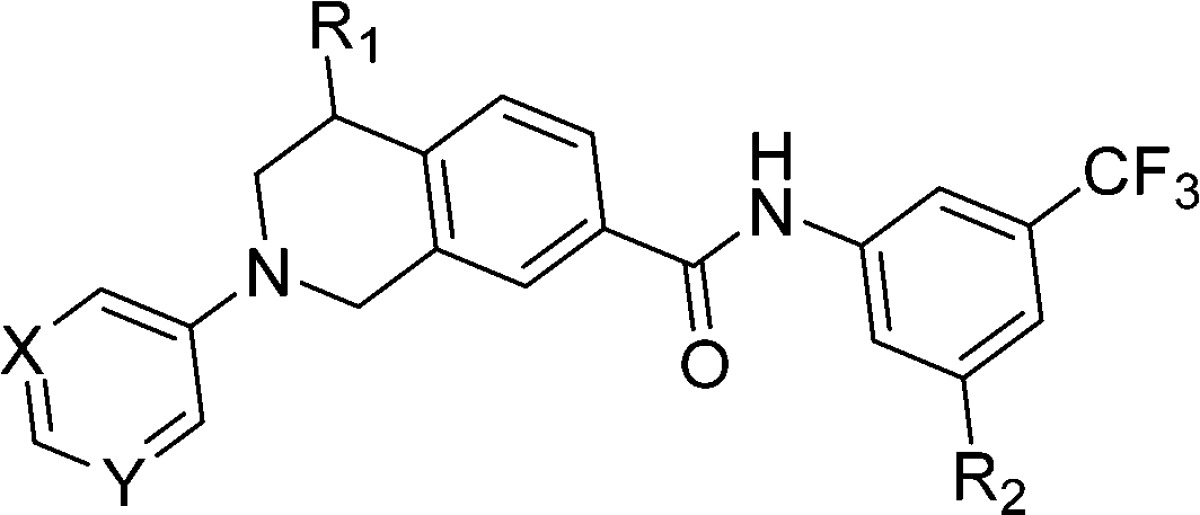
DDR1 shares approximately 61% sequence identity with Abelson (Abl) kinase in its adenosine triphosphate (ATP) binding domain, and most reported selective DDR1 inhibitors are derivatized from Abl antagonists.19 Previous investigations revealed that a π–π stacking interaction between the chemical molecule and Tyr253 of Abl is critical for most of the reported Abl inhibitors (Supporting Information (SI), Figure S1A,B,C),20−22 but the corresponding interaction is unnecessary for DDR1 binding. Indeed, DDR1-IN-1 achieved selective DDR1 inhibition because its hinge binding moiety is oriented away from the P-loop, avoiding the potential π–π interactions with Tyr253 in Abl.23 However, the molecule could form an additional π–π interaction with Phe382 in Abl, which contributed greatly to its relatively low selectivity between DDR1 and Abl (SI, Figure S1D).21,22 Diminishing this interaction may further improve DDR1 selectivity. On the basis of this hypothesis, a series of 1, 2, 3, 4-tetrahydroisoquinoline derivatives were designed as novel, selective DDR1 inhibitors in which a pyrimidinyl group was utilized as the potential hinge binding moiety. A N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl) phenyl)carboxamide group was also introduced based on our previous investigation (Figure 2A).13
Figure 2.
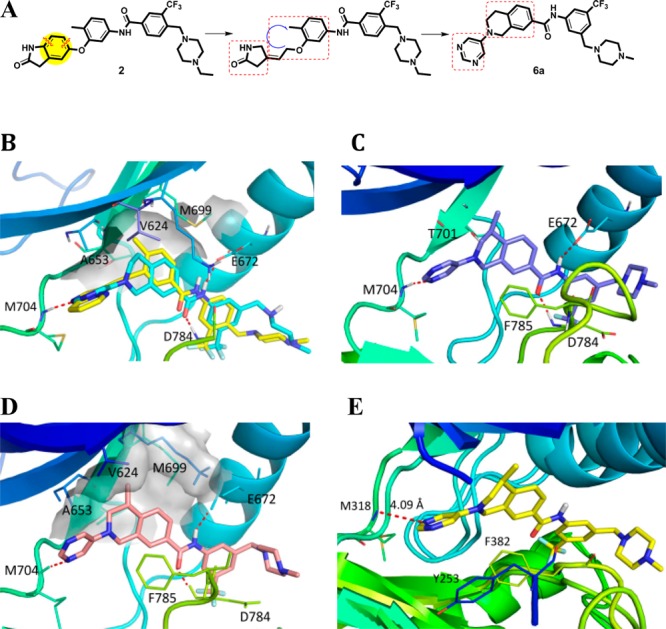
(A) Design of new DDR1 inhibitor 6a. (B) Molecular docking of 6a (cyan) into the DDR1-ponatinib (yellow) costructure (PDB ID: 3ZOS). (C) Co-crystal structure of 6c with DDR1 (PDB ID: 5FDP). (D) Molecular docking of 6b into DDR1. (E) Superposition of 6c with Abl (PDB ID: 3IK3).
