

ABSTRACT
The SREBP transcription factors are major regulators of lipid metabolism. Disturbances in lipid metabolism are at the core of several health issues facing modern society, including cardiovascular disease, obesity and diabetes. In addition, the role of lipid metabolism in cancer cell growth is receiving increased attention. Transcriptionally active SREBP molecules are unstable and rapidly degraded in a phosphorylation-dependent manner by Fbw7, a ubiquitin ligase that targets several cell cycle regulatory proteins for degradation. We have previously demonstrated that active SREBP1 is stabilized during mitosis. We have now delineated the mechanisms involved in the stabilization of SREBP1 in mitotic cells. This process is initiated by the phosphorylation of a specific serine residue in nuclear SREBP1 by the mitotic kinase Cdk1. The phosphorylation of this residue creates a docking site for a separate mitotic kinase, Plk1. Plk1 interacts with nuclear SREBP1 in mitotic cells and phosphorylates a number of residues in the C-terminal domain of the protein, including a threonine residue in close proximity of the Fbw7 docking site in SREBP1. The phosphorylation of these residues by Plk1 blocks the interaction between SREBP1 and Fbw7 and attenuates the Fbw7-dependent degradation of nuclear SREBP1 during cell division. Inactivation of SREBP1 results in a mitotic defect, suggesting that SREBP1 could regulate cell division. We propose that the mitotic phosphorylation and stabilization of nuclear SREBP1 during cell division provides a link between lipid metabolism and cell proliferation. Thus, the current study provides additional support for the emerging hypothesis that SREBP-dependent lipid metabolism may be important for cell growth.
KEYWORDS: cancer, cholesterol, Fbw7, lipids, mitosis, Plk1, SREBP
Introduction
Members of the sterol regulatory element-binding protein (SREBP) family of transcription factors control cholesterol and fatty acid metabolism.1,2 The SREBP family of transcription factors consists of 3 different proteins; SREBP1a, SREBP1c, and SREBP2. The SREBPs are synthesized as large precursor proteins that are inserted into the endoplasmic reticulum membranes and are transcriptionally inactive.3,4 In sterol-depleted cells, the SREBPs are transported to the Golgi by SCAP, where they are processed by 2 membrane-associated proteases that release the mature forms of the proteins.5 In addition, the proteolytic maturation of SREBP1c is enhanced in response to insulin signaling. The mature forms of the SREBPs are transcriptionally active and are translocated to the nucleus where they bind to the promoters of SREBP target genes, most of which are involved in lipid metabolism. The factors and mechanisms involved in the activation of the SREBPs have been elucidated in great detail in sophisticated biochemical and genetic studies.3 The functional importance of the SREBP pathway has been illuminated in a series of genetic studies in mice.5 However, the potential role of SREBP-dependent processes during development or in rapidly proliferating cells, such as cancer cells, have not received the same level of attention.
SREBP1a and SREBP1c originate from a single gene and the nuclear proteins only differ in the length of their N-terminal transactivation domains. SREBP1c is the dominant form of SREBP1 in most adult tissues, except some rapidly proliferating tissues.6 The relatively high level of expression of SREBP1a in these tissues could, at least in part, be explained by the ability of SREBP1a to activate genes involved in both fatty acid and cholesterol synthesis, something that may be required to meet the high demand for lipid synthesis in these tissues in order to sustain cell proliferation.
The nuclear forms of the SREBPs are highly unstable and are rapidly degraded by the ubiquitin-proteasome pathway.7 GSK3-mediated phosphorylation of the C-terminus of nuclear SREBP1 (nSREBP1) creates a docking site for Fbw7, the substrate recognition component of a specific SCF ubiquitin ligase.8,9 Fbw7 is a tumor suppressor and is involved in the degradation of a number of proteins involved in cell cycle progression.10-12 Fbw7 interacts with nSREBP1 and enhances its ubiquitination and degradation in a manner dependent on the phosphorylation of threonine 426 (T426) and serine 430 (S430) (T402 and S406 in SREBP1c) (the organization of nSREBP1 and the localization of the phosphorylation sites discussed in this report are illustrated in Fig. S1). Since Fbw7 is a tumor suppressor and targets a large number of cell cycle regulators, we were interested in the potential link between SREBP function and cell cycle progression. We reported that nSREBP1 was hyperphosphorylated and stabilized during mitosis, and that the expression of many SREBP target genes was induced in mitotic cells.13 Subsequently, we identified one of the phosphorylated residues, serine 439 (S439), in SREBP1a (S415 in SREBP1c), located in the C-terminal domain of the protein, and demonstrated that Cdk1 was responsible for the modification of this residue during mitosis.14 When the phosphorylation of this residue was inhibited the hyperphosphorylation and stabilization of nSREBP1 during mitosis were attenuated. However, the mechanisms involved in the stabilization of nSREBP1 during mitosis remained unresolved.
The protein kinase Polo-like kinase (Plk) 1 is involved in a number of mitotic events, including entry into mitosis, assembly of the bipolar spindle, and mitotic exit.15-17 The activity of Plk1 starts to increase during G2 and peaks during mitosis. Plk1 consists of a kinase domain and 2 polo-box regions. Activation of Plk1 in G2 requires Aurora A to phosphorylate threonine 210 in the activation loop of its kinase domain. Following activation, the 2 polo-box regions fold together to form a polo-box domain (PBD), which binds phosphorylated target proteins. Cdk1 often provides the initial phosphorylation of substrate proteins recognized by the PBD in Plk1. As mentioned above, Cdk1 phosphorylates S439/415 in the C-terminal domain of nSREBP1 during mitosis. The sequence surrounding S439/415 fits the preferred binding site of the PBD of Plk1, suggesting that nSREBP1 could be a target for this kinase in mitosis. We now confirm this hypothesis, demonstrating that the Cdk1-mediated phosphorylation of S439/415 creates a functional binding site for Plk1 in the C-terminal domain of nSREBP1. Plk1 interacts with nSREBP1 during mitosis, resulting in the Plk1-mediated phosphorylation of at least 3 Thr/Ser residues in the C-terminal domain of the protein. The phosphorylation of these residues contributes to the hyperphosphorylation and stabilization nSREBP1 during mitosis. Mechanistically, we demonstrate that the phosphorylation of these residues interferes with the interaction between nSREBP1 and Fbw7, thereby preventing the Fbw7-dependent degradation of nSREBP1 during mitosis. Finally, we demonstrate that inactivation of SREBP1 results in mitotic defects, suggesting that SREBP1 could regulate cell division.
Results
Nuclear SREBP1 interacts with Plk1 during mitosis
Cdk1 phosphorylates S439/415 in the C-terminus of nSREBP1 during mitosis and the sequence surrounding S439/415 fits the preferred binding site for the PBD found in Plk1. To determine if Plk1 could interact with nSREBP1, cells were transfected with nSREBP1a, either wild-type (WT) or a mutant lacking the C-terminal domain, including S439. Endogenous Plk1 interacted with WT nSREBP1a, but failed to interact with the mutant lacking the C-terminal domain (Fig. 1A). Endogenous Plk1 also failed to interact with nSREBP1a in which S439 was mutated to alanine (S439A in Fig. 1B), suggesting that S439 is important for the interaction between nSREBP1 and Plk1. In order to directly test this hypothesis, we used peptide pull-down assays utilizing peptides containing the sequence surrounding S439, either unmodified or phosphorylated on S439. The phosphorylated peptide interacted with endogenous Plk1 in cell extracts, while the unmodified peptide failed to do so (Fig. 1C, upper panel). The phosphorylated peptide also interacted with the PBD of Plk1 in extracts prepared from cells expressing this domain, while it failed to interact with a mutant form of PBD unable to interact with target proteins (Fig. 1C, lower panel). In addition, recombinant nSREBP1a interacted with Plk1 following Cdk1-mediated phosphorylation, and mutation of S439 blocked this interaction (Fig. 1D). Taken together, these results suggest that the phosphorylation of S439 in the C-terminus of nSREBP1 creates a binding site for the PBD of Plk1.
Figure 1.

Plk1 interacts with nuclear SREBP1 during mitosis. (A) HEK293 cells were transfected with FLAG-tagged nuclear SREBP1a (nSREBP1a), either wild-type (WT, amino acids 1–490) or the ΔC mutant (amino acids 1–417), lacking the entire C-terminal domain of nSREBP1a. Cell lysates were inmunoprecipitated with anti-FLAG antibodies. The amounts of immunoprecipitated Plk1 and nSREBP1a, and the levels of nSREBP1a and Plk1 in whole-cell lysates (WCL) were determined by Western blotting, with α-tubulin serving as a loading control. (B) HEK293 cells were transfected with FLAG-nSREBP1a, either WT or the S439A mutant. Cell lysates were immunoprecipitated with anti-FLAG antibodies. The amounts of immunocrecipitated Plk1 and nSREBP1a, and the levels of nSREBP1a, Plk1 and α-tubulin in cell lysates were determined by Western blotting. (C) Cell lysates from HeLa cells were used in peptide pull-down assays, using 2 peptides corresponding to residues 436–442 of human SREBP1a, either non-phosphorylated (Ref) or the same peptide phosphorylated on S439 (P-pept) (upper panel). Myc-PBD, either WT or a binding-deficient mutant (MT), was expressed in HEK293 cells, and the cell lysates were used in peptide pull-down assays, using the same 2 peptides as those described above (lower panel). The bound proteins were subjected to SDS/PAGE and Western blotting using 20% of input as control. (D) Recombinant nSREBP1a, either WT or the S439A mutant, was used in in vitro kinase assays in the absence or presence of recombinant Cdk1/cyclin B. The phosphorylated proteins were mixed with lysates of HEK293 cells expressing GFP-Plk1. The His-tagged nSREBP1a proteins were captured on NiTA-agarose, washed and resolved by SDS/PAGE, followed by Western blotting. The phosphorylation of S439 in nSREBP1a was monitored with a phosphorylation-specific antibody (pS439). (E) HeLa cells were synchronized at the G1/S transition by a double-thymidine protocol. Cells were collected after the second thymidine block (G1/S) or 14 h after the release into either nocodazole-containing media (Mit) or normal media (G1). Cell lysates were incubated with recombinant GST-PBD, either WT or binding-deficient mutant (MT), in the presence of glutathione beads. The amount of nSREBP1 associated with the glutathione beads was determined by Western blotting (left panel). The GST-PBD proteins were detected by coomassie brilliant blue staining (CBB). The levels of nSREBP1 and α-tubulin in cell lysates were determined by Western blotting (right panel) (F) HeLa cells were synchronized at the G1/S transition by a double-thymidine protocol. Cells were collected after the second thymidine block (G1/S) or 14 h after the release into either nocodazole-containing media (Mit) or normal media (G1). Cell lysates were immunoprecipitated with either a control antibody (Gal4, lanes 1–3) or SREBP1 antibody (lanes 4–6). The amounts of immunoprecipitated Plk1 and nSREBP1 were determined by Western blotting (left panel). The levels of nSREBP1, Plk1 and α-tubulin in cell lysates were determined by Western blotting (right panel).
