

ABSTRACT
Recent studies have demonstrated that cell cycle plays a central role in development and carcinogenesis. Thus, the use of big databases and genome-wide high-throughput data to unravel the genetic and epigenetic mechanisms underlying cell cycle progression in stem cells and cancer cells is a matter of considerable interest.
Real genetic-and-epigenetic cell cycle networks (GECNs) of embryonic stem cells (ESCs) and HeLa cancer cells were constructed by applying system modeling, system identification, and big database mining to genome-wide next-generation sequencing data. Real GECNs were then reduced to core GECNs of HeLa cells and ESCs by applying principal genome-wide network projection. In this study, we investigated potential carcinogenic and stemness mechanisms for systems cancer drug design by identifying common core and specific GECNs between HeLa cells and ESCs. Integrating drug database information with the specific GECNs of HeLa cells could lead to identification of multiple drugs for cervical cancer treatment with minimal side-effects on the genes in the common core. We found that dysregulation of miR-29C, miR-34A, miR-98, and miR-215; and methylation of ANKRD1, ARID5B, CDCA2, PIF1, STAMBPL1, TROAP, ZNF165, and HIST1H2AJ in HeLa cells could result in cell proliferation and anti-apoptosis through NFκB, TGF-β, and PI3K pathways. We also identified 3 drugs, methotrexate, quercetin, and mimosine, which repressed the activated cell cycle genes, ARID5B, STK17B, and CCL2, in HeLa cells with minimal side-effects.
KEYWORDS: big database mining, cell cycle projection method, cell cycle, carcinogenic mechanism, genetic-and-epigenetic cell cycle network, NGS data, stemness mechanism, systems pharmacology
Introduction
In general, the properties of stem cells, called “stemness,” include self-renewal and differentiation;1,2 whereas those of cancer non-stem cells, referred to as “cancerness,” comprise high mutation rates and cell cycle dysregulation that promotes cell proliferation. In 2011, Okita and Yamanaka3 proposed that the cell cycle played an important role in generating induced pluripotent stem cells. Stem cells regulate their cell cycle to undergo periods of rapid division or quiescence, maintain self-renewal, and avoid senescence.4 The cell cycle appears to influence stemness, cancerness, or both. Furthermore, recent evidence suggests that microRNAs (miRNAs) may control cell proliferation by influencing the level of a manifold of cell cycle regulators. Additionally, miRNAs are critical for human carcinogenesis and have been used as drug targets to control critical genes in cancer.5 It is now apparent that miRNAs play an important role in human carcinogenesis and aging.6-8 However, it is difficult to analyze genetic and epigenetic regulation simultaneously by applying traditional experimental analysis. Therefore, the switch between stemness and cancerness strategies in genetic and epigenetic regulation during cell cycle progression remains an unresolved issue.
Empirical evidence shows that telomeres, the segments of DNA at the ends of chromosomes, shorten slightly after each new cell division until they achieve a critical length, which triggers apoptosis; this phenomenon is called the Hayflick limit. Embryonic stem cells (ESCs) and cervical carcinoma HeLa cells present a highly active telomerase during cell division, which allows them to bypass the Hayflick limit and become immortal.9,10 In normal cells, anti-oncogenes or tumor suppressor genes regulate cell proliferation and cell cycle progression. It has been observed that cell cycle progression becomes aberrant or deregulated in nearly all transformed and neoplastic cells.11 Furthermore, recent studies have shown that miRNAs promote cell cycle progression in ESCs, thereby enabling their rapid proliferation.12 Testis-specific Y-encoded protein enhances cell proliferation and tumorigenesis by promoting cell cycle progression in HeLa cells.13 In addition, it has been suggested that cell cycle plays a major role in driving cervical cancer (i.e., HeLa cells).14 Both ESCs and HeLa cells are capable of promoting cell cycle progression through genetic and epigenetic regulation. This opens the possibility that a similar association could exist between ESCs and HeLa cells during cell cycle progression, which could be a crucial event to control cell properties, i.e., stemness or cancerness.
To date, the mechanisms underlying a number of complicated systems, such as ecosystems, biological, economic, and social systems, remain poorly understood owing to a fragmented distribution, and inconsistency in data reporting and literature. Big data has been defined as a set of techniques and technologies aimed at retrieving hidden information from large datasets that are diverse, complex, and of massive scale.15 An important issue that still needs to be addressed is the mining and integration of information from big data and expanding databases (big database) to construct an explanatory pattern or model (big mechanism) with which to uncover the overall mechanisms of these complicated systems.7,8,16-18 Next-generation sequencing (NGS) and microarray analysis, which can be regarded as big data techniques to a certain extent, still contain considerable amounts of information and hidden mechanisms that have never been extracted. Owing to the accuracy of NGS and the low cost of microarrays, they are commonly used for gene expression measurements. However, their simultaneous use in cellular network analysis has been rarely advocated. Moreover, the predictive value of cellular transcription regulation networks can be used to effectively find useful information and the corresponding mechanisms from gene expression data.17,19-21 Recent evidence indicates that the pathology of most cancers is a consequence of small abnormalities in many genes, whose added effect is to perturb healthy genetic-and-epigenetic networks.22 Accordingly, and in terms of treatment, cancer should be considered a systemic disease. Network analysis can provide scientists with a systematic approach to understand such complex and systemic diseases, which could not otherwise be understood in terms of a single target or pathway model.23
At present, correlation between gene expression coefficients remains the most widely used method to identify gene networks.24-26 However, correlation networks have undirected edges and no causality between genes. In this study, we constructed a dynamic system model that could characterize causal molecular mechanisms, such as the genetic regulation exerted by transcription factors (TF) and miRNAs on target genes. With this model, we applied a system identification method to identify real genetic-and-epigenetic cell cycle networks (GECNs) of ESCs and HeLa cells. This approach allowed us to further investigate carcinogenesis in cervical cancer and the stemness mechanism in stem cells using genome-wide microarray and NGS data. Furthermore, a systems cancer drug design method was also applied to the carcinogenic mechanism to select multiple drugs with minimal side-effects.
In order to investigate carcinogenesis and stemness mechanisms from a cell cycle perspective, the corresponding genes should first be identified. Hierarchical clustering of gene expression is the most popular method for identifying genes expressed during each cell cycle phase.27,28 Since these genes could be expressed in multiple cell cycle phases, in this study, we apply the cell cycle projection method to the genome-wide expression data to quantify the phase-specific ability of each gene at each cell cycle phase. By applying the cell cycle projection method to 4-phase high-throughput data, we obtained the cell cycle genes of the GECNs in ESCs and HeLa cells and used them toward big mechanism analysis of carcinogenic and stemness mechanisms.
Because the further analysis of the genome-wide network is complex to get more insight into carcinogenic and stemness mechanisms, we should extract the core networks from the real GECNs in ESCs and HeLa cells, respectively. Up to now, the core network was still identified by the connecting numbers of its nodes.29-31 As we could identify the connection weights of the real GECN using genome-wide expression data, we applied the principle genome-wide network projection (PGNP) to extract the core network from the viewpoint of significant genome-wide network structure. To investigate carcinogenesis and stemness mechanisms, the cancer-specific GECN, common core GECN, and stem-specific GECN were obtained through complexity reduction by applying PGNP to real GECNs of ESCs and HeLa cells. We could then identify cervical carcinogenic mechanisms by applying big mechanism analysis to help design potential multiple drugs for cervical cancer treatment. The flowchart to identify real GECNs of ESCs and HeLa cells and their core GECNs is shown in Fig. 1.
Figure 1.
