

ABSTRACT
Asthma is a chronic respiratory disease characterized by reversible airway obstruction with persistent airway inflammation and airway remodeling. Features of airway remodeling include increased airway smooth muscle (ASM) mass. A disintegrin and metalloproteinase (ADAM)–33 has been identified as playing a role in the pathophysiology of asthma. ADAM-33 is expressed in ASM cells and is suggested to play a role in the function of these cells. However, the regulation of ADAM-33 is not fully understood. Vascular endothelial growth factor (VEGF) has been implicated in inflammatory and airway blood vessel remodeling in asthmatics. Although VEGF was initially thought of as an endothelial-specific growth factor, recent reports have found that VEGF can promote proliferation of other cell types, including ASM cells. To investigate the precise mechanism of VEGF's effect on ASM cell proliferation, we tested the expression of ADAM-33, phospho-extracellularsignal-regulated kinase 1/2 (ERK1/2), and phospho-Akt in VEGF-stimulated ASM cells. We found that VEGF up-regulates ADAM-33 mRNA and protein levels in a dose- and time-dependent manner as well as phosphorylation of ERK1/2 and Akt. We also found that VEGF-induced ASM cell proliferation is inhibited by both ADAM-33 knockdown and a selective VEGF receptor 2 (VEGFR2) inhibitor (SU1498). Furthermore, VEGF-induced ADAM-33 expression and ASM cell proliferation were suppressed by inhibiting ERK1/2 activity, but not by inhibiting Akt activity. Collectively, our findings suggest that VEGF enhances ADAM-33 expression and ASM cell proliferation by activating the VEGFR2/ERK1/2 signaling pathway, which might be involved in the pathogenesis of airway remodeling. Further elucidation of the mechanisms underlying these observations might help develop therapeutic strategies for airway diseases associated with smooth muscle hyperplasia such as asthma.
KEYWORDS: Akt, airway remodeling, ADAM-33, ERK1/2, VEGF
Introduction
Asthma is a chronic respiratory disease characterized by reversible airway obstruction with persistent airway inflammation and airway remodeling. Airway remodeling and airway obstruction have several features in common, including airway smooth muscle (ASM) cell hyperplasia and hypertrophy, as well as an increase in vascular permeability and angiogenesis,1,2 both of which have been the target for many therapeutic regimens.
A disintegrin and metalloproteinase (ADAM)–33, a recently discovered ADAM family member, has been identified as playing a role in the pathophysiology of asthma.3 Similar to other protease-type ADAM members, the active site sequence of ADAM-33 is in the metalloprotease domain, implying that it can promote the processing of growth factors, various adhesion molecules, cytokines, and cytokine receptors.4 ADAM-33 is preferentially expressed in smooth muscle cells, fibroblasts, and myofibroblasts, but not in T cells, epithelial cells, or inflammatory cells.5 ADAM-33 has been linked to allergic airway inflammation; however, its role in the pathophysiology of asthma remains to be proven. Recent studies show that ADAM-33 is a critical factor for VEGF-D-induced proliferation of human gastric cancer cell lines.6
Recently, a number of inflammatory cell-derived growth factors and cytokines have been implicated in ASM cell division and growth. Among them, VEGF-A (hereafter referred to as VEGF) has been implicated in asthma related inflammation and remodeling.7,8 VEGF receptor (VEGFR)-2 is the primary receptor controlling VEGF-stimulated growth of blood vessels. Mechanistically, VEGF/VEGFR2signaling induces hemangiogenesis by promoting blood endothelial cell survival, migration, and proliferation in part through the activation of the phosphatidylinositol3-kinase (PI3K)/Akt signal transduction pathways and mitogen-activated protein kinase/extracellular-signal-regulated kinase-1/2 (MAPK/ERK1/2).9 In contrast, the mechanisms underlying VEGF-induced ASM cell proliferation remain poorly defined and controversial.
Therefore, in the present study, we aimed to investigate the effect of ADAM-33 on VEGF-induced ASM cell proliferation and subsequent intracellular signaling pathways.
Results
VEGF up-regulated ADAM33 expression in ASM cells
To evaluate the effect of VEGF on expression of ADAM33, we performed real-time PCR in ASM cells. When ASM cells were treated with various doses of VEGF for different times, ADAM33 mRNA expression was upregulated in a dose- and time-dependent manner (Fig. 1A, B), suggesting that VEGF up-regulates ADAM33 mRNA expression in ASM cells. In addition, we performed western blot analysis to evaluate whether VEGF also regulates ADAM33protein expression in ASM cells. Interestingly, VEGF also up-regulated ADAM33 protein expression in ASM cells in dose- and time-dependent manners (Fig. 1C, D).We did not find any cytotoxicity by VEGF in our experimental conditions (data not shown).
Figure 1.