In order to determine if the PBD of Plk1 could interact with endogenous SREBP1, we used recombinant GST-tagged Plk1-PBD in GST pull-down assays with extracts from HeLa cells arrested at different phases of the cell cycle. The PBD of Plk1 pulled out nSREBP1 specifically from mitotic extracts (Fig. 1E). Only a small amount of nSREBP1 associated with the binding-deficient mutant version of the PBD under these conditions, suggesting that the interaction is specific. In addition, both nSREBP1a and nSREBP1c interacted with endogenous Plk1 in mitotic cells following transfection into HEK293 cells (Fig. S2). Importantly, endogenous Plk1 interacted with endogenous nSREBP1 in mitotic HeLa cells (Fig. 1F). Thus, Plk1 interacts with nSREBP1 during mitosis following phosphorylation of S439/415 in its C-terminal domain.
Plk1 phosphorylates the C-terminal domain of nuclear SREBP1
Having demonstrated that nSREBP1 interacts with Plk1 during mitosis, we wanted to determine if Plk1 could phosphorylate nSREBP1. To test this hypothesis, we performed in vitro kinase assays with recombinant nSREBP1a and Plk1 in the presence of 32P-labeled ATP, followed by phosphopeptide mapping and Edman degradation. Plk1 was able to phosphorylate nSREBP1 on both Ser and Thr residues in vitro (Fig. S3). Edman degradation of individual phosphopeptides identified 3 potential phosphorylation sites in nSREBP1a, threonine 424 (T424), serine 467 (S467) and serine 486 (S486) (Fig. S3), all contained within the C-terminal domain of nSREBP1 (Fig. S1). The sequence of the C-terminal domains of nSREBP1a and nSREBP1c are identical and the residues targeted by Plk1 correspond to T400, S443 and S462 in human SREBP1c. In order to analyze the phosphorylation of these residues further, we generated phosphorylation-specific antibodies to all 3 residues and evaluated their specificity (Fig. S4). Recombinant Plk1 was able to phosphorylate all 3 residues in vitro (Fig. 2A). Furthermore, all 3 residues were phosphorylated when recombinant nSREBP1a was used in in vitro kinase assays with extracts from mitotic HeLa cells (Fig. 2B). Importantly, a specific Plk1 inhibitor, BTO-1, reduced the phosphorylation of all 3 residues, suggesting that endogenous Plk1 may target all 3 residues. This possibility was supported by our observation that the phosphorylation of all 3 residues was significantly reduced when mitotic extracts from cells treated with Plk1 siRNA were used in the in vitro kinase assay (Fig. 2C).
Figure 2.

Plk1 phosphorylates T424, S467 and S486 in nuclear SREBP1 during mitosis. (A) In vitro kinase assay with recombinant nSREBP1a and Plk1. The levels and phosphorylation (pT424, pS467 and pS486) of nSREBP1a were monitored by Western blotting. (B) Recombinant nSREBP1a was used in in vitro kinase assays with mitotic HeLa extracts (Mit) in the absence or presence of the Plk1 inhibitor BTO-1 (5, 10 and 25 μM). The levels and phosphorylation (pT424, pS467 and pS486) of nSREBP1a were monitored by Western blotting. (C) Recombinant nSREBP1a was used in in vitro kinase assays with extracts from HeLa cells transfected with either control or Plk1 siRNA. The levels and phosphorylation (pT424, pS467 and pS486) of nSREBP1a were monitored by Western blotting (left panel). The efficiency of the Plk1 knockdown was monitored by Western blotting, with α-tubulin serving as a loading control (right panel). (D) Recombinant nSREBP1a, either WT or the S439A mutant, was used in in vitro kinase assays with mitotic HeLa extracts. The levels and phosphorylation (pT424, pS467 and pS486) of nSREBP1a were monitored by Western blotting. (E) HEK293 cells were transfected with nSREBP1a or nSREBP1c and left untreated or treated with nocodazole. After immunoprecipitation, the levels and phosphorylation (pT424, pS467 and pS486) of the respective nSREBP1 protein were determined by Western blotting. (F) HeLa cells were synchronized at the G1/S transition by a double-thymidine protocol. Cells were collected after the second thymidine block (G1/S) or 14 h after the release into either nocodazole-containing media (Mit) or normal media (G1). The levels and phosphorylation (pT424, pS467 and pS486) of nSREBP1 were determined by Western blotting following immunoprecipitation of SREBP1.
The results described in Fig. 1 suggested that the phosphorylation of S439 should be crucial for the interaction between nSREBP1 and Plk1 and thereby for the Plk1-mediated phosphorylation of nSREBP1. To test this hypothesis, we repeated the in vitro kinase assay with mitotic HeLa extracts using recombinant nSREBP1a, either WT or the S439A mutant. As seen in Fig. 2D, the phosphorylation of the potential Plk1 target sites were significantly reduced in the S439A mutant, suggesting that the phosphorylation of S439/415 is critical for the subsequent Plk1-dependent phosphorylation of T424, S467 and S486. Both nSREBP1a and nSREBP1c were phosphorylated on all 3 residues when expressed in cells, especially in cells arrested in mitosis in response to nocodazole treatment (Fig. 2E). Importantly, all 3 residues were phosphorylated in endogenous nSREBP1 during mitosis (Fig. 2F).
Plk1 controls the phosphorylation of nuclear SREBP1 during mitosis
We next asked if Plk1 contributed to the hyperphosphorylation and stabilization of nSREBP1 during mitosis. To answer this question, HCT116 cells were arrested at the G1/S transition by a double-thymidine block, transfected with either control or Plk1 siRNA, and released from the second thymidine block in media containing nocodazole. Nuclear SREBP1 was phosphorylated on the 3 potential Plk1 phosphorylation sites as control cells entered mitosis (Fig. 3A). The phosphorylation of all 3 residues was reduced in the Plk1 knockdown cells. In addition, the accumulation of nSREBP1 was attenuated in response Plk1 knockdown. Although the hyperphosphorylation of nSREBP1 was delayed in response to Plk1 knockdown, a substantial proportion of the protein was still highly phosphorylated. This could mean that Plk1 is not required for the hyperphosphorylation of nSREBP1 during mitosis. However, it could also mean that the low levels of Plk1 remaining in the knockdown cells are sufficient to phosphorylate nSREBP1. To address this issue, HeLa cells arrested in mitosis were treated for a short period of time in the absence or presence of a specific Plk1 inhibitor, BI 2536. Addition of the inhibitor resulted in a rapid loss of the phosphorylation of the 3 potential Plk1 phosphorylation sites (Fig. 3B). Importantly, it also resulted in a loss of nSREBP1, and the remaining protein migrated at the same apparent molecular weight as that in non-mitotic cells. Similar results were obtained with BTO-1, a separate Plk1 inhibitor (Fig. S5). These results indicate that the kinase activity of Plk1 is important for both the hyperphosphorylation and accumulation of nSREBP1 in mitotic cells. This possibility was supported by the fact that transfected Plk1 enhanced the levels of cotransfected nSREBP1a and 1c, whereas a kinase-deficient mutant of Plk1 failed to have the same effect (Fig. 3C). In order to test if the Plk1-dependent accumulation of nSREBP1a was the result of reduced turnover, cells were transfected with nSREBP1a in the absence and presence of Plk1 and used in protein degradation assays. As illustrated in Fig. 3D, the turnover of nSREBP1a was attenuated in the presence of Plk1, confirming that Plk1 stabilizes nSREBP1 by reducing its degradation.
Figure 3.