Our preliminary modeling suggested that the initial lead, 6a, could fit nicely into the DDR1 binding pocket and maintain the key interactions with DDR1. The pyrimidinyl moiety of 6a could form an essential hydrogen bond with the NH of Met704 in the hinge region of DDR1. Two additional hydrogen bonds have also formed between the linker amide and Glu672 in the C-helix and Asp784 in the Asp-Phe-Gly (DFG) motif, respectively (Figure 2B). Encouragingly, compound 6a failed to dock into the Abl binding pocket, suggesting its potential selectivity among kinases. Compound 6a and its derivatives were readily synthesized using Buchwald–Hartwig amination as the key step (Scheme 1).24 Briefly, the substituted methyl 4-(2-(2,2,2-trifluoroacetamido)ethyl)benzoate (8) was prepared by the trifluoroacetylation of methyl 4-(2-aminoethyl)benzoate (7) and then underwent a classical Pictet–Spengler reaction to yield the protected tetrahydroisoquinoline derivatives (9), which were deprotected and reacted with a hydrochloric acid solution in MeOH to form the key intermediate (10). Compound 10 was coupled with 5-bromopyrimidine, 3-bromopyridine, or bromobenzene through a Buchwald–Hartwig amination reaction to provide compound 11. The final products were obtained by treating intermediate 11 with different anilines under basic conditions.
Scheme 1. Synthesis of Compound 6a and its Derivatives.

Reagents and conditions: (a) trifluoroacetic anhydride, 0 °C to rt, 58–63%; (b) (HCHO)n, conc H2SO4, 0 °C to rt, 51–89%; (c) (i) K2CO3, MeOH/H2O (2:1), rt, (ii) HCl·MeOH, MeOH, rt, 86–95% (two steps); (d) 5-bromopyrimidine or 3-bromopyridine or bromobenzene, Pd(dba)2, Ruphos, Cs2CO3, toluene, 80 °C, 42–89%; (e) substituted aniline, t-BuOK, THF, −20 °C to rt, 69–86%.
DDR1 inhibition of the compounds was determined using a well-established Lance Ultra kinase assay.25 The potential target selectivity was additionally evaluated by monitoring their inhibition against Abl. Compound 1 was included as a positive control, which displayed similar IC50 values to the previously reported data.13 It was shown that 6a exhibited modest DDR1 inhibitory activity, with an IC50 value of 442 nM, while its potency against Abl was strikingly inferior (IC50 > 10.0 μM). These results are consistent with our computational prediction.
Further computational investigation suggested that a small hydrophobic recess formed by Val624, Ala653, and Met699 was available in the ATP binding pocket of DDR1 (Figure 2B). A lipophilic substituent at R1 may occupy this pocket to achieve improved potency. The (R)-methyl (6b) and (R)-ethyl derivatives (6d) displayed a 20-fold and 12-fold potency improvement, respectively. The (S)-methyl compound (6c), in which the methyl moiety was oriented away from the pocket, as confirmed by a 2.3 Å cocrystal structure with DDR1 (Figure 2C), displayed a similar inhibitory potency to that of the R1 unsubstituted 6a. A large group at the R1 position was predicted to be detrimental to the binding of DDR1. As expected, R1-(R)-isopropyl (6f) caused a substantial potency loss for DDR1. Additionally, compounds 6e and 6g, which featured (S)-ethyl and (S)-isopropyl, respectively, displayed almost no DDR1 inhibition. The X-ray crystal structure also confirmed the presence of a strong hydrogen bonding network between the new inhibitor and DDR1 (Figure 2C). The deletion of a hydrogen bond by eliminating the N atoms in the pyrimidinyl group (6i) totally abolished DDR1 inhibitory potency. Not surprisingly, when the pyrimidinyl group was replaced by a pyridinyl moiety, the resulting compound, 6h, exhibited an almost identical IC50 value to that of 6a.