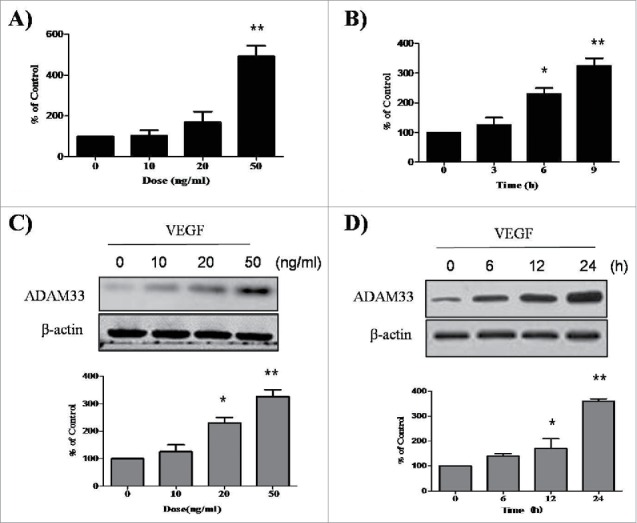
VEGF promotes ADAM-33 expression at both mRNA and protein level. ASM cells were incubated with indicated doses of VEGF for 9 h, and then real-time PCR performed. The values are normalized relative to the GAPDH standard (A). ASM cells were incubated at indicated times of VEGF (50 ng/ml), and then real-time PCR performed (B). ASM cells were incubated with indicated doses of VEGF for 24 h (C). ASM cells were incubated at indicated times of VEGF (50 ng/ml), and then western blotting analysis for ADAM-33 was performed. β-actin was used as a loading control (D). All data are representative of 3 independent experiments. Values represent the means ± SEM. *P < 0.05, **P < 0.005 vs. control.
ADAM33 siRNA transfection inhibits ASM cell proliferation
It has been reported that VEGF-D-enhanced ADAM33 plays an important role in tumor cell proliferation in the gastric cancer cell line SNU-601.6 We initially tested the effect of VEGF on the proliferation of ASM cells. When ASM cells were treated with various doses of VEGF for different times, VEGF enhanced BrdU incorporation in a dose- and time-dependent manner in ASM cells (Fig. 2A, B).
Figure 2.
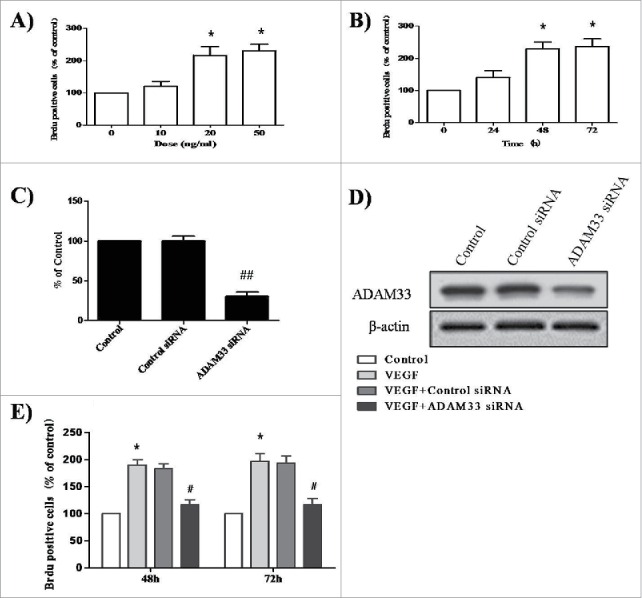
ADAM-33 siRNA transfection inhibits VEGF-induced cell proliferation. ASM cells were incubated with indicated doses of VEGF for 48 h (A). ASM cells were incubated at indicated times of VEGF (50 ng/ml), and then cell proliferation was determined by BrdU incorporation (B). ASM cells were transfected with negative siRNA or ADAM-33 siRNA, and then real-time PCR performed. The values are normalized relative to the GAPDH standard (C). ASM cells were transfected with negative siRNA or ADAM-33 siRNA, and then western blotting analysis for ADAM-33 was performed. β-actin was used as a loading control (D). ASM cells were transfected with negative siRNA or ADAM-33 siRNA in the presence of VEGF (50 ng/ml) for 48 or 72 h, and then cell proliferation was determined by BrdU incorporation (E). All experiments were done at least twice. Values represent the means ± SEM. *P < 0.05, **P < 0.005 vs. control; # P < 0.05 vs. control siRNA.
Next, to elucidate the effect of ADAM33 on the proliferation of ASM cells, we constructed an ADAM33 siRNA transfection reagent. As shown in Fig. 2C and D, we confirmed ADAM33 gene silencing at the mRNA and protein level. When ASM cells were transfected with ADAM33 siRNA or control siRNA for 48 and 72 h in the presence of 50 ng/ml VEGF, BrdU incorporation was decreased in ADAM33 siRNA transfected cells compared to negative control siRNA transfected cells (Fig. 2E). These data indicate that ADAM33 is required for VEGF-induced ASM cell proliferation.
ADAM33siRNA induces G1-phase cell-cycle arrest in VEGF-induced ASM cell proliferation
Flow cytometry analysis was used to assess whether the antiproliferative effect of ADAM33siRNA was due to cellcycle arrest in a specific phase. As shown in Figure 3, VEGF treatment significantly increased the proportion of ASM cells in the S and G2/M phases of the cell cycle, with a concomitant decrease in the proportion in G1 phaseas compared to control cells. However, ADAM33 siRNA transfection in the presence of VEGF markedly reduced the percentage of cells in the S and G2/M phases, resulting in a significant accumulation of cells in G1 phase, as compared to negative control siRNA transfected ASM cells.