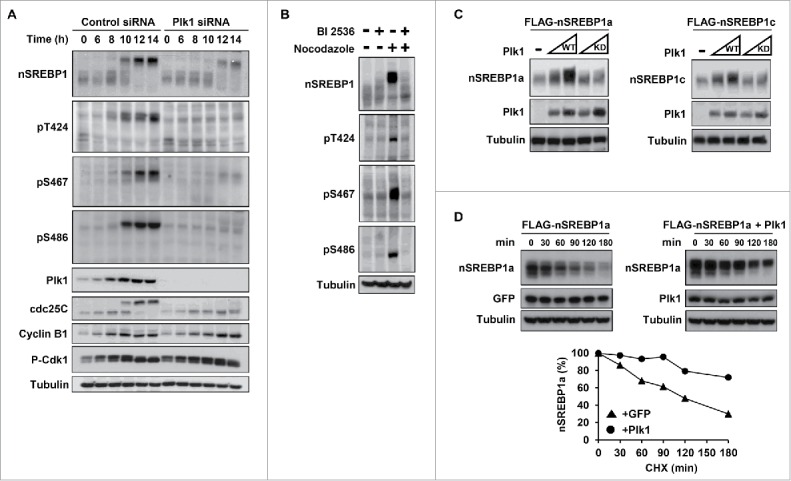
Plk1 phosphorylates SREBP1 in vivo. (A) HCT116 cells were synchronized at the G1/S transition by a double-thymidine protocol. After the first thymidine block, cells were transfected with control or Plk1 siRNA. Cells were collected at the indicated time points after release from the second thymidine block in media containing nocodazole. Cell lysates were analyzed by Western blotting with antibodies against the indicated proteins. The levels and phosphorylation (pT424, pS467 and pS486) of nSREBP1 were determined by Western blotting following immunoprecipitation of SREBP1. (B) HeLa cells were left untreated or treated with nocodazol to induce G2/M arrest. Where indicated, BI-2536 (250 nM) was added for the last 2 hours. The levels and phosphorylation (pT424, pS467 and pS486) of nSREBP1 were determined by Western blotting following immunoprecipitation of SREBP1. (C) HEK293 cells were transfected with nSREBP1a (left panel) or nSREBP1c (right panel) in the absence or presence of increasing amounts of WT or kinase-deficient (KD) Plk1. The levels of nSREBP1a, nSREBP1c, Plk1 and α-tubulin (loading control) were determined by Western blotting. (D) HEK293 cells were transfected with nSREBP1a in the presence of GFP (left panel) or GFP-Plk1 (right panel). Forty-eight hour after transfection, cells were treated with cycloheximide (CHX, 100 μgr/ml) for the indicated times. The levels of nSREBP1a, Plk1 and α-tubulin (loading control) were determined by Western blot analysis. The relative amount of nSREBP1a at each time point is plotted as percentage of the amount at the start of the assay (lower panel).
Plk1-mediated phosphorylation controls the levels and activity of nuclear SREBP1 during mitosis
We next asked if the phosphorylation of the 3 residues in nSREBP1 targeted by Plk1 could affect the expression or activity of nSREBP1. To address this question, we initially analyzed the steady-state levels of nSREBP1a following transfection into HEK293 cells. When the potential Plk1 sites were mutated to non-phosphorylated alanine residues (3A), the steady-state levels were reduced compared to WT SREBP1a (Fig. 4A). However, the opposite was true when all 3 residues were substituted with phosphomimetic residues (3D and 3E in Fig. 4A), indicating that phosphorylation of these residues could enhance the expression of nSREBP1. In agreement with this hypothesis, the levels of transfected nSREBP1a were reduced in response to siRNA-mediated Plk1 inactivation (Fig. 4B, left panel). The levels of the 3A mutant were already low in cells transfected with control siRNA and were unresponsive to Plk1 inactivation. This correlated well with the transcriptional activities of WT and 3A nSREBP1a under the same conditions, i.e. the activity of the WT protein was reduced in response to Plk1 inactivation whereas the activity of the 3A mutant was low in control cells and did not respond to Plk1 inactivation (Fig. S6A). In addition, the transcriptional activity of nSREBP1a was enhanced when coexpressed with WT Plk1, but not with the kinase-deficient version of the kinase (Fig. S6B). The transcriptional activity of the 3A mutant was insensitive to Plk1 coexpression. In contrast to the 3A version of nSREBP1a, the levels of the 3D and 3E mutants were higher than the WT protein in control cells and were not reduced in response to Plk1 inactivation (Fig. 4B, right panel). Similar results were obtained when this experiment was repeated with nSREBP1c (Fig. S7).
Figure 4.
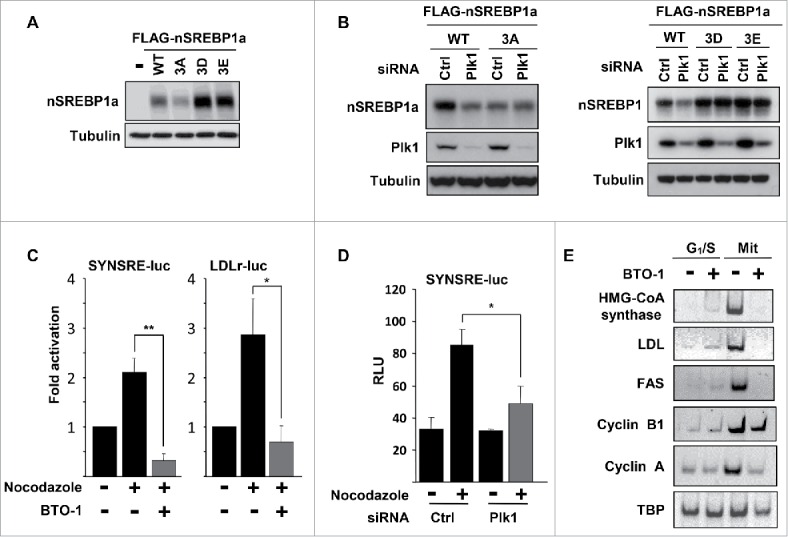
Plk1 regulates the stability and transcriptional activity of nuclear SREBP1. (A) HEK293 cells were transfected with nSREBP1a, either WT or the indicated mutants. The levels of nSREBP1a were determined by Western blotting, with α-tubulin serving as a loading control. (B) HEK293 cells were transfected with nSREBP1a, either WT or the 3A mutant (left panel) or the 3D and 3E mutants (right panel), in the presence of control or Plk1 siRNA. The levels of nSREBP1a, Plk1 and α-tubulin were monitored by Western blotting. (C) HepG2 cells were transfected with the indicated promoter reporter construct. Twenty-four hours after transfection, cells were treated with nocodazole for 16h and luciferase activity was measured. Where indicated, cells were treated with BTO-1 (50 μM) for the last 4h. The data represent the averages ± SD of three independent experiments. Statistical analyses were performed using a 2-tailed Student's t-test. For all tests, P-values lower than 0.05 were considered statistically significant. *P < 0 .05, **P < 0 .01 and ***P < 0 .001. (D) HepG2 cells were transfected with SYNSRE-luc and either control or Plk1 siRNA. Twenty-four hours after transfection, cells were treated with nocodazole and luciferase activity was measured. The data represent the averages ± SD of three independent experiments. Statistical analyses were performed using a 2-tailed Student's t-test. For all tests, P-values lower than 0.05 were considered statistically significant. *P < 0 .05, **P < 0 .01 and ***P < 0 .001. (E) HeLa cells were synchronized at the G1/S transition by a double-thymidine protocol. Cells were collected after the second thymidine block (G1/S) or 14 h after the release into nocodazole-containing media (Mit). Where indicated, cells were treated with BTO-1 (50 μM) for the last 4 h. Total RNA was used to determine the expression of the indicated genes by semi quantitative RT-PCR analysis.
We next asked if Plk1 could modulate the transcriptional activity of endogenous SREBP1. To address this question, HepG2 cells were transfected with 2 SREBP-responsive promoter-reporter genes and treated in the absence or presence of nocodazole to arrest the cells in mitosis. As reported previously, the expression of both reporter genes was enhanced in mitotic cells (Fig. 4C). The induction of both reporter genes was reversed if the cells were treated for a short time with the Plk1 inhibitor BTO-1, suggesting that the kinase activity of Plk1 is important for the activation of SREBP1 during mitosis, potentially by enhancing the steady-state levels of the nuclear protein (Fig. 3B). The mitotic induction of the HMG-CoA synthase promoter-reporter gene was also attenuated in cells transfected with Plk1 siRNA (Fig. 4D), although not to the same extent as with the Plk1 inhibitor. As discussed previously, the inability of Plk1 siRNA to completely block the mitotic activation of SREBP1 could be explained by the presence of residual Plk1 protein in the knockdown cells. However, we cannot exclude the possibility that other factors are also involved. In order to determine if Plk1 could modulate the expression of endogenous SREBP target genes, HeLa cells were either arrested at the G1/S boundary by a double-thymidine block protocol or arrested in mitosis by a double-thymidine nocodazole release protocol. Both sets of cells were treated for 4 hours with the Plk1 inhibitor BTO-1, followed by RNA isolation and RT-PCR analysis of the indicated mRNAs. The expression levels of all SREBP target genes analyzed were significantly higher in the mitotic cells compared to the G1/S cells (Fig. 4E). Importantly, the expression of these genes was reduced in response to the Plk1 inhibitor in mitotic cells, whereas the same inhibitor had no effect in G1/S cells.
Plk1-mediated phosphorylation of nuclear SREBP1 prevents its degradation by Fbw7 during mitosis
Nuclear SREBP1 is highly unstable and is degraded by the ubiquitin ligase Fbw7 following the phosphorylation of T426 and S430 within its Fbw7-specific phosphodegron.9 Fbw7 is active during mitosis and has been shown to degrade cyclin E during this phase of the cell cycle.18 Thus, we were interested in determining if the Plk1-mediated phosphorylation of nuclear SREBP1 during mitosis could interfere with its Fbw7-dependent degradation. We were able to confirm that Fbw7 degrades cyclin E during mitosis using WT and Fbw7 knockout HCT116 cells (Fig. 5A). In contrast to cyclin E, the levels of nSREBP1 were low in asynchronous WT cells and increased during mitosis (Fig. 5A). However, the levels of nSREBP1 were already high in asynchronous Fbw7 knockout cells and there was no further accumulation of the protein during mitosis. In fact, the levels of nSREBP1 in mitotic WT cells were very similar to those in the asynchronous Fbw7 knockout cells, suggesting a potential link between Fbw7 and the accumulation of nSREBP1 during mitosis. Further support for such a link was provided by the results presented in Fig. 5B. The levels of nSREBP1 were greatly enhanced when asynchronous cells were treated with the proteasome inhibitor MG-132, whereas the same inhibitor only had a minor effect on the levels of nSREBP1 in mitotic cells. These results suggest that nSREBP1 is not a substrate for the proteasome during mitosis, despite the fact that its phosphodegron is highly phosphorylated during this phase of the cell cycle (Fig. S8).