It was also noteworthy that all the new DDR1 inhibitors exhibited excellent DDR1 selectivity over the structurally related Abl kinase (Table 1). To rationalize this target selectivity, the inhibitor 6c was superimposed into the Abl structure (PDB ID: 3IK3) (Figure 2E). It was shown that the 1, 2, 3, 4-tetrahydroisoquinoline scaffold in 6c forced the pyrimidinyl moiety to adopt a different dihedral angle that prevented the formation of critical interactions with Tyr253 and Phe382 in Abl. Moreover, the distance between the N atom in the pyrimidinyl group and Met318 in Abl was predicted to be 4.09 Å, which exceeds the limit to form a potential hydrogen bond.
Table 1. In Vitro Kinase Inhibition of Compounds 6a–6k against DDR1a and Abl1b.
| kinase inhibition (IC50, nM) |
||||||
|---|---|---|---|---|---|---|
| compd | X | Y | R1 | R2 | DDR1 | Abl1 |
| 6a | N | N | H | (4-methylpiperazin-1-yl)methyl | 442 ± 69 | >10000 |
| 6b | N | N | (R)-Me | (4-methylpiperazin-1-yl)methyl | 24.3 ± 4.1 | >10000 |
| 6c | N | N | (S)-Me | (4-methylpiperazin-1-yl)methyl | 309 ± 44 | >10000 |
| 6d | N | N | (R)-Et | (4-methylpiperazin-1-yl)methyl | 36.4 ± 5.7 | >10000 |
| 6e | N | N | (S)-Et | (4-methylpiperazin-1-yl)methyl | >2000 | >10000 |
| 6f | N | N | (R)-i-Pr | (4-methylpiperazin-1-yl)methyl | >1000 | >10000 |
| 6g | N | N | (S)-i-Pr | (4-methylpiperazin-1-yl)methyl | >3000 | >10000 |
| 6h | N | C | H | (4-methylpiperazin-1-yl)methyl | 328 ± 35 | >10000 |
| 6i | C | C | H | (4-methylpiperazin-1-yl)methyl | >3000 | >10000 |
| 6j | N | N | (R)-Me | 4-methyl-1H-imidazol-1-yl | 9.4 ± 1.7 | >10000 |
| 6k | N | N | (S)-Me | 4-methyl-1H-imidazol-1-yl | 326 ± 43 | >10000 |
| 1(13) | 9.7 ± 2.3 | 308 ± 42 | ||||
DDR1 experiments were performed using the LANCE ULTRA kinase assay, according to the manufacturer’s instructions. The data are the means from at least two independent experiments.
Abl1 activity experiments were performed using the FRET-based Z-Lyte assay, according to the manufacturer’s instructions. The data are the means from at least 3 independent experiments.
Further structural optimization of the inhibitor 6b yielded 6j as a promising candidate, with an IC50 value of 9.4 nM against DDR1 (Table 1). The compound also exhibited reasonable pharmacokinetic (PK) properties, with an oral bioavailability of 66.8% and a T1/2 value of 1.25 h at an oral dose of 20 mg/kg (Table 2) in rats. The PK profile of compound 6j was also investigated in ICR mice, which showed that the compound had a similar oral bioavailability to that of rats. However, the area under concentration–time curve (AUC) value of the compound in mice was obviously higher than that in rats, suggesting its good absorption property in mice.