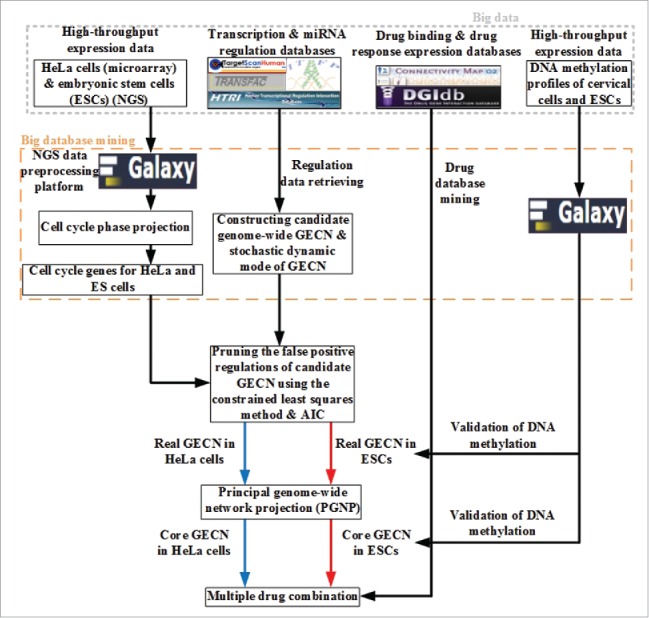
Flowchart depicting the strategy to identify core GECN in HeLa and ES cells. In this study, we mined omics information from big databases, such as transcription and miRNA regulation interactions, to construct real GECNs using genome-wide high-throughput data, a dynamic model, and AIC. For drug screening, information was integrated from cervical carcinogenic mechanisms in cancer-specific GECN and drug databases for cervical cancer treatment.
Systems pharmacology employs network analysis at multiple biological levels to understand both the therapeutic benefits and side-effects of drugs, and to design potential multiple drugs for cancer treatment.23,32 Based on the concept of systems pharmacology, we further designed multiple drugs for the decomposition of cancer-specific GECNs by integrating cervical carcinogenic mechanisms and drug databases.
In summary, we investigated the carcinogenic mechanisms of cervical cancer cells and the stemness of ES cells by applying big mechanism analysis to cell cycle progression. Based on these carcinogenic mechanisms, we designed multiple drugs for the treatment of cervical cancer. The proposed systematic design method may lead to drugs with high target specificity and minimal side-effects.
Material and methods
Data retrieval and pre-processing of NGS data in ESCs
Singh et al.33 performed RNA-seq with NGS of ESCs in early G1 (DN), late G1 (KO2), S (AzLo), and G2/M (AzHi) phases. NGS data were obtained from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/), accession number GSE53481, in Sequence Read Archive (SRA) format. Reads were aligned to the human reference genome hg19 assembly. Alignment of RNA-seq reads against known genomic annotations downloaded from the University of California Santa Cruz web site (http://www.genome.ucsc.edu) was performed using Bowtie version 0.12.7 34 and TopHat version 1.5.0 software.35 We mapped these to gene and miRNA expression data employing Cufflinks 0.0.7 gene annotation 36 on the Galaxy platform. 37-39 We used cubic spline interpolation to obtain the expression of genes and miRNAs during G1, S, G2, and M phases.
Data retrieval and pre-processing of microarray data from HeLa cells
Sadasivam et al.40 reported the microarray gene and miRNA expression profiles of cells released from phase synchronization at 0 (G1), 2 (S), 4 (S), 6 (G2), 8 (M), and 12 h (G1). In this study, 3 replicates were obtained from 3 independent experiments. The microarray data were obtained from the GEO database with accession number GSE26922. Gene and miRNA expression of the 4 cell cycle phases from 3 replications were obtained also by cubic spline interpolation. The mean expression of the replicates was then used to identify cell cycle genes and the real GECN of HeLa cells based on the cell cycle projection and system identification methods. The strategy to identify common core and specific GECNs between HeLa and ES cells based on big data mining, cell cycle projection, and system identification is summarized in Figure 1.
Data retrieval and pre-processing of methylation profiles in both HeLa and ES cells
To validate the proposed epigenetic regulations, we looked at the genome-wide methylation profile of 307 human cervical cancer cell samples from The Cancer Genome Atlas (TCGA; http://cancergenome.nih.gov/) and 5 human embryonic stem cell lines (GSE24676) from the GEO database.41 By applying one-way ANOVA, we calculated the p-value for each gene (or miRNA) and confirmed the significance of expression change between human cervical cancer and ES cells. We used Matlab's text file and string manipulation tools in text mining to unify the gene names based on the gene symbols in the GEO database.
Cell cycle projection method to determine the cell cycle phase of each gene
We first applied the cell cycle projection method to the genome-wide raw data of HeLa and ES cells to determine the cell cycle phase of each gene (or miRNA). Based on this, we chose the phase-specific cell cycle genes. Finally, we applied system identification and system order detection methods to the dynamic model of GECN to identify real GECNs.
In order to determine cell cycle phase of the i-th gene (or miRNA), the projection model of cell cycle phase was characterized by the following expression:
| (1) |
where xi(0), xi(1), xi(2), and xi(3) denote the expression of the i-th gene (or miRNA) during G1, S, G2, and M phases, respectively; the cell cycle period T = 4 indicates 4 cell cycle phases; k = 1, 2, 3, 4 correspond to G1, S, G2, and M phases, respectively; ai,k represents the phase-specific ability of gene i during the k-th phase; bi denotes the basal level of the i-th gene (or miRNA); and sin(π/2–2πt/T), sin(π–2πt/T), sin(3π/2–2πt/T), and sin(2π–2πt/T) represent the basic functions of G1, S, G2, and M phases, respectively. Equation (1) can be scaled up as follows:
| (2) |
where Xi denotes the vector of the i-th gene (or miRNA) expression during G1, S, G2, and M phases. Accordingly, it was possible to obtain 4 phase-specific abilities for each gene (or miRNA). In order to determine the phase of each gene (or miRNA), we designed the following criterion:
| (3) |
where ai,k* denotes the maximal phase-specific ability of the i-th gene (or miRNA). Accordingly, the phase criteria in (3) allow us to determine the cell cycle phase of each gene (or miRNA) in the human genome. Moreover, for each gene (or miRNA) we could obtain 12 (4 × 3) phase-specific abilities from 3 biological replicates in HeLa cells. The maximal phase-specific ability of each gene (or miRNA) was used to determine the cell cycle phase. The top 5 most significant phase-specific genes in HeLa and ES cells are shown in Table 1. Furthermore, we defined an upper threshold (≥ ath) of the phase-specific ability to choose the significant cell cycle genes. Tables S1 and S2 show 303 cell cycle genes (ath = 0.2) in HeLa cells and 299 cell cycle genes (ath = 5.2) in ESCs. These genes were validated by taking into account their expression Z scores (Fig. 2A and B, respectively).
Table 1.