Figure 5.
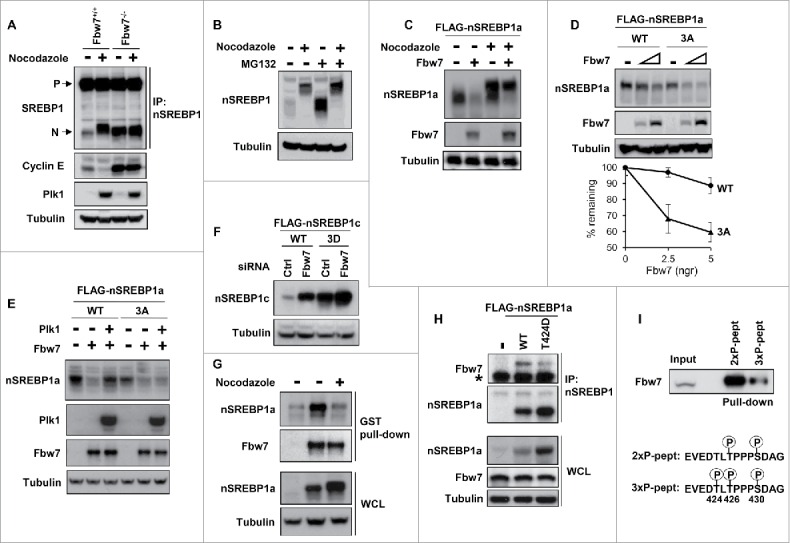
Plk1-mediated phosphorylation of nuclear SREBP1 prevents its degradation by Fbw7. (A) WT (Fbw+/+) or Fbw7 knockout (Fbw7−/−) HCT116 cells were treated in the absence or presence of nocodazole. The levels of SREBP1, Cyclin E, Plk1 and α-tubulin (loading control) were determined by Western blotting. The membrane-associated precursor (P) and nuclear (N) forms of SREBP1 are indicated. (B) HeLa cells were left untreated or treated with nocodazole to induce mitotic arrest. Where indicated, cells were treated with the proteasome inhibitor MG-132 (25 μM) for the last 4 h. The levels of nSREBP1 and α-tubulin were determined by Western blotting. (C) HEK293 cells were transfected with nSREBP1a in the absence or presence of Fbw7α and the cells were left untreated or treated with nocodazole. The levels of nSREBP1a, Fbw7α and α-tubulin were determined by Western blotting. (D) HEK293 cells were transfected with nSREBP1a, either WT or the 3A mutant, in the absence or presence of increasing amounts of Fbw7α. The levels of nSREBP1a, Fbw7α and α-tubulin were determined by Western blotting (upper panel). The percentage of nSREBP1a remaining is plotted against the concentration of Fbw7α (lower panel). The data represent the averages ± SD of three independent experiments. (E) HEK293 cells were transfected with nSREBP1a, either WT or the 3A mutant, in the absence or presence of Plk1 and Fbw7α. The levels of nSREBP1a, Plk1, Fbw7α and α-tubulin were monitored by Western blotting. (F) HEK293 cells were transfected with nSREBP1c, either WT or the 3D mutant, together with control or Fbw7 siRNA. The levels of nSREBP1c and α-tubulin were determined by Western blotting. (G) HEK293 cells were transfected with nSREBP1a and either left untreated or treated with nocodazole. Lysates from the transfected cells were mixed with recombinant GST-Fbw7α. Following capture of GST-Fbw7α and extensive washing, the bound proteins were subject to SDS/PAGE and the amount of nSREBP1a and Fbw7α were determined by Western blotting (upper panel). The amount of nSREBP1a and α-tubulin in whole cell lysates (WCL) were determined by Western blotting (lower panel). (H) HEK293 cells were transfected with FLAG-nSREBP1a, either WT or the T424D mutant. Cell lysates were immunoprecipitated with anti-FLAG antibodies. The amounts of immunoprecipitated Fbw7 and nSREBP1a (upper panel), and the levels of nSREBP1a, Fbw7 and α-tubulin in cell lysates (Input, lower panel) were determined by Western blotting. The band indicated by the asterisk corresponds to the IgG chain. (I) HeLa cell lysates were used in peptide pull-down assays, using 2 peptides corresponding to residues 422–442 of human SREBP1a, either phosphorylated on T426 and S430 (2xP-pept) or the same peptide phosphorylated on T424, T426 and S430 (3xP-pept). The bound proteins were subjected to SDS/PAGE and Western blotting using 20% of input as control.
In order to directly test if nSREBP1 is a substrate for Fbw7-dependent degradation during mitosis, cells were transfected with nSREBP1a in the absence or presence of Fbw7α. The transfected cells were left untreated or treated with nocodazole to promote mitotic arrest. Nuclear SREBP1a was degraded in the presence of Fbw7α in asynchronous cells (Fig. 5C). However, Fbw7α only had a limited effect on the levels of nSREBP1a in mitotic cells, suggesting that the highly phosphorylated form of the protein found during this phase of the cell cycle is not a substrate for Fbw7α (compare the upper bands in lanes 3 and 4 of Fig. 5C). We next wanted to determine if the residues phosphorylated by Plk1 could affect the Fbw7-dependent degradation of nSREBP1. The non-phosphorylated 3A mutant of nSREBP1a was more sensitive to Fbw7-dependent degradation compared to the WT protein when cotransfected with increasing amount of Fbw7α (Fig. 5D). In addition, cotransfected Plk1 attenuated the Fbw7-dependent degradation of WT nSREBP1a, while having no effect on the Fbw7-dependent degradation of the 3A mutant under the same conditions (Fig. 5E). Furthermore, the levels of WT nSREBP1c were enhanced in response to Fbw7 inactivation, whereas the levels of the phosphomimetic 3D mutant were high in control cells and only minimally enhanced in response to Fbw7 inactivation (Fig. 5F). Taken together, these results indicate that the Plk1-mediated phosphorylation of nSREBP1 could attenuate its Fbw7-dependent degradation.
In order to address the mechanisms involved in the Plk1-dependent stabilization of nSREBP1, we decided to test if Plk1 could interfere with the interaction between nSREBP1 and Fbw7. Recombinant GST-tagged Fbw7α was able to pull out transfected nSREBP1a from extracts prepared from asynchronous cells (Fig. 5G). However, Fbw7α failed to interact with nSREBP1a in extracts prepared from mitotic cells, despite the enhanced amounts of nSREBP1a in these extracts (Fig. 5G), suggesting that Fbw7α is unable to interact with mitotic nSREBP1. Since one of the residues targeted by Plk1, T424, is located in close proximity to the Fbw7 phosphodegron in nSREBP1, it was possible that phosphorylation of this residue could interfere with the interaction between the 2 proteins. To test this possibility, we performed co-immunoprecipitation assays between endogenous Fbw7 and transfected nSREBP1a, either WT or a mutant containing a phosphomimetic residue at position 424 (T424D). The interaction between nSREBP1 and Fbw7 was reduced in the T424D mutant, despite the fact that this mutant accumulated to higher levels compared to the WT protein (Fig. 5H). We obtained the opposite result when this experiment was repeated with a mutant containing a non-phosphorylated residue at position 424 (T424A), i.e., the T424A mutant pulled out more endogenous Fbw7 compared to the WT protein, despite displaying lower steady-state levels (Fig. S9).
In order to directly determine if the phosphorylation of T424 interfered with the interaction between Fbw7 and the phosphodegron in nSREBP1, we used peptide pull-down assays utilizing 2 separate peptides containing amino acids surrounding T426 and S430 in SREBP1a. One of these peptides was phosphorylated on T426 and S430, an absolute requirement for Fbw7 binding.9 The second peptide was also phosphorylated on T424. As expected, the peptide phosphorylated on T426/S430 was able to pull out a significant amount of endogenous Fbw7 from HeLa extracts (Fig. 5I). In agreement with our hypothesis, phosphorylation of T424 significantly reduced the ability of Fbw7 to interact with the phosphodegron, suggesting that Plk1-mediated phosphorylation of T424 during mitosis could attenuate the interaction between Fbw7 and the phosphodegron in nSREBP1, thereby promoting the accumulation of nSREBP1 observed during this phase of the cell cycle.