Figure 3.
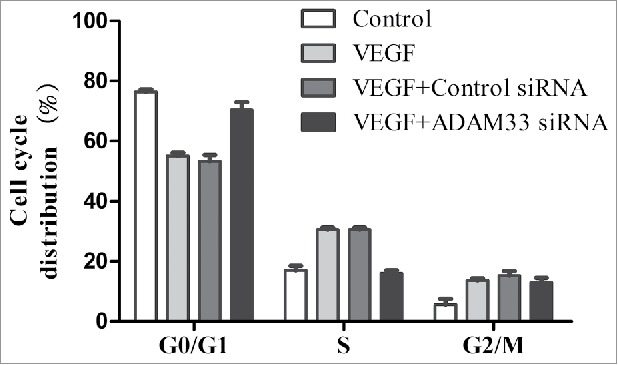
ADAM-33 siRNA transfection inhibits cell cycle in ASM cells. ASM cells were transfected with negative siRNA or ADAM-33 siRNA in the presence of VEGF (50 ng/ml) for 48 h, and then Flow cytometric analysis for cell cycle was performed. All experiments were done at least twice. Values represent the means ± SEM.
VEGF-induced ADAM33 expression and ASM cell proliferation is dependent on VEGFR2 (KDR/Flk1)
Several studies have reported that VEGF-induced cell proliferation is mediated by the interaction of VEGF with VEGFR2 (also known as KDR or FLK1).11 To determine whether blocking the VEGF-VEGFR2interaction will prevent VEGF-mediated ADAM33 up-regulation and ASM cell proliferation, we used SU1498, an inhibitor of the tyrosine kinase activity of VEGFR2, that blocks interaction of VEGF with VEGFR2 but not with VEGFR1 (FLK1). As shown in Figure 4A, VEGF-increased ADAM33 expression was inhibited by SU1498 in a dose-dependent manner. In addition, SU1498 also blocked VEGF-induced BrdU incorporation in ASM cells (Fig. 4B). These data indicate that ADAM33 is regulated by VEGF/VEGFR2 interaction.
Figure 4.
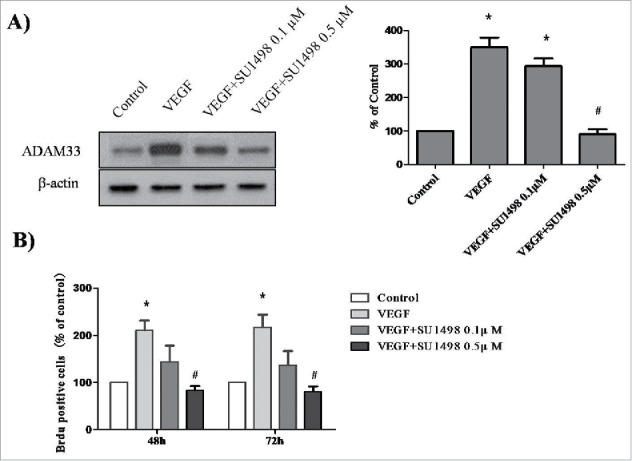
Effect of SU1498 on VEGF-induced ADAM-33 expression and cell proliferation in ASM cells. ASM cells were incubated with indicated doses of SU1498 for 2 h before treatment with VEGF (50 ng/ml) for 24 h, and then western blotting analysis for ADAM-33 was performed. β-actin was used as a loading control (A). ASM cells were incubated with indicated doses of SU1498 for 2 h before treatment with VEGF (50 ng/ml) for 48 or 72 h, and then cell proliferation was determined by BrdU incorporation (B). All experiments were done at least twice. Values represent the means ± SEM. *P < 0.05 vs. control; # P < 0.05 vs. VEGF alone.
VEGF/VEGFR2 interaction increases the phosphorylation of ERK1/2 and Akt
It has been reported that the interaction of VEGF with VEGFR2 can activate the PI3-K/Akt-dependent signaling pathway and the MAPK/ERK1/2 signal transduction pathway.12
To evaluate whether VEGF activates Akt and ERK1/2 in ASM cells, the effects of VEGF were investigated by western blotting analysis. When the cells were incubated in the absence or presence of 50 ng/ml VEGF for the indicated times, an increase in the phosphorylation of ERK1/2 and Akt was observed, with no effect on the total protein levels of these molecules (Fig. 5A, B).In addition, we observed that SU1498 inhibited VEGF-induced phosphorylation of ERK1/2 and Akt (Fig. 5C, D). These results indicate that the VEGF/VEGFR2 interaction can activate the MAPK/ERK1/2and PI3K/Akt signaling pathways in ASM cells.
Figure 5.