Top 5 phase-specific genes in HeLa and ES cells.A complete list of identified cell cycle genes in HeLa cells (303) and ESCs (299) can be found in Tables S1 and S2, respectively.
| HeLa cells |
ES cells |
||
|---|---|---|---|
| Cell cycle gene | Cell cycle phase | Cell cycle gene | Cell cycle phase |
| CCNE2 | G1-phase | RNU86 | G1-phase |
| CCNE1 | G1-phase | HSP90AB1 | G1-phase |
| HIST2H4A | G1-phase | TMSB4X | G1-phase |
| MAB21L3 | G1-phase | CD24 | G1-phase |
| HIST1H2BG | G1-phase | NCL | G1-phase |
| HIST1H2BM | S phase | HMGA1 | S phase |
| HIST1H1B | S phase | RNA45S5 | S phase |
| HIST1H4B | S phase | RPS6 | S phase |
| HIST2H2AB | S phase | ENO1 | S phase |
| HIST1H2AB | S phase | GAPDH | S phase |
| TROAP | G2 phase | ACTB | G2 phase |
| PSRC1 | G2 phase | KPNA2 | G2 phase |
| CDCA8 | G2 phase | UBE2C | G2 phase |
| FAM83D | G2 phase | ACTG1 | G2 phase |
| UBE2C | G2 phase | TOP2A | G2 phase |
| FNDC7 | M phase | HSPA8 | M phase |
| RNU4-85P | M phase | VTRNA1-3 | M phase |
| ZNF382 | M phase | PCBP2 | M phase |
| RNPS1 | M phase | ARL6IP1 | M phase |
| LINC00652 | M phase | SERBP1 | M phase |
Figure 2.

Identification of HeLa and ESC cell cycle genes after applying the cell cycle projection method. HeLa and ES cells cell cycle genes were selected according to the maximal phase-specific ability value, i.e., ai,k* in (3). The right bar represents the Z score, from maximum (white) to minimum value (black). The phase-shift of cell cycle genes can be observed as white blocks along the diagonal.
Construction of candidate GECN by the big database mining method
In order to construct the candidate GECN of human cells, including transcription and miRNA regulation during cell cycle progression, we mined the integrated information from the big database. This encompassed TF-gene association data from the Integrated Transcription Factor Platform (ITFP),42 the Human Transcriptional Regulation Interactions database (HTRIdb),43 the TRANScription FACtor (TRANSFAC) database,44 and miRNA-target association data from TargetScan.45 TF-gene association and miRNA-target association data provided 152,828 and 2,477 candidate associations, respectively, including experimental and computational results. To evaluate each association and prune false positives we applied system identification and system order detection methods to the dynamic model of candidate GECN (see below).
Construction of the dynamic model in candidate GECN by big database mining
To identify regulation mechanisms in the candidate GECN, we constructed a dynamic model of the GECN in human cells. The latter characterized the transcription and miRNA regulation of the i-th cell cycle gene by the stochastic discrete equation as follows:
| (4) |
where xi[t], ym[t], and zn[t] denote the i-th gene, the m-th TF, and the n-th miRNA expressions at time t, respectively; aim and -bin indicate the regulatory abilities of the m-th TF and the n-th miRNA to the i-th target gene (-bin ≤ 0), respectively; MTF I and NMIR I are the numbers of candidate TF and miRNA associations with cell cycle gene i obtained from the constructed candidate GECN, respectively; M represents the number of cell cycle genes identified by the cell cycle projection method; -λi denotes the degradation effect of the present state on the next state (-λi ≤ 0); κi is the basal level of target gene i (κi ≥ 0); and vi[t] represents the stochastic noise due to model uncertainty and fluctuation.
The biological meaning of (4) is that the mRNA expression level of the cell cycle gene i at time t+1 is affected by its own mRNA expression level at time t, the transcription level regulation from the associated TFs , the repression exerted by associated miRNAs , its own degradation effect (-λixi[t]) at time t, the basal level from other sources, such as DNA methylation and histone modification among others. We assumed that the basal level change of the i-th gene between 2 cells determined the epigenetic alterations of gene i. Considering that large-scale measurement of protein activity has yet to be realized and 73% of the variance in protein abundance can be explained by mRNA abundance,46 we employed mRNA expression profiles in lieu of protein expression profiles.
Identification of the real GECN by pruning false positives in candidate GECN via the akaike information criterion (AIC) system order detection method
After constructing the dynamic model of GECN in (4), we applied the system identification and system order detection methods to the model to prune the false-positive associations in HeLa (or ES) cells using their corresponding cell cycle expression data. To identify the regulatory parameters in (4), i.e., aim and bin, we rewrote the stochastic dynamic model in (4) as the following stochastic linear regression form:
| (5) |
where ϕi[t] denotes the regression vector of target gene i; ϕi[t] and xi[t+1] can be obtained from the gene (or miRNA) expression data; and θi indicates the parameter vector of the cell cycle gene i to be estimated. Moreover, taking the cubic spline method to interpolate expression data can effectively prevent parameter overfitting in the parameter estimation process. The inequality constraint in (5) guarantees that -λi ≤ 0, -bin ≤ 0 and κi ≥ 0.
Furthermore, the stochastic linear regression equation (5) can be scaled up along each time point as the following form:
| (6) |
where T denotes the number of expression data time points after using the cubic spline interpolation method.
For convenience, (6) is represented by the following equation:
| (7) |
The constrained least square estimation problem in (5) or (7) relating to the parameter vector of the target gene i was formulated as follows:
| (8) |
The problem in (8) can be optimized using the constrained least-square solver based on a reflective Newton method for minimizing a quadratic function to identify the parameter θi using the MATLAB optimization toolbox.47
When the regulatory parameters in the candidate GECN could be identified by solving the problem in (8) one gene at a time, we applied AIC 48 as a system order detection method to prune false-positive regulations from the candidate GECN. AIC can simultaneously consider the estimated residual error and model complexity, and it can estimate the system order of the dynamic model (i.e., the number of regulations MTF i+ NMIRi in this case). For a stochastic discrete equation in (4) with MTF i+ NMIRi regulatory parameters, AIC could be written as follows:
| (9) |
where denotes the estimated expression of the i-th target gene, i.e., , and the estimated residual error . As the residual error decreases, AIC decreases. In contrast, the number of TF and miRNA regulations, i.e., MTF i+ NMIRi, increases and AIC increases. Therefore, AIC constitutes a trade-off between residual error and model order, and will achieve the minimum at the true order, i.e., the true regulation number. If AIC in (9) were minimized, the real GECN 48 could be obtained by deleting insignificant TF and miRNA regulations (i.e., the so-called false-positive regulations) out of the true regulations identified by AIC. In addition, Student's t-test was applied to calculate the p-value of each regulation parameter under the null hypothesis H0: aim = 0 or H0: bin= 0.49
By applying the regulation parameter estimation in (8) and the false-positive regulation deletion using AIC in (9) one gene at a time, we could identify the real GECNs of HeLa and ES cells in each cell cycle phase (Fig. 3A and B, respectively) from a candidate GECN using the classified high-throughput expression data in each cell cycle phase of HeLa and ES cells, respectively.
Figure 3.

Real GECNs of HeLa and ES cells. The number of G1 and S phase TFs and genes, and G2 phase TFs decreased significantly in HeLa compared to that in ES cells; whereas the number of M phase TFs and genes increased significantly. In addition, the numbers of G1, S, and G2 phase TF regulations in HeLa cells also decreased compared to ES cells; whereas the number of M phase TF regulations increased. These results support the immortal nature of HeLa cells being due to dysregulation of genes at the G1/G2 checkpoint and cell cycle exit.
Identification of core GECNs in the real GECNs of HeLa and ES cells for the investigation of carcinogenic and stemness mechanisms and multiple drug design via PGNP
To investigate the carcinogenic and stemness mechanisms in cervical cancer and ES cells, respectively, we applied PGNP to the real GECNs (Fig. 3) to extract the core GECNs. This included specific GECNs in HeLa and ES cells, and common core GECN between the 2 cell types. The extracted specific GECNs in HeLa cells were regarded as a cervical cancer-specific network and the principal regulatory network in the real GECN of HeLa cells, which should be useful for multiple drug design of cervical cancer. A PGNP approach based on singular value decomposition 50 was used for the extraction of principal network structure and reduction of network dimension. First, the regulation matrix R of GECNs, consisting of the regulatory parameters in (4), i.e., aim and bin, was set as follows:
| (10) |
The regulation matrix R can be decomposed by singular value decomposition method as follows 50:
| (11) |
where the orthogonal matrices and with IM and ; with decreasing singular values d1 ≥ d2 ≥ ··· ≥ dm ≥ ··· ≥ dM ≥ 0; diag(d1,···,dM) denotes a square diagonal matrix with the elements d1, ···, dM; representing zeros matrix with dimension M by MTF + NMIR; and indicates the MTF + NMIR by MTF + NMIR identity.