Table 2. Pharmacokinetic Profile of Compound 6j in Micea and Ratsb.
| mice |
rats |
|||
|---|---|---|---|---|
| oral 4 mg/kg | iv 1 mg/kg | oral 20 mg/kg | iv 4 mg/kg | |
| AUC(0–∞) (ng/mL·h) | 2554.1 | 1211.9 | 788.3 ± 41.5 | 236.0 ± 53.1 |
| T1/2 (h) | 1.1 | 0.2 | 1.3 ± 0 | 1.5 ± 0.3 |
| Tmax (h) | 0.5 | 1 ± 0 | ||
| Cmax (ng/mL) | 2193.9 | 2246.8 | 341.7 ± 115.5 | 473.3 ± 114.7 |
| CLz (L/h/kg) | 0.8 | 17.6 ± 4.4 | ||
| BA (%) | 41.6 | 66.8 | ||
ICR mice (male, 24 animals per group) weighing 18–30 g were used for the study.
SD rats (male, 3 animals per group) weighing 180–220 g were used for the study.
The DDR1 inhibition of 6j was further validated by determining its binding affinity with the DDR1 protein (conducted by DiscoveRx, San Diego, CA).26 It was shown that 6j bound tightly to DDR1, with a binding constant (Kd) value of 4.7 nM. The target specificity of 6j was also investigated by conducting a kinase selectivity profiling study against a panel of 468 kinases (including 403 nonmutated kinases) at 1.0 μM, which is approximately 210-fold above its Kd value against DDR1 using the DiscoveRx screening platform. It was shown that 6j displayed excellent target selectivity, with S(10) and S(1) scores of 0.022 and 0.012, respectively (Table S5).26 The potential “off-target” kinases tested included cyclin-dependent kinase 11 (CDK 11), DDR2, ephrin type-B receptor 8 (EPHB8), muscle-specific receptor tyrosine kinase (MUSK), nerve growth factor receptor A (TrkA), TrkB, and TrkC. However, the IC50 value of 6j against DDR2 was determined to be 188 nM in our Lance Ultra kinase assay, indicating that 6j was 20-fold less potent against DDR2. Further determination of the binding affinities (Kd values) revealed that 6j exhibited an approximately 21–120-fold less potency against the majority of the other “off target” kinases, with the exception of TrkB and TrkC, which displayed Kd values of 22 and 18 nM, respectively (Figure 3). These results collectively supported the extraordinary target selectivity of 6j against DDR1.
Figure 3.
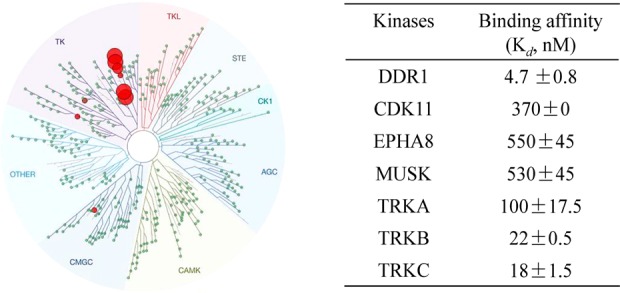
(A) KinomeScan kinase selectivity profiles for 6j. Compound 6j was profiled at a concentration of 1.0 μM against a diverse panel of 468 kinases by DiscoveRx. (B) Binding constants (Kd values) of compound 6j against the top hits. The data are the means from at least three independent experiments.
Further investigation revealed that the activation of DDR1 as well as its downstream signaling intermediate, p38,27 were both dose-dependently suppressed by 6j in primary human lung fibroblasts (Figure 4), suggesting the efficacy of this compound against DDR1-induced signaling. In light of the critical role of DDR1 in BLM-induced pulmonary fibrosis,12 we treated mice with compound 6j after the onset of a BLM challenge. Inhibitor 6j was orally administered at 10 and 50 mg/kg twice daily (BID) for 2 weeks based on its PK properties (Table 2). Unlike those from the phosphate buffered saline (PBS) treated mice, which had large alveolar spaces and were weakly stained by Masson’s trichrome, the lungs from BLM-challenged animals exhibited a reduction in alveolar spaces and were stained blue by Masson’s trichrome, demonstrating typical fibrotic features. The compound prevented these BLM-induced pathological changes in a dose-dependent manner (Figure 5A). These results agreed with the expression levels of fibrotic markers in lung tissue lysates, including fibronectin and α-smooth muscle actin (SMA) (Figure 5B).28 Further analyses also revealed that the administration of compound 6j caused a dose-dependent suppression in the content of hydroxyproline (Figure 5C), a unique amino acid found in collagen.29 The above data collectively indicate the promising therapeutic potential of 6j against the BLM-induced pulmonary fibrosis.