Inactivation of SREBP1 affects mitotic progression
Our results suggest that Plk1-mediated phosphorylation of nSREBP1 stabilizes the protein during mitosis. In order to analyze if SREBP1 could influence mitosis, HCT116 cells were arrested at the G1/S transition, transfected with either control or SREBP1 siRNA and then released from the G1/S arrest and allowed to proceed through the cell cycle. Cells were collected at different time points after the release and their progression through the cell cycle was monitored by FACS analysis. Based on the cell cycle profiles, both sets of cells appeared to enter mitosis in a timely manner (Fig. 6A). However, the SREBP1 knockdown cells displayed a delay in mitotic exit. Although limited, the mitotic defect in SREBP1 knockdown was statistically significant (Fig. S10). In order to confirm this observation, the experiment was repeated, but this time the phosphorylation of Ser10 in histone H3 was monitored in the 2 sets of cells. Phosphorylation at Ser10 of histone H3 is tightly correlated with chromosome condensation during mitosis and this modification is rapidly removed as cells leave this phase of the cell cycle. The phosphorylation of histone H3 increased in both sets of cells as they entered mitosis (Fig. 6B). As cells exited mitosis, the phosphorylation of H3 was rapidly reduced in control cells. However, the phosphorylation of H3 Ser10 declined only slightly over time in SREBP1 knockdown cells and remained high throughout the timeframe of the experiment. Although not as striking, a similar trend was seen in the levels of Plk1 in the SREBP1 knockdown cells. These results suggest that SREBP1 could play a role during mitosis, potentially by regulating mitotic exit.
Figure 6.

SREBP1 inactivation results in a delay in mitotic exit. (A) HCT116 cells were synchronized at the G1/S transition by a double-thymidine protocol. After the first thymidine block, cells were transfected with control (Ctrl) or SREBP1 siRNA. Cells were collected at the indicated time points after release from the second thymidine block and used for FACS analysis. The cell cycle distribution at each time point corresponds to the average of 3 independent experiments. (B) HCT116 cells were treated as in (A) and collected at the indicated time points after the second thymidine block. Cell lysates were analyzed by Western blotting with antibodies to the indicated proteins. (C) Phosphorylation-dependent stabilization of nSREBP1 during mitosis. Cdk1 phosphorylates the C-terminus of nSREBP1 thereby creating a docking site for Plk1. Plk1 binds nSREBP1 during mitosis, resulting in the phosphorylation of at least 3 residues in the C-terminus of nSREBP1. Phosphorylation of these residues attenuates the interaction between nSREBP1 and Fbw7, thereby blocking Fbw7-dependent degradation of nSREBP1 during mitosis.
In our previous work we demonstrated that Cdk1 phosphorylates nSREBP1 on S439 (S415 in SREBP1c) during mitosis, resulting in the hyperphosphorylation and stabilization of the protein.14 The results presented in the current investigation suggest that the phosphorylation of S439 creates a docking site for a separate mitotic protein kinase, Plk1 (Fig. 6C). Plk1 interacts with nSREBP1 during mitosis in a manner dependent on the phosphorylation of S439. Plk1 subsequently phosphorylates at least 3 separate Thr/Ser residues in the C-terminal domain of nSREBP1, i.e. T424, S467 and S486 (T400, S443 and S462 in SREBP1c). The phosphorylation of these residues interferes with the Fbw7-dependent degradation of nSREBP1 during mitosis. Furthermore, we demonstrate that the Plk1-mediated phosphorylation of one of these residues, T424, located in close proximity of the Fbw7 binding site in nSREBP1 reduces the interaction between Fbw7 and nSREBP1. Finally, we demonstrate that inactivation of SREBP1 affects mitotic progression, possibly by delaying mitotic exit. Based on these observations, we propose that the mitotic phosphorylation and stabilization of nSREBP1 could provide a link between lipid metabolism and cell proliferation, something that could be of importance for cell growth.
Discussion
The renewed interest in metabolism, especially cancer metabolism, has rejuvenated interest in the functional importance of lipid synthesis for cell proliferation. As a result, several recent studies have probed the importance of SREBP-regulated lipid metabolism during cell proliferation, especially cancer cell growth, both in vitro and in vivo. Taken together, these studies suggest that the SREBP pathway supports the enhanced demand for de novo lipogenesis, as well as lipid uptake, in rapidly dividing cells.19-22 It has been clearly demonstrated that SREBP-dependent lipid metabolism is induced in cancer cells through the PI3K/AKT/mTOR axis downstream of growth factor signaling.23-26 It has also been demonstrated that SREBP1 is activated in response to transformation of cells with oncogenic mutants of PI3K and Ras.27 Furthermore, inactivation of SREBP1 in cancer cells attenuates cell proliferation.14,23,24,27,28 These observations are in agreement with reports demonstrating that inactivation of SREBP1 attenuates tumor growth in xenograph tumor models in mice.28-30 SREBP1 may be especially important to protect cancer cells from metabolic stress and lipotoxicity by regulating the ratio between saturated and unsaturated fatty acids, primarily by controlling the expression of the fatty acid desaturase SCD1.28,30,31
Although it is now clear that SREBP1 is activated through the PI3K/AKT/mTOR pathway in cancer cells, much less is known about the mechanisms involved. Transcription of the SREBP genes is controlled by both feedback and feed-forward mechanisms, some of which are responsive to growth factor signaling.3,5 In addition, the cleavage and activation of the SREBPs are under stringent control by sterols and growth factor signaling.32,33 Furthermore, the stability of the nuclear forms of these transcription factors are controlled by their phosphorylation-dependent degradation, and some of these phosphorylation events are regulated by growth factor signaling.8,9,34 The relative contribution of these regulatory mechanisms to the enhanced activity of the SREBP pathway in cancer cells is currently unclear. While investigating the SREBP pathway during the cell cycle, we found that nSREBP1 accumulated as a highly phosphorylated form during mitosis as a result of Cdk1-mediated phosphorylation of S439 in the C-terminal domain of the protein.13,14 We also found that a number of SREBP target genes were induced during this phase of the cell cycle, presumably as a result of the stabilization of nSREBP1. In the current investigation, we demonstrate that the Cdk1-mediated phosphorylation of nSREBP1 creates a binding site for Plk1, a separate mitotic kinase. Plk1 binds nSREBP1 during mitosis, resulting in further phosphorylation of its C-terminal domain. We identify 3 residues in nSREBP1 targeted by Plk1, T424, S467 and S486. Phosphorylation of these residues contributes to the hyperphosphorylation and stabilization of nSREBP1 during mitosis. Consequently, inactivation of Plk1 attenuated both the accumulation of nSREBP1 and the expression of SREBP target genes during this phase of the cell cycle. In agreement with these observations, it was recently reported that the expression of several SREBP target genes was attenuated when a panel of prostate cancer cells were treated with an inhibitor of Plk1.35,36 Although these studies did not resolve the mechanisms involved, the authors suggested that the reduced expression of these genes could attenuate cell proliferation, potentially by impacting on androgen receptor signaling.
Plk1 phosphorylates a large number of proteins, especially in late G2 and mitosis.15,17 In some cases, the Plk1-mediated phosphorylation has been found to affect the stability of its substrates. However, in most of these instances the substrates are targeted for degradation by the ubiquitin-proteasome pathway following phosphorylation. More specifically, it has been demonstrated that Plk1-mediated phosphorylation of substrate proteins often generate binding sites for β-TrCP, the substrate recognition component of a specific SCF ubiquitin ligase.37-42 One exception is FoxM1, a transcription factor that plays important roles for mitotic progression. It was demonstrated that Plk1-mediated phosphorylation of FoxM1 during mitosis enhanced its transcriptional activity.43 Interestingly, FoxM1 is a target of Fbw7, the same ubiquitin ligase that targets nSREBP1 for degradation.44 It will be interesting to learn if FoxM1 escapes Fbw7-dependent degradation during mitosis as a result of its phosphorylation by Plk1.
We have previously demonstrated that nSREBP1 is degraded by Fbw7 following phosphorylation of 2 residues in its C-terminal domain, T426 and S430 in SREBP1a.8,9 One of the residues targeted by Plk1, T424, is located in close proximity of the Fbw7 binding site in nSREBP1. Using various protein-protein interaction assays, we were able to demonstrate that phosphorylation of T424 attenuates the interaction between Fbw7 and nSREBP1. Consequently, mutation of this residue to a non-phosphorylated residue destabilized nSREBP1, while the opposite was true if T424 was substituted with a phosphomimetic residue, suggesting that the phosphorylation of T424 during mitosis interferes with its interaction with Fbw7. This hypothesis was further strengthened by our observation that Fbw7 was unable to interact with the highly phosphorylated form of nSREBP1 found in mitotic cells. Fbw7 is found as a dimer in vivo, and it was recently suggested that dimerization of Fbw7 could affect its substrate specificity.45,46 The authors proposed that high affinity substrates could be targeted for degradation by both monomers and dimers, while the degradation of low affinity substrates required Fbw7 dimerization. Interestingly, the authors found that the degradation of nSREBP1 required the dimerization of Fbw7, suggesting that nSREBP1 is a low affinity substrate. Based on their analysis, the authors suggested that one explanation for the apparent weak binding between Fbw7 and nSREBP1 could be the presence of polar residues in close proximity of its Fbw7 binding site, especially T424. In support of this hypothesis, our data suggest that the phosphorylation of this residue in nSREBP1 by Plk1 may disrupt its interaction with Fbw7 during mitosis, resulting in the stabilization of nSREBP1 seen during this phase of the cell cycle. However, Plk1 also phosphorylates 2 other residues in the C-terminal domain of nSREBP1, S467 and S486. Although it is possible that the phosphorylation of these residues contributes to the stabilization of nSREBP1 during mitosis, the functional importance of these phosphorylation events remains to be elucidated. However, it is becoming increasingly clear that the C-terminus of nSREBP1 should be regarded as a regulatory domain, with crosstalk between different phosphorylation events and interacting proteins. This possibility could be important to the SREBP field, since various C-terminal truncations of nSREBP1 have been used in several studies. Depending on the extent of these truncations, such constructs may lack one or more regulatory motifs, something that could affect the function of the corresponding nSREBP1 proteins.