Moreover, the eigen expression fraction was defined as Em = dm2/ (i.e., the normalization of singular values). We could then select the top L singular vectors VL of V such that ≥ 0.85 with the minimal L. Therefore, the L principal components contained 85% of the principal structure of the network from the energy viewpoint. Projecting the regulation matrix R to the top L principal singular vectors VL was performed as follows:
| (12) |
where ri and vl denote the i-th row vectors of R and the l-th column vectors of the top L principal singular vectors VL, respectively.
Finally, we calculated the dependence (or projection) of the i-th gene relative to the top L principal singular vectors VL by the following 2-norm projection value (or PGNP projection value):
| (13) |
If the i-th gene has a large P(i), the gene i is more related to the top L principal singular vectors VL (i.e., more principal in the GECN).
We defined an upper threshold (≥ dth) of the PGNP projection value to choose the core cell cycle genes of the core GECN. The TFs and miRNAs that regulated core cell cycle genes, were also considered as the core TFs and miRNAs of the core GECN, respectively. When we defined dth = 0.001 and 0.1 in HeLa and ES cells, respectively, we obtained the specific GECNs and the common core GECN (Fig. 4). According to the specific GECNs in HeLa and ES cells, we could unravel the carcinogenic mechanism in cervical cells and stemness mechanism in ES cells. The specific GECN in HeLa cells also allowed us to propose potential multiple drugs for the treatment of cervical cancer.
Figure 4.

Specific and common core GECNs of HeLa cells and ESCs. Specific and common core networks were selected by PGNP with 0.001 (HeLa cells) and 0.1 (ESCs) thresholds. For each core network, inner circles contain miRNAs; middle circles correspond to cell cycle genes; and outer circles include TFs.
Drug mining and design through the specific GECN of HeLa cells
To design multiple drugs with minimal side-effects for the treatment of cervical cancer based on the core GECN in HeLa cells, we considered 2 databases, the Connectivity Map (CMap)51 and the Drug Gene Interaction Database (DGIdb).52 CMap contains the genome-wide microarray data of 5 cell lines (HL60, MCF7, PC3, SKMEL5, and ssMCF7) in response to 1,327 drugs; while DGIdb comprises a drug–gene interaction database. Multiple drug therapy induces a genome-wide response. The strategy of multiple drug screening is that multiple drugs should inhibit the highly expressed cell cycle genes in the principal GECN of HeLa cells and activate the lowly expressed cell cycle genes, but have no influence on common core cell cycle genes in the core GECN. The binding protein substrates of the designed multiple drugs could also be obtained using the DGIdb. This strategy leads to improved drug safety and efficacy in the treatment of cervical cancer.
Results and discussion
Results of cell cycle projection
By applying the cell cycle projection method in (3) to genome-wide high-throughput expression data of HeLa and ES cells (see Material and Methods), we identified 303 cell cycle genes (46 in G1; 76 in S; 96 in G2; and 85 in M phase) in HeLa cells (Fig. 2A and Table S1) and 299 (105 in G1; 108 in S; 73 in G2; and 13 in M phase) in ESCs (Fig. 2B and Table S2). HeLa and ES cell cycle genes were validated by their expression Z scores (Fig. 2A and B, respectively). The five phase-specific genes with top maximal phase-specific ability, i.e., ai,k* in (3) in HeLa and ES cells are shown in Table 1. The identified phase-specific genes (or cell cycle genes) of HeLa and ES cells are shown in Tables S1 and S2, respectively. We only identified 13 genes in the M phase of ESCs. Of these, HSPA8 has been reported as a constitutively expressed gene involved in M phase regulation.53 Therefore, we suggest that M phase-specific genes, which may be the constitutively expressed, could play an important role in promoting properly timed cell cycle exit.
Real and core GECNs in HeLa and ES cells
We applied system identification and AIC to the dynamic model of GECN using genome-wide high-throughput data in HeLa and ES cells. We identified the real GECNs in HeLa and ES cells based on cell cycle genes resulting from cell cycle projection (Fig. 3A and B, respectively). We have removed 94 and 24 genes, which were neither regulated by TFs nor by miRNAs in the real GECNs of HeLa (Fig. 3A) and ES (Fig. 3B) cells, respectively. The results showed that the numbers of G1 and S phase TFs and genes, and G2 phase TFs in HeLa cells decreased significantly compared to ES cells, while the numbers of M phase TFs and genes in HeLa increased significantly with respect to ES cells. Additionally, the numbers of G1, S, and G2 phase TF regulations in HeLa cells decreased, while the number of M phase TF regulations increased compared to ES cells. These results support the notion that the immortal nature of HeLa cells depends on dysregulation of genes controlling the G1/G2 checkpoint and cell cycle exit. Moreover, we identified 3 cell cycle genes expressed in the same phase in HeLa and ES cells: CCNE1/2 in G1, CDK1 in G2, and CDC20 in M phase. This finding is supported by cell cycle studies of human cancer 54,55 and human stem cells.33
By applying PGNP to the real GECNs (Fig. 3 and Material and Methods), we identified the core GECNs, including the specific GECNs of HeLa and ES cells and the core GECN common to HeLa and ES cells. These networks (Fig. 4) represent the significant regulation networks of real GECNs (Fig. 3). As such, they could offer a lot of useful information for carcinogenesis studies. In the common core GECN, we identified 10 common TFs and 6 common cell cycle genes: PLK1, TOP2A, AURKB, FAM83D, HJURP, and HMMR, which are involved in chromosome instability and G2/M checkpoint in HeLa and ES cells. As a result, we suggest that they are housekeeping genes for cell cycle progression. The dysregulation of the common core genes in HeLa cells could promote carcinogenesis during cellular reprogramming. The common core genes can be considered in the multiple drug design of cervical cancer to help avoid side-effects. Additionally, this result showed that the number of TFs and their regulation increased significantly in HeLa cells compared to ESCs, while at the same time the number of miRNA regulations decreased. Therefore, we suggest that the dysregulations of miRNAs play an important role in cervical carcinogenesis.