Figure 4.

Effects of DDR1 inhibition by 6j on signaling in primary human lung fibroblasts. 6j inhibited DDR1-mediated signaling in a concentration-dependent manner in primary human lung fibroblasts (24 h treatment). Lysates were probed for the indicated targets by Western blot analysis.
Figure 5.
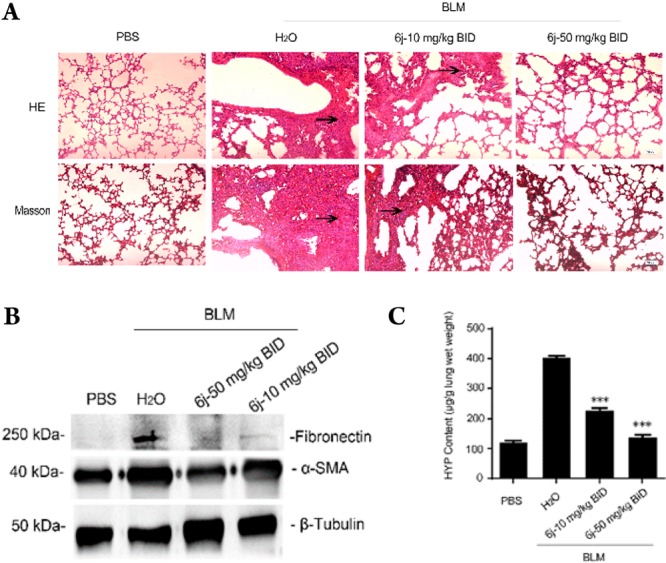
Compound 6j prevents BLM-induced lung fibrosis. Fourteen days after the onset of BLM injury, C57BL/6 mice (five animals each group) received an oral gavage of 6j twice daily at the indicated dosages, and the lungs were collected on day 28. The upper panels in (A) display the hematoxylin and eosin (H&E) staining images of the dissected lungs. The bottom panels in (A) represent Masson’s trichrome staining. The arrows indicate the fibrotic areas of the tissues. The images in (B) show the results of the immunoblotting with the indicated antibodies. The histogram in (C) shows the determined hydroxyproline content. ***P < 0.001.
Conclusion
In summary, a series of 1, 2, 3, 4-tetrahydroisoquinoline derivatives were designed as novel highly selective DDR1 inhibitors. Compound 6j strongly suppressed DDR1, with a single digital nM IC50 value, but it is significantly less potent in a panel of 400 nonmutated kinases. Thus, to the best of our knowledge, this compound represents one of the most selective DDR1 inhibitors to date. The compound also demonstrated reasonable PK properties and a promising oral therapeutic effect in a BLM-induced mouse pulmonary fibrosis model. Its strong DDR1 inhibitory potency and extraordinary target specificity make compound 6j not only a promising lead compound for new drug discovery but also a valuable research probe for further biological investigation of its target.
Experimental Section
General Chemistry
Reagents and solvents were obtained from commercial suppliers and used without further purification. Flash chromatography was performed using silica gel (200–300 mesh). 1H and 13C NMR spectra were recorded on a Bruker AV-400 spectrometer at 400 MHz and Bruker AV-500 spectrometer at 125 MHz. The low or high resolution of ESI-MS was recorded on an Agilent 1200 HPLC-MSD mass spectrometer or Applied Biosystems Q-STAR Elite ESI-LC-MS/MS mass spectrometer, respectively. The purity of compounds was determined to be over 95% (>95%) by reverse-phase high performance liquid chromatography (HPLC) analysis. HPLC instrument: Dionex Summit HPLC (column, Diamonsil C18, 5.0 μm, 4.6 mm × 250 mm (Dikma Technologies); detector, PDA-100 photodiode array; injector, ASI-100 autoinjector; pump, p-680A). Elution: 85% MeOH in water with 0.1% modifier (ammonia, v/v); flow rate, 1.0 mL/min.