Since we found that nSREBP1 escaped Fbw7-dependent degradation during mitosis, we asked whether SREBP1 could play a role during cell division. Inactivation of SREBP1 resulted in a partial, but reproducible, mitotic defect. Cells appeared to remain in mitosis for longer times, suggesting that SREBP1 could contribute to mitotic progression, potentially by regulating mitotic exit. The regulation of lipid metabolism during the cell cycle has not been investigated in great detail. However, it was recently demonstrated that cells modulate the levels and localization of specific lipids during cell division.47 In addition, the authors demonstrated that siRNA-mediated inactivation of several genes coding for enzymes involved in lipid metabolism, including the SREBP1 target gene SCD1, resulted in mitotic defects. Thus, it is possible that SREBP1 could influence mitotic progression by regulating one or more of its classical target genes involved in lipid metabolism. This model is in agreement with a recent report suggesting that de novo fatty acid synthesis during mitosis is required to complete cell division.48 However, it is also possible that SREBP1 controls other genes related to cell division. It was recently shown that nSREBP1 represses primary cilium assembly through its regulation of the gene encoding phospholipase A2 group III.49 Loss of the primary cilium is often seen in cancer cells, and the assembly and disassembly of this structure is tightly linked to the cell cycle.50 Specifically, the primary cilium needs to be disassembled prior to mitosis and reassembled once cell division is completed. Importantly, Plk1 has been shown to regulate the disassembly of the primary cilium.51,52 Thus, it is possible that SREBP1 could influence cell cycle progression, including cell division, through its regulation of primary ciliogenesis. Another possibility is that nSREBP1 is stabilized during mitosis to ensure that cells retain a pool of active SREBP1 molecules once they leave mitosis and enter G1.14 This type of epigenetic mechanism to retain transcriptional regulators during mitosis, known as mitotic bookmarking, has been reported for other transcription factors53-55. The latter model is in agreement with the observation that nSREBP1 is associated with target promoters during mitosis.13 It is also in accord with observations that long-term inactivation of SREBP1 in asynchronous cells results in a partial G1 arrest.14,28 In the case of nSREBP1, such a mechanism could be important since the membrane structures controlling the activation of SREBP1 are fragmented during mitosis. Further work is needed to determine whether the mitotic defect reported here in response to acute inactivation of SREBP1 during this phase of the cell cycle contributes to the G1/S arrest observed following chronic SREBP1 inactivation.
The current study demonstrates that Plk1-mediated phosphorylation of nSREBP1 protects it from Fbw7-dependent degradation during mitosis, and suggest that SREBP1 could influence cell division. However, additional work is required to identify the exact role(s) of SREBP1 during this phase of the cell cycle. Plk1 is overexpressed in several human cancers, and the kinase activity of Plk1 is therefore an attractive cancer drug target.56-58 Further work will be required to determine whether the Plk1-mediated regulation of nSREBP1 during cell division impacts on the enhanced activity of this transcription factor seen in cancer cells. In addition, inactivating mutations in Fbw7 are common in a wide range of human cancers.11,59 Thus, the role of Fbw7 in the activation of SREBP1 in cancer cells will be an important area of future work, something that was highlighted by a recent report demonstrating that inactivation of mTor complex 2 (mTORC2) suppressed lipogenesis in cancer cells by enhancing the Fbw7-dependent degradation of nSREBP1.34
Materials and methods
Cell culture and treatments
All tissue culture media and antibiotics were obtained from Sigma. Human embryonic kidney 293 (HEK293), HepG2 and HeLa cells were from American Type Culture Collection. Fbw7-positive and Fbw7-negative HCT116 cells were kind gifts of B. Vogelstein.60 To arrest cells in G2/M, cells were treated with nocodazole (100 ng/ml) for 16 hours. The double-thymidine block protocol was performed with a first block of 16 h, a 10 h release, and a second block of 15 h. The final concentration of thymidine used in the block medium was 2 mM.
Reagents and antibodies
HRP-conjugated anti-mouse and anti-rabbit IgG were from Invitrogen. Anti-FLAG M2 affinity gel, Anti-FLAG antibody (M5), nocodazole, BTO-1, MG-132 (Z-Leu-Leu-Leu-al) and standard chemicals were from Sigma. BI-2536 was from Selleck Chemicals. Monoclonal anti-SREBP1 (2A4), anti-tubulin (TU-02), anti-cyclin B1 (GNS1), anti-c-myc (9E10), anti-Plk1 (F-8), anti-GFP (MB-2), anti-GST (B-14), and rabbit cyclin E (C-19), anti-SREBP1 (H160), anti-p-Histone H3 (Ser10)-R and anti-His (H-15) were from Santa Cruz Biotechnology. Anti-phospho-Cdk1 (Thr161) and anti-Cdc25C were from Cell Signaling Technology. Recombinant human Plk1 was from Invitrogen and recombinant Cdk1/cyclin B1 was a kind gift from David O. Morgan.
Plasmids, siRNA and DNA transfections
The expression vectors for FLAG-tagged nSREBP1a (amino acid residues 1–490), nSREBP1c (amino acid residues 1–466) and nSREBP1aΔC (amino acid residues 1–417) have been described.8,14 Point mutations in nSREBP1 were generated by site-directed mutagenesis (QuickChange, Stratagene). The HMG-CoA synthase (SYNSRE-luc) and LDL receptor (LDLr-luc) promoter-reporter constructs have been described.9 Expression vectors for HA-Plk1, WT and kinase-deficient (K82M), were kind gifts of KS Lee.61 Expression vectors for GST-PBD and myc-PBD, WT and binding-deficient, were kind gifts of HH Silljé.62 All other expression vectors have been described previously.8,13,14 ON-TARGETplus SMARTpool control, SREBP1, Plk1 and Fbw7 siRNA were from GE Dharmacon. Transient transfections were performed using the MBS transfection kit (Stratagene).
Cell lysis and immunoblotting
Cells were lysed in buffer A (50 mM HEPES (pH 7.2), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 20 mM NaF, 2 mM Na3VO4, 10 mM β-glycerophosphate, 1% (w/v) Triton X-100, 10% (w/v) glycerol, 1 mM PMSF, 10 mM sodium butyrate, 1% aprotinin, 0.1% SDS and 0.5% sodium deoxycholate) and cleared by centrifugation. For in vitro kinase assays and protein-protein and protein-peptide interactions cell lysates were prepared in the absence of SDS and sodium deoxycholate. Proteins were resolved by SDS-PAGE and transferred to nitrocellulose membranes (GE Healthcare). To ensure that equal amounts of protein were loaded in each well, the levels of α-tubulin in the samples were estimated by Western blotting.
Luciferase and β-galactosidase assays
Cells were transiently transfected with the indicated promoter-reporter gene in the absence or presence of the indicated expression vectors and/or siRNA. Luciferase activities were determined in duplicate samples as described by the manufacturer (Promega). Cells were also transfected with the β-galactosidase gene as an internal control for transfection efficiency. Luciferase values (relative light units, RLU) were calculated by dividing the luciferase activity by the β-galactosidase activity. The data represent the average −/+ SD of three independent experiments performed in duplicates.
Kinase assays
Recombinant 6xHis-nSREBP1a, either WT or the indicated mutants, was incubated with whole cell lysates in kinase buffer (10 mM Hepes (pH 7.4), 3.5 mM MgCl2, 0.2 mM DTT, 1 mM ATP and 10 mM β-glycerolphosphate) for 1 hour at 30°C. In other experiments, recombinant 6xHis-nSREBP1a was incubated with recombinant Plk1 or Cdk1/cyclin B1 in kinase buffer for 30 min at 30°C. The 6xHis-tagged proteins were captured on NiTA-agarose (Qiagen), washed with buffer A and resolved by SDS-PAGE. The phosphorylation of nSREBP1a was monitored with phosphorylation-specific antibodies. When indicated, the reactions were performed in the presence of [γ-32P] ATP (Amersham).
Peptide pull-down assays
Peptides corresponding to sequences in human SREBP1, either unmodified or phosphorylated on the indicated residues, were synthesized using Fmoc chemistry. The peptides were coupled to Sulpholink (Pierce) and used in peptide pull-down assays with cell lysates from HEK293 or HeLa cells in buffer A, and the bound proteins were subjected to SDS-PAGE and Western blotting using 20% of input as controls.
In situ trysin digestion, 2-dimensional phosphopeptide mapping and phosphoamino acid analysis
Phosphorylated proteins were separated by SDS-PAGE, using NuPAGE 4–12% Bis-Tris gels (Invitrogen) and transferred to nitrocellulose membranes (GE Healthcare). Samples were processed for tryptic digestion, 2-dimensional phosphopeptide mapping, phosphoamino acid analysis, and Edman degradation, as described.63
Generation of phosphorylation-specific SREBP1 antibodies
Synthetic phosphopeptides corresponding to residues 420–433 (T424 phosphorylated), 463–472 (S467 phosphorylated) and 483–490 (S486 phosphorylated) in human SREBP1a were coupled to keyhole limpet haemocyanin before being injected into rabbits. The phosphopeptides and the corresponding non-phosphorylated peptides, as well as phospho-serine (anti-pS467 and anti-pS486) and phospho-threonine (anti-pT424), were coupled to Sulpholink (Pierce) and used as affinity matrixes to purify the individual antibodies from rabbit sera.
Generation of recombinant proteins
The GST and 6xHis fusion proteins were expressed in Escherichia coli BL21 and purified according to standard protocols.