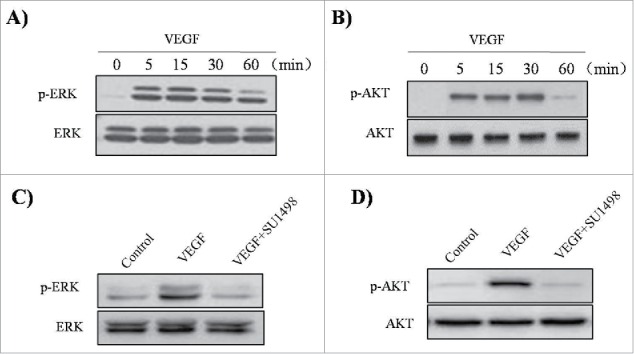
Effect of VEGF and SU1498 on phosphorylation of ERK1/2 and Akt in ASM cells. ASM cells were incubated at indicated times of VEGF (50 ng/ml), and then western blotting analysis for phospho-ERK 1/2 (A) and phospho-Akt (B) was performed. ASM cells were incubated with 0.5µM SU1498 for 2 h before treatment with VEGF (50 ng/ml) for 15 min, and then western blotting analysis for phospho-ERK 1/2 was performed (C). ASM cells were incubated with 0.5µM SU1498 for 2 h before treatment with VEGF (50 ng/ml) for 30 min, and then western blotting analysis for phospho-Akt was performed (D). The total ERK1/2 and Akt was used as a loading control. All experiments were done at least twice.
VEGF-induced cellular proliferation involves the MAPK/ ERK1/2 pathway
To determine which signaling pathways are playing a role in VEGF-induced ADAM33 expression and ASM cell proliferation, we used specific inhibitors of ERK1/2 and Akt signaling pathways. The inhibitors tested included U0126 (a MAPK/ERK1/2 inhibitor) and LY294002 (a PI3K inhibitor). Pretreatment of ASM cells with U0126 was found to significantly decrease ADAM33 expression in VEGF-treated ASM cells compared to control (Fig. 6A). However, VEGF-induced ADAM33 expression was not affected by the addition of LY294002 (Fig. 6A). These results demonstrate that VEGF increases ADAM33 expression through the activation of ERK1/2. We also investigated whether U0126 and LY294002 could inhibit the VEGF-induced proliferation of ASM cells. When the cells were incubated with U0126 or LY294002in the presence of VEGF, U0126 blocked VEGF-induced BrdU incorporation (Fig. 6B). However, LY294002 had no effect on VEGF-induced BrdU incorporation (Fig. 6B). These data show that VEGF induces proliferation of ASM cells through the activation of the ERK1/2/ADAM33 pathway.
Figure 6.
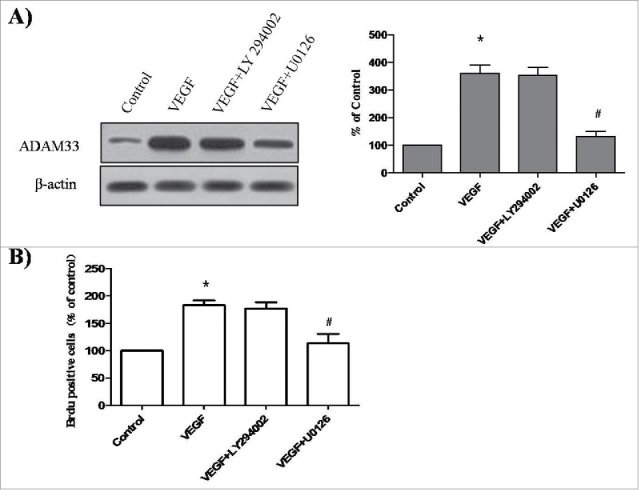
Effect of LY294002 and U0126 on ADAM-33 expression and cell proliferation in ASM cells. ASM cells were incubated with 20 µM U0126 or 20 µM LY294002 for 2 h before treatment with VEGF (50 ng/ml) for 24 h, and then western blotting analysis for ADAM33 was performed. β-actin was used as a loading control (A). ASM cells were incubated with 20 µM U0126 or 20 µM LY294002 for 2 h before treatment with VEGF (50 ng/ml) for 48 h, and then cell proliferation was determined by BrdU incorporation (B). All experiments were done at least twice. Values represent the means ± SEM. *P < 0.05 vs. control; # P < 0.05 vs. VEGF alone.
Discussion
Understanding the regulatory mechanisms of human ASM cell proliferation is of potential clinical value and can provide further insights into new treatment strategies of airway diseases associated with smooth muscle hyperplasia such as asthma.
As is well known, VEGF is a potent stimulator of angiogenesis in asthma. Studies have found that epithelial cell-secreted VEGF can promote airway remodeling in asthma.13 In addition, VEGF levels are increased in lung tissues and sputum of asthmatic patients and positively correlate with asthma disease severity. Furthermore, inhibition of VEGF can lead to a significant reduction in goblet cell hyperplasia and basement membrane thickness.14,15 A previous study found that ASM cells under strain lead to angiogenesis via secretion of VEGF and that blocking VEGF decreased these angiogenic changes.16 VEGF secreted by ASM cells is thought to play a role in extracellular matrix modulation and fibronectin secretion, as well as smooth muscle hypertrophy and therefore remodeling.17,18 In the present study, we investigated the effects of VEGF and relevant signal transduction pathways on cell proliferation in human ASM cells.