Acknowledgments
We appreciate the financial support from the National Natural Science Foundation of China (81425021 and 21572230) and the Natural Science Foundation of Guangdong Province (2015A03031201). We also thank Diamond Light Source for beam time (proposal mx8421) as well as the staff of beamline I04 for their assistance with crystal testing and data collection. The SGC is a registered charity (no. 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, the Canada Foundation for Innovation, the Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome Trust [092809/Z/10/Z].
Glossary
Abbreviations Used
- DDR
discoidin domain receptor
- IC50
half-maximal (50%) inhibitory concentration of a substance
- RTKs
receptor tyrosine kinases
- p38 MAPK
P38 mitogen-activated protein kinase
- Abl
abelson
- ATP
adenosine triphosphate
- Tyr
tyrosine
- Phe
phenylalanine
- Met
methionine
- Glu
glutamic acid
- Asp
aspartic acid
- DFG
Asp-Phe-Gly
- MeOH
methanol
- PDB
Protein Data Bank
- rt
room temperature
- Pd(dba)2
bis(dibenzylideneacetone)palladium
- Ruphos
2-dicyclohexyl phosphino-2′,6′-diisopropoxy-1,1′-biphenyl
- t-BuOK
potassium tert-butanolate
- THF
tetrahydrofuran
- Val
valine
- Ala
alanine
- compd
compounds
- AUC
area under concentration–time curve
- T1/2
half-life period
- ICR
Institute of Cancer Research
- SD
Sprague–Dawley
- Tmax
peak time
- Cmax
peak concentration
- CL
clearance
- BA
bioavailability
- iv
intravenous
- CDK11
cyclin-dependent kinase 11
- EPHB8
ephrin type-B receptor 8
- MUSK
muscle-specific receptor tyrosine kinase
- TrkA
nerve growth factor receptor A
- PHLF
primary human lung fibroblast
- BLM
bleomycin
- BID
twice daily
- PK
pharmacokinetic
- PBS
phosphate buffered saline
- SMA
α-smooth muscle actin
- H&E
hematoxylin and eosin
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b00140.
Synthetic procedures and compound characterization, procedures, and results for in vitro kinase assay, KINOMEscan, protein expression and purification, crystallization and structure determination, computational study, Western blot analysis, animal experiments, antitumor activity of compound 6j. The 1H and 13C NMR spectra of compounds 6a–6k (PDF)
Molecular formula strings (CSV)
Accession Codes
Atomic coordinates and experimental data for the co-crystal structure of 6c with DDR1 (PDB ID: 5FDP) will be released upon article publication.
Author Contributions
Z. Wang, H. Bian, and S. G. Bartual contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Shrivastava A.; Radziejewski C.; Campbell E.; Kovac L.; McGlynn M.; Ryan T. E.; Davis S.; Goldfarb M. P.; Glass D. J.; Lemke G.; Yancopoulos G. D. An orphan receptor tyrosine kinase family whose members serve as nonintegrin collagen receptors. Mol. Cell 1997, 1, 25–34. 10.1016/S1097-2765(00)80004-0. [DOI] [PubMed] [Google Scholar]
- Vogel W.; Gish G. D.; Alves F.; Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol. Cell 1997, 1, 13–23. 10.1016/S1097-2765(00)80003-9. [DOI] [PubMed] [Google Scholar]
- Xu H.; Raynal N.; Stathopoulos S.; Myllyharju J.; Farndale R. W.; Leitinger B. Collagen binding specificity of the discoidin domain receptors: binding sites on collagens II and III and molecular determinants for collagen IV recognition by DDR1. Matrix Biol. 2011, 30, 16–26. 10.1016/j.matbio.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel W. F.; Abdulhussein R.; Ford C. E. Sensing extracellular matrix: an update on discoidin domain receptor function. Cell. Signalling 2006, 18, 1108–1116. 10.1016/j.cellsig.2006.02.012. [DOI] [PubMed] [Google Scholar]