Carcinogenic mechanism of the specific GECN in HeLa cells
In order to unravel the cervical carcinogenic mechanism, we identified the principal GECN. We defined the principal network as that containing the cell cycle genes with the top PGNP projection values and assumed that each of the 4 cell cycle phase groups involved at least one cell cycle gene. We then identified the principal GECN of HeLa cells (Fig. 5A) from its specific GECN (Fig. 4). Owing to the direct effects of DNA methylation on the binding affinities of miRNAs, RNA polymerase, and transcription factors (TFs) on target genes,56 we assumed that the change of basal level between HeLa and ES cells in the dynamic model (4) would indicate the occurrence of methylation on gene i. Thus, we identified 5 common core cell cycle genes from the common core GECN as potentially methylated: PLK1, TOP2A, AURKB, HJURP, and HMMR (Fig. 4). In recent studies, methylation of PLK1 57 and genetic mutations of PLK1 58 and TOP2A 59 have been reported in human cancers. Furthermore, we used the genome-wide high-throughput DNA methylation profile of human cervical cancer from TCGA and ESCs 41 to validate our findings. We found that the identified genes presented a different DNA methylation pattern between HeLa and ES cells: PLK1 (p-value < 3.708 × 10−211), TOP2A (p-value < 7.65 × 10−52), AURKB (p-value < 3.18 × 10−197), HJURP (p-value < 2.73 × 10−200), and HMMR (p-value < 3.36 × 10−165). These results revealed that DNA methylation played a critical role in dysregulating chromosome stability and G2/M checkpoint in HeLa cells. The dysregulation of miR-100, miR-192, miR-124, and let-7b (p-value < 1.00 × 10−16) in HeLa cells (Fig. 4) could trigger DNA repair and cell apoptosis signals to counteract the accumulated mutations and methylations. However, cervical cancer cells do not undergo apoptosis,60 instead leading to chromosome instability and the accumulation of mutations and methylations that lead to subsequent phenotypic changes,61,62 which finally induce carcinogenesis. Interestingly, we report that the expression of HMMR changes from G2 phase in ESCs to M phase in HeLa cells. Normally, HMMR must be degraded in G2/M to prevent uncontrolled proliferation.63 However, in HeLa cells HMMR expression is enhanced in M phase, which could lead to an abnormal G2/M checkpoint. We found that miR-99A inhibited the expression of HMMR (p-value < 1.00 × 10−16) in G2 phase of ESCs whereas HIF1A up-regulated HMMR (p-value < 1.00 × 10−16) in M phase of HeLa cells. Hence, compared to ESCs, miR-99A and HIF1A dysregulation leads to phase alteration of HMMR expression in HeLa cells, which may further promote carcinogenesis of cervical cells.
Figure 5.

Principal networks of HeLa (A) and ES (B) cells. To find the principal networks, we assumed that they contained cell cycle genes with top PGNP projection values and that each of the 4 cell cycle phase groups involved at least one cell cycle gene. We identified the principal GECNs from the specific GECNs shown in Figure 4. Genes/TFs/miRNAs in blue and red denote activated expression in HeLa and ES cells, respectively (p-value ≤ 0.05).
To date, cervical cancer has been proposed to be a leading cause of death in women. Current treatment strategies, including chemotherapy and surgery, do not provide a permanent cure but prolong the patient's survival.64. To unravel carcinogenic mechanisms underlying cervical cancer and develop a new drug strategy, we applied the big mechanism analysis to the principal GECN in HeLa cells (Fig. 5A) using the gene ontology tool DAVID (https://david.ncifcrf.gov/) (Fig. 6A).65 We identified 12 cell cycle genes: HIS1H2AG, ANKRD1, ARID5B, HIST1H2AJ, HIST1H4L, CDCA2, NUF2, PIF1, STAMBPL1, CKAP2L, TROAP, and ZNF165, which could be methylated due to their basal level changes between HeLa and ES cells in (4). It has been reported that NUF2, TROAP, STAMBPL1, and ARID5B can be methylated in cancers.66-69 Furthermore, we used the DNA methylation profile of human cervical cancer and ESCs to validate our findings. Results indicated that 8 cell cycle genes presented a significantly different methylation pattern between HeLa and ES cells, including ANKRD1 (p-value < 1.70 × 10−9), ARID5B (p-value < 5.00 × 10−84), CDCA2 (p-value < 2.34 × 10−57), PIF1 (p-value < 6.08 × 10−39), STAMBPL1 (p-value < 4.31 × 10−24), TROAP (p-value < 7.65 × 10−52), ZNF165 (p-value <1.08 × 10−147), and HIST1H2AJ (p-value < 7.98 × 10−3). In addition, HIST1H4L was also characterized by a minor change in DNA methylation pattern (p-value < 2.47 × 10−1). Therefore, we concluded that the DNA methylation profile of cervical cancer and ES cells supported our results. To this end, ANKRD1, ARID5B, CKAP2L, and TTK have also been shown to accumulate genetic mutations in cancers.70-73
Figure 6.

Carcinogenic and stemness mechanisms. (A) Carcinogenic mechanism of HeLa cells and (B) stemness mechanism of ES cells. Carcinogenic and stemness mechanisms were derived by applying big mechanism analysis to the principal GECN in HeLa and ES cells, respectively (Fig. 5 A and B), using the gene ontology tool DAVID. Genes/TFs/miRNAs in blue and red denote activated expression in HeLa and ES cells, respectively (p-value ≤ 0.05).
Core TFs/miRNAs identified in the principal GECN of HeLa cells (Fig. 5A) are summarized in Table S3. By integrating the results presented in Fig. 5A and Table S3, we obtained macroscopic results (i.e., big mechanism for carcinogenesis) (Fig. 6A and Table S4). We classified these functions into 4 cancer-associated biological processes: anti-apoptosis, metastasis, DNA repair, and proliferation (Table S4).
Cervical cancer cells maintain directly or indirectly the transduction of proliferation signals during cell cycle progression. It is likely that miR-98, miR-17, miR-34A and, miR-29C (p-value < 6.70 × 10−4) do not properly transduce signals from NFκB and PI3K pathways resulting in uncontrolled proliferation. Therefore, we propose that cervical cancer cells maintain or enhance their proliferation ability by activating miR-98, miR-17, miR-34A, and miR-29C.
miR-192, which is affected by the TGF-β pathway, is involved in cell metastasis during the intermediate cell cycle phases, G1 and G2. Our results show that miR-192 could affect metastasis by regulating FAM111B (p-value < 1.00 × 10−16) and ARHGAP11A (p-value < 1.00 × 10−16) in G1 and G2 (Fig. 5A). Moreover, most genetic mutations and methylations could accumulate in S and G2 phase, leading to their dysregulation, which plays an important role in driving carcinogenesis in cervical cancer. Therefore, to accumulate genetic mutations and abnormal epigenetic regulations, cervical cancer robustly enhances cell survival in the unsTable S and G2 phases by genetic mutations and methylations.61
Furthermore, our results revealed that cancer cells enhanced cell proliferation through cell cycle progression. In normal cells, the accumulated genetic and epigenetic alterations would trigger DNA repair, which in turn induces apoptosis.74 However, accumulated alterations in S and G2 in HeLa cells trigger anti-apoptosis mechanisms to avoid cell death. Accumulated genomic mutations and methylations lead to phenotypic changes characteristic of cervical cancer such as altered metastatic activity in G1 and G2 phase, which may be related to failure of G1 and G2 checkpoints.
According to the causal and temporal cell cycle interplay, cervical cancer cells can potentially increase DNA replication through regulation of miR-98 and miR-34A (p-value < 6.70 × 10−4), as well as methylation. Accordingly, they may further enhance the cells' metastatic ability through regulation of miR-192 in G1 phase. Subsequently, in S phase, cancer cells activate miR-34A (p-value < 3.06 × 10−9) and accumulate methylations and genetic mutations 62 to alter histone regulation and modification. Taken together, these alterations can trigger cervical carcinogenesis. This is also supported by the model simulation of cancer.61 With alterations in histone modifications, methylated genes, such as STAMBPL1, TROAP, CDCA2, PIF1, CKAP2L, and NUF2, in G2 phase could execute numerous functions to promote carcinogenesis. At the same time, miR-192 regulates metastasis in carcinogenesis and mitosis. Furthermore, methylations, accumulated genetic mutations, and miR-192 regulation enhance the anti-apoptotic ability of HeLa cells, which in turn leads to the accumulation of additional genetic and epigenetic alterations caused by subsequent chromosome instability. Cervical cancer cells thus, cause abnormal methylations and numerous genetic mutations, which finally induce irreversible damage, i.e., cancer metastasis. At the last stage, activated miR-17 (p-value < 7.76 × 10−8) enhances these aberrant proliferations in M phase. Iteratively, normal cells generate the tumor and finally trigger metastasis.