RT-PCR assays
Total RNA was extracted with TRIzol reagent (Invitrogen). cDNA was synthesized with SuperScript III Reverse Transcriptase (Invitrogen) using oligo-dT, followed by PCR with target-specific primers. The PCR programs, using High Fidelity DNA polymerase (Invitrogen), were optimized for the individual target genes as described.8,9,14
FACS analysis
For FACS analysis, cells were harvested by treatment with trypsin, washed in phosphate-buffered saline, and resuspended in 70% ethanol for storage at −20°C until further analysis. Cells were stained with propidium iodide (50 μg/ml) and their DNA content was analyzed with a CYAN ADP High-Performance Flow Cytometer (DakoCytomation).
Supplementary Material
Abbreviations
- SREBP
sterol regulatory element-binding protein
- Plk1
polo-like kinase 1
- PBD
polo-box domain
- Fbw7
F-box and WD repeat domain-containing 7
- SCF
Skp1-Cullin-F-box ubiquitin ligase complex
- Cdk
Cyclin-dependent protein kinase
- mTor
mechanistic target of rapamycin
- KD
kinase-deficient
- siRNA
small interfering RNA
- FACS
fluorescence activated cell sorting
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Ulla Engström for peptide synthesis and antibody production, Christer Wernstedt for assistance with phosphopeptide mapping and identification, and Alfonso Blanco and the UCD Conway Flow Cytometry Core for assistance with FACS analysis.
Funding
This work was supported by Science Foundation Ireland (07/SK/B1242b and 10/IN.1/B2986).
References
- [1].Brown MS, Goldstein JL. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci U S A 1999; 96:11041-8; PMID:10500120; http://dx.doi.org/ 10.1073/pnas.96.20.11041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 2002; 109:1125-31; PMID:11994399; http://dx.doi.org/ 10.1172/JCI0215593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell 2006; 124:35-46; PMID:16413480; http://dx.doi.org/ 10.1016/j.cell.2005.12.022 [DOI] [PubMed] [Google Scholar]
- [4].Osborne TF, Espenshade PJ. Evolutionary conservation and adaptation in the mechanism that regulates SREBP action: what a long, strange tRIP it's been. Genes Dev 2009; 23:2578-91; PMID:19933148; http://dx.doi.org/ 10.1101/gad.1854309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Brown MS, Goldstein JL. Cholesterol feedback: from Schoenheimer's bottle to Scap's MELADL. J Lipid Res 2009; 50(Suppl):S15-27; PMID:18974038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shimomura I, Shimano H, Horton JD, Goldstein JL, Brown MS. Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. J Clin Invest 1997; 99:838-45; PMID:9062340; http://dx.doi.org/ 10.1172/JCI119247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hirano Y, Yoshida M, Shimizu M, Sato R. Direct demonstration of rapid degradation of nuclear sterol regulatory element-binding proteins by the ubiquitin-proteasome pathway. J Biol Chem 2001; 276:36431-7; PMID:11477106; http://dx.doi.org/ 10.1074/jbc.M105200200 [DOI] [PubMed] [Google Scholar]
- [8].Bengoechea-Alonso MT, Ericsson J. A phosphorylation cascade controls the degradation of active SREBP1. J Biol Chem 2009; 284:5885-95; PMID:19126544; http://dx.doi.org/ 10.1074/jbc.M807906200 [DOI] [PubMed] [Google Scholar]
- [9].Sundqvist A, Bengoechea-Alonso MT, Ye X, Lukiyanchuk V, Jin J, Harper JW, Ericsson J. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab 2005; 1:379-91; PMID:16054087; http://dx.doi.org/ 10.1016/j.cmet.2005.04.010 [DOI] [PubMed] [Google Scholar]
- [10].Cremona CA, Sancho R, Diefenbacher ME, Behrens A. Fbw7 and its counteracting forces in stem cells and cancer: Oncoproteins in the balance. Semin Cancer Biol 2016; 36:52-61; PMID:26410034; http://dx.doi.org/ 10.1016/j.semcancer.2015.09.006 [DOI] [PubMed] [Google Scholar]
- [11].Davis RJ, Welcker M, Clurman BE. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell 2014; 26:455-64; PMID:25314076; http://dx.doi.org/ 10.1016/j.ccell.2014.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Xu W, Taranets L, Popov N. Regulating Fbw7 on the road to cancer. Semin Cancer Biol 2016; 36:62-70; PMID:26459133; http://dx.doi.org/ 10.1016/j.semcancer.2015.09.005 [DOI] [PubMed] [Google Scholar]
- [13].Bengoechea-Alonso MT, Punga T, Ericsson J. Hyperphosphorylation regulates the activity of SREBP1 during mitosis. Proc Natl Acad Sci U S A 2005; 102:11681-6; PMID:16081540; http://dx.doi.org/ 10.1073/pnas.0501494102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bengoechea-Alonso MT, Ericsson J. Cdk1/cyclin B-mediated phosphorylation stabilizes SREBP1 during mitosis. Cell Cycle 2006; 5:1708-18; PMID:16880739; http://dx.doi.org/ 10.4161/cc.5.15.3131 [DOI] [PubMed] [Google Scholar]
- [15].Bruinsma W, Raaijmakers JA, Medema RH. Switching Polo-like kinase-1 on and off in time and space. Trends Biochem Sci 2012; 37:534-42; PMID:23141205; http://dx.doi.org/ 10.1016/j.tibs.2012.09.005 [DOI] [PubMed] [Google Scholar]
- [16].de Carcer G, Manning G, Malumbres M. From Plk1 to Plk5: functional evolution of polo-like kinases. Cell Cycle 2011; 10:2255-62; PMID:21654194; http://dx.doi.org/ 10.4161/cc.10.14.16494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zitouni S, Nabais C, Jana SC, Guerrero A, Bettencourt-Dias M. Polo-like kinases: structural variations lead to multiple functions. Nat Rev Mol Cell Biol 2014; 15:433-52; PMID:24954208; http://dx.doi.org/ 10.1038/nrm3819 [DOI] [PubMed] [Google Scholar]
- [18].Grim JE, Gustafson MP, Hirata RK, Hagar AC, Swanger J, Welcker M, Hwang HC, Ericsson J, Russell DW, Clurman BE. Isoform- and cell cycle-dependent substrate degradation by the Fbw7 ubiquitin ligase. J Cell Biol 2008; 181:913-20; PMID:18559665; http://dx.doi.org/ 10.1083/jcb.200802076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular fatty acid metabolism and cancer. Cell Metab 2013; 18:153-61; PMID:23791484; http://dx.doi.org/ 10.1016/j.cmet.2013.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Guo D, Bell EH, Mischel P, Chakravarti A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr Pharm Des 2014; 20:2619-26; PMID:23859617; http://dx.doi.org/ 10.2174/13816128113199990486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Peck B, Schulze A. Lipid Desaturation: the next step in targeting lipogenesis in cancer? FEBS J 2016; 283:2767-78; PMID:26881388 [DOI] [PubMed] [Google Scholar]
- [22].Porstmann T, Santos CR, Lewis C, Griffiths B, Schulze A. A new player in the orchestra of cell growth: SREBP activity is regulated by mTORC1 and contributes to the regulation of cell and organ size. Biochem Soc Trans 2009; 37:278-83; PMID:19143646; http://dx.doi.org/ 10.1042/BST0370278 [DOI] [PubMed] [Google Scholar]
- [23].Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al.. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 2010; 39:171-83; PMID:20670887; http://dx.doi.org/ 10.1016/j.molcel.2010.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Guo D, Prins RM, Dang J, Kuga D, Iwanami A, Soto H, Lin KY, Huang TT, Akhavan D, Hock MB, et al.. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci Signal 2009; 2:ra82; PMID:20009104; http://dx.doi.org/ 10.1126/scisignal.2000446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, Downward J, Schulze A. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 2005; 24:6465-81; PMID:16007182 [DOI] [PubMed] [Google Scholar]
- [26].Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, Schulze A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab 2008; 8:224-36; PMID:18762023; http://dx.doi.org/ 10.1016/j.cmet.2008.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ricoult SJ, Yecies JL, Ben-Sahra I, Manning BD. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 2016; 35:1250-60; PMID:26028026; http://dx.doi.org/ 10.1038/onc.2015.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Williams KJ, Argus JP, Zhu Y, Wilks MQ, Marbois BN, York AG, Kidani Y, Pourzia AL, Akhavan D, Lisiero DN, et al.. An essential requirement for the SCAP/SREBP signaling axis to protect cancer cells from lipotoxicity. Cancer Res 2013; 73:2850-62; PMID:23440422; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-0382-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cheng C, Ru P, Geng F, Liu J, Yoo JY, Wu X, Cheng X, Euthine V, Hu P, Guo JY, et al.. Glucose-Mediated N-glycosylation of SCAP Is Essential for SREBP-1 Activation and Tumor Growth. Cancer Cell 2015; 28:569-81; PMID:26555173; http://dx.doi.org/ 10.1016/j.ccell.2015.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Griffiths B, Lewis CA, Bensaad K, Ros S, Zhang Q, Ferber EC, Konisti S, Peck B, Miess H, East P, et al.. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab 2013; 1:3; PMID:24280005; http://dx.doi.org/ 10.1186/2049-3002-1-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lewis CA, Brault C, Peck B, Bensaad K, Griffiths B, Mitter R, Chakravarty P, East P, Dankworth B, Alibhai D, et al.. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 2015; 34:5128-40; PMID:25619842; http://dx.doi.org/ 10.1038/onc.2014.439 [DOI] [PubMed] [Google Scholar]