- Valiathan R. R.; Marco M.; Leitinger B.; Kleer C. G.; Fridman R. Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metastasis Rev. 2012, 31, 295–321. 10.1007/s10555-012-9346-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitinger B. Discoidin domain receptor functions in physiological and pathological conditions. Int. Rev. Cell Mol. Biol. 2014, 310, 39–87. 10.1016/B978-0-12-800180-6.00002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai L. K.; Luczynski M. T.; Huang P. H. Discoidin domain receptors: a proteomic portrait. Cell. Mol. Life Sci. 2014, 71, 3269–3279. 10.1007/s00018-014-1616-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borza C. M.; Pozzi A. Discoidin domain receptors in disease. Matrix Biol. 2014, 34, 185–192. 10.1016/j.matbio.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothiwale S.; Borza C. M.; Lowe E. W. Jr.; Pozzi A.; Meiler J. Discoidin domain receptor 1 (DDR1) kinase as target for structure-based drug discovery. Drug Discovery Today 2015, 20, 255–261. 10.1016/j.drudis.2014.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju G. X.; Hu Y. B.; Du M. R.; Jiang J. L. Discoidin domain receptors (DDRs): potential implications in atherosclerosis. Eur. J. Pharmacol. 2015, 751, 28–33. 10.1016/j.ejphar.2015.01.033. [DOI] [PubMed] [Google Scholar]
- Li Y.; Lu X.; Ren X.; Ding K. Small molecule discoidin domain receptor kinase inhibitors and potential medical applications. J. Med. Chem. 2015, 58, 3287–3301. 10.1021/jm5012319. [DOI] [PubMed] [Google Scholar]
- Avivi-Green C.; Singal M.; Vogel W. F. Discoidin domain receptor 1-deficient mice are resistant to bleomycin-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 420–427. 10.1164/rccm.200603-333OC. [DOI] [PubMed] [Google Scholar]
- Gao M.; Duan L.; Luo J.; Zhang L.; Lu X.; Zhang Y.; Zhang Z.; Tu Z.; Xu Y.; Ren X.; Ding K. Discovery and optimization of 3-(2-(Pyrazolo[1,5-a]pyrimidin-6-yl)ethyn yl)benzamides as novel selective and orally bioavailable discoidin domain receptor 1 (DDR1) inhibitors. J. Med. Chem. 2013, 56, 3281–3295. 10.1021/jm301824k. [DOI] [PubMed] [Google Scholar]
- Kim H. G.; Tan L.; Weisberg E. L.; Liu F.; Canning P.; Choi H. G.; Ezell S. A.; Wu H.; Zhao Z.; Wang J.; Mandinova A.; Griffin J. D.; Bullock A. N.; Liu Q.; Lee S. W.; Gray N. S. Discovery of a potent and selective DDR1 receptor tyrosine kinase inhibitor. ACS Chem. Biol. 2013, 8, 2145–2150. 10.1021/cb400430t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkamhawy A.; Park J. E.; Cho N. C.; Sim T.; Pae A. N.; Roh E. J. Discovery of a broad spectrum antiproliferative agent with selectivity for DDR1 kinase: cell line-based assay, kinase panel, molecular docking, and toxicity studies. J. Enzyme Inhib. Med. Chem. 2016, 31, 158–166. 10.3109/14756366.2015.1004057. [DOI] [PubMed] [Google Scholar]
- Richters A.; Nguyen H. D.; Phan T.; Simard J. R.; Grutter C.; Engel J.; Rauh D. Identification of type II and III DDR2 inhibitors. J. Med. Chem. 2014, 57, 4252–4262. 10.1021/jm500167q. [DOI] [PubMed] [Google Scholar]
- Terai H.; Tan L.; Beauchamp E. M.; Hatcher J. M.; Liu Q.; Meyerson M.; Gray N. S.; Hammerman P. S. Characterization of DDR2 inhibitors for the treatment of DDR2 mutated non-small cell lung cancer. ACS Chem. Biol. 2015, 10, 2687–2696. 10.1021/acschembio.5b00655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray C. W.; Berdini V.; Buck I. M.; Carr M. E.; Cleasby A.; Coyle J. E.; Curry J. E.; Day J. E.; Day P. J.; Hearn K.; Iqbal A.; Lee L. Y.; Martins V.; Mortenson P. N.; Munck J. M.; Page L. W.; Patel S.; Roomans S.; Smith K.; Tamanini E.; Saxty G. Fragment-based discovery of potent and selective DDR1/2 inhibitors. ACS Med. Chem. Lett. 2015, 6, 798–803. 10.1021/acsmedchemlett.5b00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day E.; Waters B.; Spiegel K.; Alnadaf T.; Manley P. W.; Buchdunger E.; Walker C.; Jarai G. Inhibition of collagen-induced discoidin domain receptor 1 and 2 activation by imatinib, nilotinib and dasatinib. Eur. J. Pharmacol. 2008, 599, 44–53. 10.1016/j.ejphar.2008.10.014. [DOI] [PubMed] [Google Scholar]