Additionally, accumulated genetic mutations and DNA methylation; the dysregulation of miR-29C, miR-34A, miR-98, and miR-215 during G1, S, and G2 phases; and accumulation of miR-17 during M phase result in aberrant cell proliferation. Dysregulation of miR-192 leads to metastatic cervical cancer during G1 and G2 phases. Moreover, accumulated genetic mutations and DNA methylation during S and G2 phases, dysregulation of miR-34A during S phase, and dysregulation of miR-192 during G2 phase could trigger ineffective DNA repair and inhibit apoptosis.
Stemness mechanism of specific GECNs in ESCs
To investigate the mechanism of stemness in ESCs we followed a similar strategy to that presented earlier for HeLa cells and identified the principal GECN (Fig. 5B) from the specific GECN (Fig. 4). We again applied the big mechanism analysis to the specific GECN of ESCs using the DAVID gene ontology tool. By assessing the basal changes between HeLa and ES cells in (4), we identified 17 genes in the principal GECN that could be methylated: RPS9, TMSB4X, DHCR24, OAZ1, EDARADD, PPIB, COX7C, RPS11, RPS14, RPS24, RPL11, RPL35, RPL36, RPLP1, ACTG1, TUBA1B, and FTL (Fig. 6B). The methylation of EDARADD, RPS11, and RPS14 has been reported earlier in cancer cells.75-77 We found significant changes between the DNA methylation profiles of human cervical cancer and ESCs in 14 genes: RPS9 (p-value < 1.91 × 10−5), TMSB4X (p-value < 3.77 × 10−8), DHCR24 (p-value < 2.06 × 10−44), EDARADD (p-value < 4.15 × 10−21), PPIB (p-value < 2.86 × 10−24), RPS11 (p-value < 1.01 × 10−17), RPS14 (p-value < 4.47 × 10−101), RPS24 (p-value < 4.03 × 10−84), RPL11 (p-value < 1.37 × 10−6), RPL35 (p-value < 9.37 × 10−15), RPL36 (p-value < 2.45 × 10−98), RPLP1 (p-value < 7.90 × 10−8), ACTG1 (p-value < 9.22 × 10−48), and TUBA1B (p-value < 6.32 × 10−13). In addition, COX7C also showed a minor change (p-value < 0.289) in DNA methylation.
Using DAVID, we classified miRNAs, TFs, and cell cycle genes of the principal GECN in ESCs into 3 signaling pathways: steroid hormone-mediated, fibroblast growth factor receptor (FGFR), and apoptotic; and three biological processes: cell development, cell differentiation, and cell cycle. It has been demonstrated that mammary stem cells are highly responsive to steroid hormone signaling.78 One of the earliest changes in gene expression affecting the steroid hormone-mediated signaling pathway in response to a differentiation signal has been observed in mammary stem cells, supporting our findings.79 It has also been reported that induction of human ES cell proliferation and differentiation promotes steroidogenesis.80 It has been suggested that the FGFR signaling pathway is involved in the maintenance and proliferation of undifferentiated human ESCs.73 Furthermore, it has recently been suggested that ESC differentiation is efficiently maintained by eliminating those cells that are slow to exit pluripotency, implicating a novel role for the apoptotic signaling pathway.81 Therefore, we propose that the functions of the principal GECN in ESCs should play a significant role in determining the ESCs mechanisms of stemness.
miR-100, miR-124, miR-155, and miR-221 have been found in the principal GECN of ESCs. It has been reported that their regulations play an important role in modulating cell differentiation, cell migration, and cell proliferation in mammalian ESCs.82-85 It has also been observed that genetic and epigenetic alterations play a critical role in the development of human ESCs.86 However, ESCs mechanisms of stemness cannot be uncovered without the use of a systems biology approach from a genome-wide perspective. According to our analyses we proposed that the development of ESCs through the gap G1/G2 phases, is regulated by the action of E2F1, TP53, and MIR-221, involved in apoptotic signaling, and the methylation of RPS9, TMSB4X, DHCR24, OAZ1, RPLP1, ACTG1, TUBA1B, and FTL. The methylation of S phase genes EDARADD, PPIB, COX7C, RPS11, RPS14, RPS24, RPL11, RPL35, and RPL36 may regulate the timing of locus replication, as previously described in mouse embryonic stem cells.59
Multiple drug design for cervical cancer treatment by integrating cervical carcinogenic mechanisms and drug databases
By integrating the drug CMap and DGIdb databases and the cancer-specific GECN, we identified a number of drugs: methotrexate, quercetin, and mimosine. These target primarily abnormal DNA repair (i.e., ARID5B), significantly truncating numerous signal transduction proteins (i.e., CCL2). As a result, the stimulation of cancer genes in S phase is dramatically reduced; this prevents the accumulation of genetic mutations that result from chromosome instability caused by phenotypic changes (i.e., carcinogenesis) in G2 phase. At the same time, the administration of these agents should inhibit anti-apoptotic activity in S and G2 phases (i.e., CCL2 and STK17B, respectively). Consequently, abnormal cells cannot rely on apoptosis to inhibit carcinogenesis. In HeLa cells the identified drugs repress activated cell cycle genes (ARID5B, STK17B, and CCL2) (Table 2) and stimulate inhibited cell cycle TFs (PAN4, PLXNA2, HIST1H4L, and HOXA4). The drug molecules, methotrexate, quercetin and mimosine, were identified in 3 human cell lines, HL60, MCF-7, and PC3. They showed minimal side-effects on the 6 common core GECNs of ESCs: PLK1, TOP2A, AURKB, FAM83D, HJURP, and HMMR (Fig. 4). The US Food and Drug Administration (FDA) approved Methotrexate for the treatment of acute lymphoblastic leukemia, breast cancer, gestational trophoblastic disease, head and neck cancer, lung cancer, mycosis fungoides, non-hodgkin lymphoma, and osteosarcoma. Mimosine is FDA approved for inhibiting hair growth and has been identified as a cell cycle blocker.87 We assumed that these genes may play essential roles in cell cycle progression. Additionally, all the drug molecules in the multiple drugs have exhibited anti-cancer effects.88-90
Table 2.
Proposed multiple drugs for the treatment of cervical cancer.Proposed targets and structures of methotrexate, quercetin, and mimosine derived from the ZINC database. The identified drugs suppress activated cell cycle genes, such as ARID5B, STK17B, and CCL2, without affecting 6 common core genes, PLK1, TOP2A, AURKB, FAM83D, HJURP, and HMMR. The ranked list of all drugs in CMap is shown in Table S5. Methotrexate targets abnormal methylation in S phase, quercetin inhibits anti-apoptosis in G2 phase, and mimosine blocks most signal transduction and inhibits anti-apoptosis in S phase. Apoptosis reduces accumulated genetic mutations, abnormal DNA methylations, and the dysregulation of miRNAs in normal cells; thus, achieving carcinogenesis inhibition.
| Drug molecule | Cell cycle gene (PGNP projection value) |
|---|---|
| Methotrexate | ARID5B (2.742993) |
| Quercetin | STK17B (0.210057) |
| Mimosine | CCL2 (0.013344) |
 | |
Conclusions
In this study, we first applied the cell cycle projection method to genome-wide high-throughput data to identify cell cycle genes. Next, we applied a systematic method to a dynamic model of GECN to construct and then compare real GECNs of HeLa cells and ESCs. Cancer- and stem-specific GECNs were used to investigate the genetic-and-epigenetic mechanisms of cervical cancer carcinogenesis and embryonic stemness. By applying the big mechanism analysis to specific GECNs, cervical carcinogenesis and embryonic stemness could be uncovered. Furthermore, we showed that in combination with drug design, core network projection methods could be used to develop effective anticancer treatments with minimal side-effects on normal cells. Finally, we designed multiple drugs by systems pharmacology using the integrated information of cervical mechanisms and drug databases for the treatment of cervical cancer by integrating genetic-and-epigenetic regulations, omics databases, high-throughput (NGS and microarray) data, and drug databases.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
The work was supported by the Ministry of Science and Technology of Taiwan under grant No. MOST 104-2221-E-007 -124 -MY3.