- [32].Bengoechea-Alonso MT, Ericsson J. SREBP in signal transduction: cholesterol metabolism and beyond. Curr Opin Cell Biol 2007; 19:215-22; PMID:17303406; http://dx.doi.org/ 10.1016/j.ceb.2007.02.004 [DOI] [PubMed] [Google Scholar]
- [33].Jeon TI, Osborne TF. SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol Metab 2012; 23:65-72; PMID:22154484; http://dx.doi.org/ 10.1016/j.tem.2011.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li S, Oh YT, Yue P, Khuri FR, Sun SY. Inhibition of mTOR complex 2 induces GSK3/FBXW7-dependent degradation of sterol regulatory element-binding protein 1 (SREBP1) and suppresses lipogenesis in cancer cells. Oncogene 2016; 35:642-50; PMID:25893295; http://dx.doi.org/ 10.1038/onc.2015.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhang Z, Chen L, Wang H, Ahmad N, Liu X. Inhibition of Plk1 represses androgen signaling pathway in castration-resistant prostate cancer. Cell Cycle 2015; 14:2142-8; PMID:25927139; http://dx.doi.org/ 10.1080/15384101.2015.1041689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhang Z, Hou X, Shao C, Li J, Cheng JX, Kuang S, Ahmad N, Ratliff T, Liu X. Plk1 inhibition enhances the efficacy of androgen signaling blockade in castration-resistant prostate cancer. Cancer Res 2014; 74:6635-47; PMID:25252916; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fukushima H, Ogura K, Wan L, Lu Y, Li V, Gao D, Liu P, Lau AW, Wu T, Kirschner MW, et al.. SCF-mediated Cdh1 degradation defines a negative feedback system that coordinates cell-cycle progression. Cell Rep 2013; 4:803-16; PMID:23972993; http://dx.doi.org/ 10.1016/j.celrep.2013.07.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hansen DV, Tung JJ, Jackson PK. CaMKII and polo-like kinase 1 sequentially phosphorylate the cytostatic factor Emi2/XErp1 to trigger its destruction and meiotic exit. Proc Natl Acad Sci U S A 2006; 103:608-13; PMID:16407128; http://dx.doi.org/ 10.1073/pnas.0509549102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mailand N, Bekker-Jensen S, Bartek J, Lukas J. Destruction of Claspin by SCFbetaTrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol Cell 2006; 23:307-18; PMID:16885021; http://dx.doi.org/ 10.1016/j.molcel.2006.06.016 [DOI] [PubMed] [Google Scholar]
- [40].Moshe Y, Boulaire J, Pagano M, Hershko A. Role of Polo-like kinase in the degradation of early mitotic inhibitor 1, a regulator of the anaphase promoting complex/cyclosome. Proc Natl Acad Sci U S A 2004; 101:7937-42; PMID:15148369; http://dx.doi.org/ 10.1073/pnas.0402442101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Peschiaroli A, Dorrello NV, Guardavaccaro D, Venere M, Halazonetis T, Sherman NE, Pagano M. SCFbetaTrCP-mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol Cell 2006; 23:319-29; PMID:16885022; http://dx.doi.org/ 10.1016/j.molcel.2006.06.013 [DOI] [PubMed] [Google Scholar]
- [42].Seki A, Coppinger JA, Du H, Jang CY, Yates JR 3rd, Fang G. Plk1- and β-TrCP-dependent degradation of Bora controls mitotic progression. J Cell Biol 2008; 181:65-78; PMID:18378770; http://dx.doi.org/ 10.1083/jcb.200712027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Fu Z, Malureanu L, Huang J, Wang W, Li H, van Deursen JM, Tindall DJ, Chen J. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat Cell Biol 2008; 10:1076-82; PMID:19160488; http://dx.doi.org/ 10.1038/ncb1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chen Y, Li Y, Xue J, Gong A, Yu G, Zhou A, Lin K, Zhang S, Zhang N, Gottardi CJ, et al.. Wnt-induced deubiquitination FoxM1 ensures nucleus β-catenin transactivation. EMBO J 2016; 35:668-84; PMID:26912724; http://dx.doi.org/ 10.15252/embj.201592810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Welcker M, Clurman BE. Oncoprotein ubiquitylation: dimers, degrons, and degradation. Cell Cycle 2014; 13:1829-30; PMID:24866825; http://dx.doi.org/ 10.4161/cc.29325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Welcker M, Larimore EA, Swanger J, Bengoechea-Alonso MT, Grim JE, Ericsson J, Zheng N, Clurman BE. Fbw7 dimerization determines the specificity and robustness of substrate degradation. Genes Dev 2013; 27:2531-6; PMID:24298052; http://dx.doi.org/ 10.1101/gad.229195.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Atilla-Gokcumen GE, Muro E, Relat-Goberna J, Sasse S, Bedigian A, Coughlin ML, Garcia-Manyes S, Eggert US. Dividing cells regulate their lipid composition and localization. Cell 2014; 156:428-39; PMID:24462247; http://dx.doi.org/ 10.1016/j.cell.2013.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Scaglia N, Tyekucheva S, Zadra G, Photopoulos C, Loda M. De novo fatty acid synthesis at the mitotic exit is required to complete cellular division. Cell Cycle 2014; 13:859-68; PMID:24418822; http://dx.doi.org/ 10.4161/cc.27767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gijs HL, Willemarck N, Vanderhoydonc F, Khan NA, Dehairs J, Derua R, Waelkens E, Taketomi Y, Murakami M, Agostinis P, et al.. Primary cilium suppression by SREBP1c involves distortion of vesicular trafficking by PLA2G3. Mol Biol Cell 2015; 26:2321-32; PMID:25904332; http://dx.doi.org/ 10.1091/mbc.E14-10-1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Izawa I, Goto H, Kasahara K, Inagaki M. Current topics of functional links between primary cilia and cell cycle. Cilia 2015; 4:12; PMID:26719793; http://dx.doi.org/ 10.1186/s13630-015-0021-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lee KH, Johmura Y, Yu LR, Park JE, Gao Y, Bang JK, Zhou M, Veenstra TD, Yeon Kim B, Lee KS. Identification of a novel Wnt5a-CK1varepsilon-Dvl2-Plk1-mediated primary cilia disassembly pathway. EMBO J 2012; 31:3104-17; PMID:22609948; http://dx.doi.org/ 10.1038/emboj.2012.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang G, Chen Q, Zhang X, Zhang B, Zhuo X, Liu J, Jiang Q, Zhang C. PCM1 recruits Plk1 to the pericentriolar matrix to promote primary cilia disassembly before mitotic entry. J Cell Sci 2013; 126:1355-65; PMID:23345402; http://dx.doi.org/ 10.1242/jcs.114918 [DOI] [PubMed] [Google Scholar]
- [53].Christova R, Oelgeschlager T. Association of human TFIID-promoter complexes with silenced mitotic chromatin in vivo. Nat Cell Biol 2002; 4:79-82; PMID:11744923; http://dx.doi.org/ 10.1038/ncb733 [DOI] [PubMed] [Google Scholar]
- [54].Kadauke S, Udugama MI, Pawlicki JM, Achtman JC, Jain DP, Cheng Y, Hardison RC, Blobel GA. Tissue-specific mitotic bookmarking by hematopoietic transcription factor GATA1. Cell 2012; 150:725-37; PMID:22901805; http://dx.doi.org/ 10.1016/j.cell.2012.06.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zaidi SK, Grandy RA, Lopez-Camacho C, Montecino M, van Wijnen AJ, Lian JB, Stein JL, Stein GS. Bookmarking target genes in mitosis: a shared epigenetic trait of phenotypic transcription factors and oncogenes? Cancer Res 2014; 74:420-5; PMID:24408924; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lee KS, Burke TR Jr, Park JE, Bang JK, Lee E. Recent Advances and New Strategies in Targeting Plk1 for Anticancer Therapy. Trends Pharmacol Sci 2015; 36:858-77; PMID:26478211; http://dx.doi.org/ 10.1016/j.tips.2015.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].McInnes C, Wyatt MD. PLK1 as an oncology target: current status and future potential. Drug Discov Today 2011; 16:619-25; PMID:21601650; http://dx.doi.org/ 10.1016/j.drudis.2011.05.002 [DOI] [PubMed] [Google Scholar]
- [58].Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov 2010; 9:643-60; PMID:20671765; http://dx.doi.org/ 10.1038/nrd3184 [DOI] [PubMed] [Google Scholar]
- [59].Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D, Marth C, et al.. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res 2007; 67:9006-12; PMID:17909001; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-1320 [DOI] [PubMed] [Google Scholar]
- [60].Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, Lengauer C. Inactivation of hCDC4 can cause chromosomal instability. Nature 2004; 428:77-81; PMID:14999283; http://dx.doi.org/ 10.1038/nature02313 [DOI] [PubMed] [Google Scholar]
- [61].Seong YS, Kamijo K, Lee JS, Fernandez E, Kuriyama R, Miki T, Lee KS. A spindle checkpoint arrest and a cytokinesis failure by the dominant-negative polo-box domain of Plk1 in U-2 OS cells. J Biol Chem 2002; 277:32282-93; PMID:12034729; http://dx.doi.org/ 10.1074/jbc.M202602200 [DOI] [PubMed] [Google Scholar]
- [62].Hanisch A, Wehner A, Nigg EA, Sillje HH. Different Plk1 functions show distinct dependencies on Polo-Box domain-mediated targeting. Mol Biol Cell 2006; 17:448-59; PMID:16267267; http://dx.doi.org/ 10.1091/mbc.E05-08-0801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Blume-Jensen P, Wernstedt C, Heldin CH, Ronnstrand L. Identification of the major phosphorylation sites for protein kinase C in kit/stem cell factor receptor in vitro and in intact cells. J Biol Chem 1995; 270:14192-200; PMID:7539802; http://dx.doi.org/ 10.1074/jbc.270.23.14192 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.