- Cowan-Jacob S. W.; Fendrich G.; Floersheimer A.; Furet P.; Liebetanz J.; Rummel G.; Rheinberger P.; Centeleghe M.; Fabbro D.; Manley P. W. Structural biology contributions to the discovery of drugs to treat chronic myelogenous leukaemia. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2007, 63, 80–93. 10.1107/S0907444906047287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg E.; Manley P. W.; Breitenstein W.; Bruggen J.; Cowan-Jacob S. W.; Ray A.; Huntly B.; Fabbro D.; Fendrich G.; Hall-Meyers E.; Kung A. L.; Mestan J.; Daley G. Q.; Callahan L.; Catley L.; Cavazza C.; Mohammed A.; Neuberg D.; Wright R. D.; Gilliland D. G.; Griffin J. D. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 2005, 7, 129–141. 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- O’Hare T.; Shakespeare W. C.; Zhu X.; Eide C. A.; Rivera V. M.; Wang F.; Adrian L. T.; Zhou T.; Huang W. S.; Xu Q.; Metcalf C. A. 3rd; Tyner J. W.; Loriaux M. M.; Corbin A. S.; Wardwell S.; Ning Y.; Keats J. A.; Wang Y.; Sundaramoorthi R.; Thomas M.; Zhou D.; Snodgrass J.; Commodore L.; Sawyer T. K.; Dalgarno D. C.; Deininger M. W.; Druker B. J.; Clackson T. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 2009, 16, 401–412. 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canning P.; Tan L.; Chu K.; Lee S. W.; Gray N. S.; Bullock A. N. Structural mechanisms determining inhibition of the collagen receptor DDR1 by selective and multi-targeted type II kinase inhibitors. J. Mol. Biol. 2014, 426, 2457–2470. 10.1016/j.jmb.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surry D. S.; Buchwald S. L. Dialkylbiaryl phosphines in Pd-catalyzed amination: a user’s guide. Chem. Sci. 2011, 2, 27–50. 10.1039/C0SC00331J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The assay was conducted by following the protocol from the agent manufacturer (https://perkinelmerreagents.onconfluence.com).
- Fabian M. A.; Biggs W. H. 3rd; Treiber D. K.; Atteridge C. E.; Azimioara M. D.; Benedetti M. G.; Carter T. A.; Ciceri P.; Edeen P. T.; Floyd M.; Ford J. M.; Galvin M.; Gerlach J. L.; Grotzfeld R. M.; Herrgard S.; Insko D. E.; Insko M. A.; Lai A. G.; Lelias J. M.; Mehta S. A.; Milanov Z. V.; Velasco A. M.; Wodicka L. M.; Patel H. K.; Zarrinkar P. P.; Lockhart D. J. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- Matsuyama W.; Wang L.; Farrar W. L.; Faure M.; Yoshimura T. Activation of discoidin domain receptor 1 isoform b with collagen up-regulates chemokine production in human macrophages: role of p38 mitogen-activated protein kinase and NF-kappa B. J. Immunol. 2004, 172, 2332–2340. 10.4049/jimmunol.172.4.2332. [DOI] [PubMed] [Google Scholar]
- Nagase T.; Uozumi N.; Ishii S.; Kita Y.; Yamamoto H.; Ohga E.; Ouchi Y.; Shimizu T. A pivotal role of cytosolic phospholipase A (2) in bleomycin-induced pulmonary fibrosis. Nat. Med. 2002, 8, 480–484. 10.1038/nm0502-480. [DOI] [PubMed] [Google Scholar]
- Colgrave M. L.; Allingham P. G.; Tyrrell K.; Jones A. Multiple reaction monitoring for the accurate quantification of amino acids: using hydroxyproline to estimate collagen content. Methods Mol. Biol. 2012, 828, 291–303. 10.1007/978-1-61779-445-2_23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.