References
- [1].Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC, Melton DA. “Stemness”: transcriptional profiling of embryonic and adult stem cells. Science 2002; 298(5593):597-600; PMID:12228720; http://dx.doi.org/ 10.1126/science.1072530 [DOI] [PubMed] [Google Scholar]
- [2].Zhu D, Wan X, Huang H, Chen X, Liang W, Zhao F, Lin T, Han J, Xie W. Knockdown of Bmi1 inhibits the stemness properties and tumorigenicity of human bladder cancer stem cell-like side population cells. Oncol Rep 2014; 31(2):727-36; PMID:24337040 [DOI] [PubMed] [Google Scholar]
- [3].Okita K, Yamanaka S. Induced pluripotent stem cells: opportunities and challenges. Philos Trans R Soc Lond B Biol Sci 2011; 366(1575):2198-207; PMID:21727125; http://dx.doi.org/ 10.1098/rstb.2011.0016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cai J, Weiss ML, Rao MS. In search of “stemness”. Exp Hematol 2004; 32(7):585-98; PMID:15246154; http://dx.doi.org/ 10.1016/j.exphem.2004.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bueno MJ, Malumbres M. MicroRNAs and the cell cycle. Biochim Biophys Acta 2011; 1812(5):592-601 [DOI] [PubMed] [Google Scholar]
- [6].Morishita A, Masaki T. miRNA in hepatocellular carcinoma. Hepatol Res 2015; 45(2):128-41; PMID:25040738; http://dx.doi.org/ 10.1111/hepr.12386 [DOI] [PubMed] [Google Scholar]
- [7].Li CW, Chen BS. Network biomarkers of bladder cancer based on a genome-wide genetic and epigenetic network derived from next-generation sequencing data. Dis Markers 2016; 2016:4149608, ; PMID:27034531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Li CW, Wang WH, Chen BS. Investigating the specific core genetic-and-epigenetic networks of cellular mechanisms involved in human aging in peripheral blood mononuclear cells.” Oncotarget 2016; 7:8556-79; PMID:26895224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science 1998; 282(5391):1145-7; PMID:9804556; http://dx.doi.org/ 10.1126/science.282.5391.1145 [DOI] [PubMed] [Google Scholar]
- [10].Ivankovic M, Cukusic A, Gotic I, Skrobot N, Matijasić M, Polancec D, Rubelj I. Telomerase activity in HeLa cervical carcinoma cell line proliferation. Biogerontology 2007; 8(2):163-72; PMID:16955216; http://dx.doi.org/ 10.1007/s10522-006-9043-9 [DOI] [PubMed] [Google Scholar]
- [11].Sa G, Das T. Anti cancer effects of curcumin: cycle of life and death. Cell Div 2008; 3:14; PMID:18834508; http://dx.doi.org/ 10.1186/1747-1028-3-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang Y, Baskerville S, Shenoy A, Babiarz JE, Baehner L, Blelloch R. Embryonic stem cell-specific microRNAs regulate the G1-S transition and promote rapid proliferation. Nat Genet 2008; 40(12):1478-83; PMID:18978791; http://dx.doi.org/ 10.1038/ng.250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Oram SW, Liu XX, Lee TL, Chan WY, Lau YF. TSPY potentiates cell proliferation and tumorigenesis by promoting cell cycle progression in HeLa and NIH3T3 cells. BMC Cancer 2006;6:154; PMID:16762081; http://dx.doi.org/ 10.1186/1471-2407-6-154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mine KL, Shulzhenko N, Yambartsev A, Rochman M, Sanson GF, Lando M, Varma S, Skinner J, Volfovsky N, Deng T, et al.. Gene network reconstruction reveals cell cycle and antiviral genes as major drivers of cervical cancer. Nat Commun 2013;4:1806; PMID:23651994; http://dx.doi.org/ 10.1038/ncomms2693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hashem IAT, Yaqoob I, Anuar NB et al.. The rise of “big data” on cloud computing: Review and open research issues. Information Systems 2015;47:98-115; http://dx.doi.org/ 10.1016/j.is.2014.07.006. [DOI] [Google Scholar]
- [16].Cohen PR. DARPA's Big Mechanism program. Phys Biol 2015; 12(4):045008; PMID:26178259; http://dx.doi.org/ 10.1088/1478-3975/12/4/045008 [DOI] [PubMed] [Google Scholar]
- [17].Chen BS, Li CW. Measuring information flow in cellular networks by the systems biology method through microarray data. Front Plant Sci 2015; 6; PMID:26082788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Li CW, Chen BS. Identifying functional mechanisms of gene and protein regulatory networks in response to a broader range of environmental stresses. Comp Funct Genomics 2010; Article ID 408705; PMID:20454442; http://dx.doi.org/ 10.1155/2010/408705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Przytycka TM, Kim YA. Network integration meets network dynamics. BMC Biol 2010; 8:48, ; PMID:20513250; http://dx.doi.org/ 10.1186/1741-7007-8-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chen BS, Li CW. On the interplay between entropy and robustness of gene regulatory networks. Entropy 2010; 12(5):1071-101http://dx.doi.org/ 10.3390/e12051071 [DOI] [Google Scholar]
- [21].Chen BS, Wong SW, Li CW. On the calculation of system entropy in nonlinear stochastic biological networks. Entropy 2015; 17(10):6801-33; http://dx.doi.org/ 10.3390/e17106801 [DOI] [Google Scholar]
- [22].Schadt EE, Friend SH, Shaywitz DA. OPINION A network view of disease and compound screening. Nat Rev Drug Discov 2009; 8(4):286-95; PMID:19337271; http://dx.doi.org/ 10.1038/nrd2826 [DOI] [PubMed] [Google Scholar]
- [23].Berger SI, Iyengar R. Network analyses in systems pharmacology. Bioinformatics 2009; 25(19):2466-72; PMID:19648136; http://dx.doi.org/ 10.1093/bioinformatics/btp465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Godoy P, Schmidt-Heck W, Natarajan K, Lucendo-Villarin B, Szkolnicka D, Asplund A, Björquist P, Widera A, Stöber R, Campos G, et al.. Gene networks and transcription factor motifs defining the differentiation of stem cells into hepatocyte-like cells. J Hepatol 2015; 63(4):934-42; PMID:26022688; http://dx.doi.org/ 10.1016/j.jhep.2015.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].McKinney EF, Lee JC, Jayne DR, Lyons PA, Smith KG. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 2015; 523(7562):612-6; PMID:26123020; http://dx.doi.org/ 10.1038/nature14468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Parikshak NN, Gandal MJ, Geschwind DH. Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat Rev Genet 2015; 16(8):441-58; PMID:26149713; http://dx.doi.org/ 10.1038/nrg3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Banyai G, Baidi F, Coudreuse D, Szilagyi Z. Cdk1 activity acts as a quantitative platform for coordinating cell cycle progression with periodic transcription. Nat Commun 2016; 7:11161; PMID:27045731; http://dx.doi.org/ 10.1038/ncomms11161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Young TR, Fernandez B, Buckalew R, Moses G, Boczko EM. Clustering in cell cycle dynamics with general response/signaling feedback. J Theor Biol 2012; 292:103-15; PMID:22001733; http://dx.doi.org/ 10.1016/j.jtbi.2011.