ADAM-33 was investigated as a potentially important molecule in ASM cell proliferation for a number of reasons. ADAM-33 has been shown to be expressed in ASM cells and airway fibroblasts, suggesting a role of this gene in modifying cellular functions such as proliferation, migration, and differentiation.3,5 Indeed, ADAM-33 expression was significantly higher in airways from human subjects with asthma compared to controls. Further, increased expression correlated with asthma severity progression from mild to severe lung function.19,20 As ADAM-33 is predominantly expressed in ASM cells, Lin et al. investigated whether ADAM-33 protein expression correlates with ASM cell mechanics in an ovalbumin- (OVA-) sensitized rat model. ADAM-33 expression was increased in ASM cells from the OVA-sensitized rats when compared to non-sensitized rats. Importantly, ADAM-33 expression positively correlated with cell traction force, stiffness, and expression of F-actin and vinculin, suggesting that ADAM-33 is a mediator of ASM cell dysfunction in asthma.21 Although the mechanisms for ADAM-33-involved remodeling are not clear, it has been reported that a soluble form of ADAM-33 causes a rapid induction of neovascularization both ex vivo and in vivo and also endothelial cell differentiation in vitro, suggesting that ADAM-33 can promote angiogenesis and lead to airway remodeling.22 Ito et al. have investigated ADAM-33 expression in ASM cells and found thatADAM-33 mRNA and protein are significantly higher in these cells from subjects with asthma compared to normal control subjects.5 In this study, we demonstrated that VEGF enhances ADAM-33 expression at both the mRNA and protein level, suggesting that expression may be primarily regulated at the mRNA level. To understand this further it will be necessary to study the ADAM-33 promoter region as well as the transcription factors that are predicted to interact with it in order to get a handle on the pathways capable of regulating ADAM-33 expression.
The majority of in vitro reports support PI3K and ERK1/2 activation as the major signal transduction pathways for cytokine-stimulated, G-protein-coupled-receptor (GPCR)-mediated, or receptor tyrosine kinase (RTK)-mediated proliferation of ASM cells.23 It has been found that ERK1/2 activation is involved in cell growth, morphogenesis, and migration of endothelial cells stimulated by angiogenic factors.24,25 Moreover, the activation of PI3K/Akt has also been connected to a variety of biological functions, including cell growth, vascular remodeling, angiogenesis, and survival.25,26 It has been reported that dual ERK1/2 and PI3K pathways could control ASM cell proliferation.27 VEGF is able to promote endothelial cell growth and survival via the ERK1/2 and PI3K/Akt pathways, respectively.9,28,29 Walker et al.30 compared the extent to which the ERK1/2 or PI3K cascades contributed to α-thrombin-stimulated or PDGF-stimulated proliferation of bovine tracheal smooth muscle. They found that although the PI3K pathway was essential, the ERK1/2 pathway was required for a full mitogenic response. Such findings suggest that although active PI3K is sufficient to stimulate ASM DNA synthesis, either by GPCR-coupled or RTK-coupled pathways, parallel ERK1/2-dependents signaling events are required for maximal proliferation. In our present study, we observed that ERK inhibition, but not PI3K inhibition, suppressed ADAM-33 expression induced by VEGF. Further research is needed to elucidate the mechanism by which the ERK1/2 pathway enhances transcription of ADAM-33.
Taken together, we provide the first evidence that ADAM-33 plays an important role in the process of VEGF-induced ASM cell proliferation, suggesting an effective therapeutic target in inflammatory diseases such as asthma.
Materials and methods
Antibodies and reagents
Antibodies against phospho-ERK1/2 (Thr202/Tyr204), phospho-Akt (Ser473), ERK1/2, and Akt were purchased from cell signaling technology (Danvers, MA). Antibody against ADAM-33 and β-actin was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Secondary antibodies was obtained from (Jackson Immunoresearch, West Grove, PA). VEGF were purchased from Sigma-Aldrich (St. Louis, MO). The VEGFR2 inhibitor SU1498, the PI3K inhibitor LY294002, and the MAPK/ERK1/2 inhibitor U0126 were obtained from CalBiochem (La Jolla, CA).
Cell culture
Human ASM cells were obtained from ScienCell Research laboratories. Cells were cultured in 6-well plates in Smooth Muscle Cell Medium (SMCM) containing 10% FBS and were maintained at 5% CO2 at 37°C as previously described.10 Cells from passage 3–6 maintained their SMC phenotype and were used in all experiments. Cells were characterized for smooth muscle cell markers including smooth muscle α-actin and smooth muscle heavy chain by immunofluorescence. In inhibition experiments, inhibitors of signal transduction pathways were added 2 h before the addition of VEGF. VEGF, LY294002 and U0126 were purchased from Sigma-Aldrich (St Louis, Mo). SU1498 was from CalBiochem (La Jolla, CA). These inhibitors were dissolved in dimethyl sulfoxide (DMSO; final concentration of 0.1%, vol/vol) and added to the medium. Vehicle controls contained the same amount of DMSO.
Transfection of small interfering RNA (siRNA)
ADAM-33 small interfering (si) RNAs were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). ADAM-33 was transfected into ASM cells according to a siRNA transfection protocol provided by Santa Cruz Biotechnology. Briefly, after culturing ASM cells in antibiotic-free SMCM at 37 °C in a humidified atmosphere of 5% CO2 for 24 h, the siRNA duplex solution, which was diluted in siRNA transfection medium (Santa Cruz Biotechnology), was added to the ASM cells. After transfection for 24 h, the medium was replaced with normal SMCM, and ASM cells were treated with VEGF. Scrambled siRNA, purchased from Santa Cruz Biotechnology, was transfected to ASM cells as a negative standard.
Real-time reverse transcriptase-PCR
Total RNA was isolated from ASM cells using a TRIzol regent (Invitrogen) after exposure to VEGF or ADAM-33 siRNA. Total RNA (2 μg) was reverse transcribed using the oligo (dT) primer and MMLV reverse transcriptase (Promega, Madison, WI) at 42°C for 90 min. Real-time PCR was performed using an ABI Prism 7500 instrument according to the manufacturer's instructions (Applied Biosystems, Foster City, CA). The following primer pairs were used: ADAM33, forward 5′- CAGGAATGCCAGCTATTATC −3′ and reverse, 5′-GTTTGGTGTGGTTCAAGTTT-3′; and GAPDH, forward 5′-GGCCAAAAGGGTCATCA TC −3′ and reverse, 5′-GTGATGGCATGGACTGTGG-3′. After an initial hot start for 10 min, amplification was performed for 40 cycles consisting of denaturation for 10 s at 94°C, annealing for 30 s at 56°C, and extension for 40 s at 72°C. The amplification kinetics was recorded as sigmoid progress curves for which fluorescence was plotted against the number of amplification cycles. The threshold cycle number (CT) was used to define the initial amount of each template. The CT was the first cycle for which a detectable fluorescent signal was observed. The mRNA expression levels were determined and compared with the GAPDH standard.
Western blot analysis
The cell extracts were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose membrane. The membranes were blocked in blocking solution [5% non-fat dried milk in phosphate buffered saline (PBS)] for 2 h at room temperature and then probed with anti-ADAM-33, anti- phospho-ERK1/2, anti-ERK1/2, anti-phospho-Akt, anti-Akt, and anti-β-actin for 1 h at room temperature. After washing 3 times in phosphate-buffered saline (PBS) containing 0·1% Tween-20 (PBS-T), the membranes were incubated with secondary antibodies for 1 h at room temperature. After washing an additional 3 times in PBS-T, the membranes were developed using an electrochemiluminescence (ECL) solution (Pierce, Rockford, IL, USA) and exposed to Kodak X-ray film.
Bromodeoxyuridine (BrdU) incorporation assay
ASM cells were seeded in 96-well plates and treated with various drugs as indicated in each experiment for 48 or 72 h. At the end of treatment, BrdU incorporation was assayed by incubating the cells with BrdU for 0.5–1 h using a BrdU Cell Proliferation Assay Kit (Calbiochem, San Diego, CA) according to the manufacturer's instructions.
Cell cycle analysis
After siRNA transfection for 24 h, the medium was replaced with normal SMCM, and ASM cells were treated with VEGF for 48 h or 72 h in SMCM containing 10% FBS. All the cells were collected, and 1×106 cells were centrifuged, resuspended in ice-cold 70% ethanol and stored at −20°C until further analysis. Washed cells were stained by 0.1% Triton X-100 in 0.01 M phosphate-buffered saline (pH 7.2) with 50 μg/ml propidium iodide (Sigma-Aldrich) and 1 mg/ml RNase A (Invitrogen), and incubated at 37°C for 30 min in the dark. Samples of the cells were then analyzed for their DNA content using FACScan flow cytometry (Beckman, Miami, FL), and cell cycle phase distributions were analyzed by the Cell Quest acquisition software (BD Biosciences, Franklin Lanes, NJ). All experiments were performed in duplicate and repeated twice.
Statistical analysis
All results are expressed as the mean ± SEM. The statistical evaluation of the results was performed by an independent t-test and an ANOVA with a Tukey post-hoc test. The results were significant with a value of p < 0.05.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
Acquisition of data: Yang, Qian, Zhao, Liu, and Zheng. Analysis and interpretation of data: Kim, Jiang. Drafting of manuscript: Pei, Kim.
Funding
This work was supported by Science and Technology Fund of Tianjin Public Health Bureau (Grant No. 2015KR13).
References
- [1].Shin JH, Shim JW, Kim DS, Shim JY. TGF-β effects on airway smooth muscle cell proliferation, VEGF release and signal transduction pathways. Respirology 2009; 14:347-53; PMID:19192227; http://dx.doi.org/ 10.1111/j.1440-1843.2008.01469.x [DOI] [PubMed] [Google Scholar]
- [2].Kim SH, Yang M, Xu JG, Yu X, Qian XJ. Role of licochalcone A on thymic stromal lymphopoietin expression: implications for asthma. Exp Biol Med (Maywood) 2015; 240:26-33; PMID:25055998; http://dx.doi.org/ 10.1177/1535370214545020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Van Eerdewegh P, Little RD, Dupuis J, Del Mastro RG, Falls K, Simon J, Torrey D, Pandit S, McKenny J, Braunschweiger K, et al.. Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature 2002; 418:426-30; PMID:12110844; http://dx.doi.org/ 10.1038/nature00878 [DOI] [PubMed] [Google Scholar]
- [4].Yoshinaka T, Nishii K, Yamada K, Sawada H, Nishiwaki E, Smith K, Yoshino K, Ishiguro H, Higashiyama S. Identification and characterization of novel mouse and human ADAM33s with potential metalloprotease activity. Gene 2002; 282:227-36; PMID:11814695; http://dx.doi.org/ 10.1016/S0378-1119(01)00818-6 [DOI] [PubMed] [Google Scholar]
- [5].Ito I, Laporte JD, Fiset PO, Asai K, Yamauchi Y, Martin JG, Hamid Q. Downregulation of a disintegrin and metalloproteinase 33 by IFN-gamma in human airway smooth muscle cells. J Allergy Clin Immunol 2007; 119:89-97; PMID:17208589; http://dx.doi.org/ 10.1016/j.jaci.2006.08.038 [DOI] [PubMed] [Google Scholar]
- [6].Kim KE, Song H, Hahm C, Yoon SY, Park S, Lee HR, Hur DY, Kim T, Kim CH, Bang SI, et al.. Expression of ADAM33 is a novel regulatory mechanism in IL-18-secreted process in gastric cancer. J Immunol 2009; 182:3548-55; PMID:19265133; http://dx.doi.org/ 10.4049/jimmunol.0801695 [DOI] [PubMed] [Google Scholar]
- [7].Hirst SJ, Barnes PJ, Twort CH. PDGF isoform-induced proliferation and receptor expression in human cultured airway smooth muscle cells. Am J Physiol 1996; 270:L415-28; PMID:8638734 [DOI] [PubMed] [Google Scholar]
- [8].Zou H, Xu YJ, Zhang ZX. Effect of vascular endothelial growth factor and its receptor KDR on human airway smooth muscle cells proliferation. Chin Med J (Engl) 2005; 118:591-4; PMID:15820091 [PubMed] [Google Scholar]
- [9].Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol 2006; 7:359-71; PMID:16633338; http://dx.doi.org/ 10.1038/nrm1911 [DOI] [PubMed] [Google Scholar]
- [10].Agrawal T, Gupta GK, Agrawal DK. Calcitriol decreases expression of importin alpha3 and attenuates RelA translocation in human bronchial smooth muscle cells. J Clin Immunol 2012; 32:1093-103; PMID:22526597; http://dx.doi.org/ 10.1007/s10875-012-9696-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dellinger MT, Brekken RA. Phosphorylation of Akt and ERK1/2 is required for VEGF-A/VEGFR2-induced proliferation and migration of lymphatic endothelium. PLoS One 2011; 6:e28947; PMID:22174934; http://dx.doi.org/ 10.1371/journal.pone.0028947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liang Y, Brekken RA, Hyder SM. Vascular endothelial growth factor induces proliferation of breast cancer cells and inhibits the anti-proliferative activity of anti-hormones. Endocr Relat Cancer 2006; 13:905-19; PMID:16954439; http://dx.doi.org/ 10.1677/erc.1.01221 [DOI] [PubMed] [Google Scholar]
- [13].Lopez-Guisa JM, Powers C, File D, Cochrane E, Jimenez N, Debley JS. Airway epithelial cells from asthmatic children differentially express proremodeling factors. J Allergy Clin Immunol 2012; 129:990-7; e6; PMID:22227417; http://dx.doi.org/ 10.1016/j.jaci.2011.11.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chetta A, Zanini A, Foresi A, D'Ippolito R, Tipa A, Castagnaro A, Baraldo S, Neri M, Saetta M, Olivieri D. Vascular endothelial growth factor upregulation and bronchial wall remodelling in asthma. Clin Exp Allergy 2005; 35:1437-42; PMID:16297139; http://dx.doi.org/ 10.1111/j.1365-2222.2005.02360.x [DOI] [PubMed] [Google Scholar]
- [15].Yuksel H, Yilmaz O, Karaman M, Bagriyanik HA, Firinci F, Kiray M, Turkeli A, Karaman O. Role of vascular endothelial growth factor antagonism on airway remodeling in asthma. Ann Allergy Asthma Immunol 2013; 110:150-5; PMID:23548522; http://dx.doi.org/ 10.1016/j.anai.2012.12.015 [DOI] [PubMed] [Google Scholar]
- [16].Hasaneen NA, Zucker S, Lin RZ, Vaday GG, Panettieri RA, Foda HD. Angiogenesis is induced by airway smooth muscle strain. Am J Physiol Lung Cell Mol Physiol 2007; 293:L1059-68; PMID:17693481; http://dx.doi.org/ 10.1152/ajplung.00480.2006 [DOI] [PubMed] [Google Scholar]
- [17].Kazi AS, Lotfi S, Goncharova EA, Tliba O, Amrani Y, Krymskaya VP, Lazaar AL. Vascular endothelial growth factor-induced secretion of fibronectin is ERK dependent. Am J Physiol Lung Cell Mol Physiol 2004; 286:L539-45; PMID:14633511; http://dx.doi.org/ 10.1152/ajplung.00130.2003 [DOI] [PubMed] [Google Scholar]
- [18].Ribatti D, Puxeddu I, Crivellato E, Nico B, Vacca A, Levi-Schaffer F. Angiogenesis in asthma. Clin Exp Allergy 2009; 39:1815-21; PMID:20085597; http://dx.doi.org/ 10.1111/j.1365-2222.2009.03385.x [DOI] [PubMed] [Google Scholar]
- [19].Lee JY, Park SW, Chang HK, Kim HY, Rhim T, Lee JH, Jang AS, Koh ES, Park CS. A disintegrin and metalloproteinase 33 protein in patients with asthma: Relevance to airflow limitation. Am J Respir Crit Care Med 2006; 173:729-35; PMID:16387804; http://dx.doi.org/ 10.1164/rccm.200409-1175OC [DOI] [PubMed] [Google Scholar]
- [20].Foley SC, Mogas AK, Olivenstein R, Fiset PO, Chakir J, Bourbeau J, Ernst P, Lemiere C, Martin JG, Hamid Q. Increased expression of ADAM33 and ADAM8 with disease progression in asthma. J Allergy Clin Immunol 2007; 119:863-71; PMID:17339047; http://dx.doi.org/ 10.1016/j.jaci.2006.12.665 [DOI] [PubMed] [Google Scholar]
- [21].Lin F, Song A, Wu J, Jiang X, Long J, Chen J, Duan Y, Shi Y, Deng L. ADAM33 protein expression and the mechanics of airway smooth muscle cells are highly correlated in ovalbumin-sensitized rats. Mol Med Rep 2013; 8:1209-15; PMID:23934418 [DOI] [PubMed] [Google Scholar]
- [22].Puxeddu I, Pang YY, Harvey A, Haitchi HM, Nicholas B, Yoshisue H, Ribatti D, Clough G, Powell RM, Murphy G, et al.. The soluble form of a disintegrin and metalloprotease 33 promotes angiogenesis: implications for airway remodeling in asthma. J Allergy Clin Immunol 2008; 121:1400-6; 6 e1-4; PMID:18410963; http://dx.doi.org/ 10.1016/j.jaci.2008.03.003 [DOI] [PubMed] [Google Scholar]
- [23].Hirst SJ, Martin JG, Bonacci JV, Chan V, Fixman ED, Hamid QA, Herszberg B, Lavoie JP, McVicker CG, Moir LM, et al.. Proliferative aspects of airway smooth muscle. J Allergy Clin Immunol 2004; 114:S2-17; PMID:15309015; http://dx.doi.org/ 10.1016/j.jaci.2004.04.039 [DOI] [PubMed] [Google Scholar]
- [24].Lee SJ, Namkoong S, Kim YM, Kim CK, Lee H, Ha KS, Chung HT, Kwon YG. Fractalkine stimulates angiogenesis by activating the Raf-1/MEK/ERK- and PI3K/Akt/eNOS-dependent signal pathways. Am J Physiol Heart Circ Physiol 2006; 291:H2836-46; PMID:16877565; http://dx.doi.org/ 10.1152/ajpheart.00113.2006 [DOI] [PubMed] [Google Scholar]
- [25].Chung BH, Kim JD, Kim CK, Kim JH, Won MH, Lee HS, Dong MS, Ha KS, Kwon YG, Kim YM. Icariin stimulates angiogenesis by activating the MEK/ERK- and PI3K/Akt/eNOS-dependent signal pathways in human endothelial cells. Biochem Biophys Res Commun 2008; 376:404-8; PMID:18789310; http://dx.doi.org/ 10.1016/j.bbrc.2008.09.001 [DOI] [PubMed] [Google Scholar]
- [26].Somanath PR, Razorenova OV, Chen J, Byzova TV. Akt1 in endothelial cell and angiogenesis. Cell Cycle 2006; 5:512-8; PMID:16552185; http://dx.doi.org/ 10.4161/cc.5.5.2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Burgess JK, Lee JH, Ge Q, Ramsay EE, Poniris MH, Parmentier J, Roth M, Johnson PR, Hunt NH, Black JL, et al.. Dual ERK and phosphatidylinositol 3-kinase pathways control airway smooth muscle proliferation: differences in asthma. J Cell Physiol 2008; 216:673-9; PMID:18338817; http://dx.doi.org/ 10.1002/jcp.21450 [DOI] [PubMed] [Google Scholar]
- [28].Matsumoto T, Mugishima H. Signal transduction via vascular endothelial growth factor (VEGF) receptors and their roles in atherogenesis. J Atheroscler Thromb 2006; 13:130-5; PMID:16835467; http://dx.doi.org/ 10.5551/jat.13.130 [DOI] [PubMed] [Google Scholar]
- [29].Kowanetz M, Ferrara N. Vascular endothelial growth factor signaling pathways: therapeutic perspective. Clin Cancer Res 2006; 12:5018-22; PMID:16951216; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-1520 [DOI] [PubMed] [Google Scholar]
- [30].Walker TR, Moore SM, Lawson MF, Panettieri RA Jr, Chilvers ER. Platelet-derived growth factor-BB and thrombin activate phosphoinositide 3-kinase and protein kinase B: role in mediating airway smooth muscle proliferation. Mol Pharmacol 1998; 54:1007-15; PMID:9855629 [DOI] [PubMed] [Google Scholar]