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bell R, Hubbard A, Chettier R, Chen D, Miller JP, Kapahi P, Tarnopolsky M, Sahasrabuhde S, Melov S, Hughes RE. A human protein interaction network shows conservation of aging processes between human and invertebrate species. Plos Genet 2009; 5(3): PMID:19293945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Drozdov I, Tsoka S, Ouzounis CA, Shah AM. Genome-wide expression patterns in physiological cardiac hypertrophy. BMC Genomics 2010; 11:557; PMID:20937113; http://dx.doi.org/ 10.1186/1471-2164-11-557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sandmann T, Girardot C, Brehme M, Tongprasit W, Stolc V, Furlong EE. A core transcriptional network for early mesoderm development in Drosophila melanogaster. Genes Dev 2007; 21(4):436-49; PMID:17322403; http://dx.doi.org/ 10.1101/gad.1509007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhao S, Iyengar R. Systems pharmacology: network analysis to identify multiscale mechanisms of drug action. Annu Rev Pharmacol Toxicol 2012; 52:505-21; PMID:22235860; http://dx.doi.org/ 10.1146/annurev-pharmtox-010611-134520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Singh AM, Chappell J, Trost R, Lin L, Wang T, Tang J, Matlock BK, Weller KP, Wu H, Zhao S, et al.. Cell-cycle control of developmentally regulated transcription factors accounts for heterogeneity in human pluripotent cells.” Stem Cell Rep 2013; 1(6):532-44; PMID:24371808; http://dx.doi.org/ 10.1016/j.stemcr.2013.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10(3):R25; PMID:19261174; http://dx.doi.org/ 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013; 14(4):R36; PMID:23618408; http://dx.doi.org/ 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010; 28(5):511-5; PMID:20436464; http://dx.doi.org/ 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Goecks J, Nekrutenko A, Taylor J, Galaxy Team . Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 2010; 11(8):R86; PMID:20738864; http://dx.doi.org/ 10.1186/gb-2010-11-8-r86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol 2010; vol. Chapter 19, pp. Unit 19 10 1–21; PMID:20069535; http://dx.doi.org/ 10.1002/0471142727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, et al.. Galaxy: a platform for interactive large-scale genome analysis. Genome Res 2005; 15(10):1451-5; PMID:16169926; http://dx.doi.org/ 10.1101/gr.4086505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sadasivam S, Duan S, DeCaprio JA. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev 2012; 26(5):474-89; PMID:22391450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nishino K, Toyoda M, Yamazaki-Inoue M, Fukawatase Y, Chikazawa E, Sakaguchi H, Akutsu H, Umezawa A. DNA Methylation Dynamics in Human Induced Pluripotent Stem Cells over Time. Plos Genet 2011; 7(5): ; PMID:21637780; http://dx.doi.org/ 10.1371/journal.pgen.1002085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zheng G, Tu K, Yang Q, Xiong Y, Wei C, Xie L, Zhu Y, Li Y. ITFP: an integrated platform of mammalian transcription factors. Bioinformatics 2008; 24(20):2416-7; PMID:18713790; http://dx.doi.org/ 10.1093/bioinformatics/btn439 [DOI] [PubMed] [Google Scholar]
- [43].Bovolenta LA, Acencio ML, Lemke N. HTRIdb: an open-access database for experimentally verified human transcriptional regulation interactions. BMC Genomics 2012; 13:405; PMID:22900683; http://dx.doi.org/ 10.1186/1471-2164-13-405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al.. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res 2006; 34:D108-10; PMID:16381825; http://dx.doi.org/ 10.1093/nar/gkj143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015; 4:e05005; PMID:26267216; http://dx.doi.org/ 10.7554/eLife.05005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat Biotechnol 2007; 25(1):117-24; PMID:17187058; http://dx.doi.org/ 10.1038/nbt1270 [DOI] [PubMed] [Google Scholar]
- [47].Coleman TF, Li YY. A reflective Newton method for minimizing a quadratic function subject to bounds on some of the variables. Siam J Opt 1996; 6(4):1040-1058; http://dx.doi.org/ 10.1137/S1052623494240456 [DOI] [Google Scholar]
- [48].Johansson R. System modeling and identification, Englewood Cliffs, NJ: Prentice Hall, 1993. [Google Scholar]
- [49].Pagano M, Gauvreau K. Principles of biostatistics, 2nd ed., Pacific Grove, CA: Duxbury, 2000. [Google Scholar]
- [50].Alter O, Brown PO, Botstein D. Singular value decomposition for genome-wide expression data processing and modeling. Proc Natl Acad Sci U S A 2000; 97(18):10101-6; PMID:10963673; http://dx.doi.org/ 10.1073/pnas.97.18.10101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et al.. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 2006; 313(5795):1929-35; PMID:17008526; http://dx.doi.org/ 10.1126/science.1132939 [DOI] [PubMed] [Google Scholar]
- [52].Griffith M, Griffith OL, Coffman AC, Weible JV, McMichael JF, Spies NC, Koval J, Das I, Callaway MB, Eldred JM, et al.. DGIdb: mining the druggable genome. Nat Methods 2013; 10(12):1209-10; PMID:24122041; http://dx.doi.org/ 10.1038/nmeth.2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Geng YJ, Zhao YF, Schuster LC, Feng B, Lynn DA, Austin KM, Stoklosa JD, Morrison JD. A chemical biology study of human pluripotent stem cells unveils HSPA8 as a key regulator of pluripotency. Stem Cell Rep 2015; 5(6):1143-54; PMID:26549849; http://dx.doi.org/ 10.1016/j.stemcr.2015.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Park HJ, Costa RH, Lau LF, Tyner AL, Raychaudhuri P. Anaphase-promoting complex/cyclosome-CDH1-mediated proteolysis of the forkhead box M1 transcription factor is critical for regulated entry into S phase. Mol Cell Biol 2008; 28(17):5162-71; PMID:18573889; http://dx.doi.org/ 10.1128/MCB.00387-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zafonte BT, Hulit J, Amanatullah DF, Albanese C, Wang C, Rosen E, Reutens A, Sparano JA, Lisanti MP, Pestell RG. Cell-cycle dysregulation in breast cancer: breast cancer therapies targeting the cell cycle. Front Biosci 2000; 5:D938-61; PMID:11102317; http://dx.doi.org/ 10.2741/zafonte [DOI] [PubMed] [Google Scholar]
- [56].Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, Schübeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 2007; 39(4):457-66; PMID:17334365; http://dx.doi.org/ 10.1038/ng1990 [DOI] [PubMed] [Google Scholar]
- [57].Ward A, Sivakumar G, Kanjeekal S, Hamm C, Labute BC, Shum D, Hudson JW. The deregulated promoter methylation of the Polo-like kinases as a potential biomarker in hematological malignancies. Leuk Lymphoma 2015; 56(7):2123-33; PMID:25347426; http://dx.doi.org/ 10.3109/10428194.2014.971407 [DOI] [PubMed] [Google Scholar]
- [58].Kamaraj B, Rajendran V, Sethumadhavan R, Purohit R. In-silico screening of cancer associated mutation on PLK1 protein and its structural consequences. J Mol Model 2013; 19(12):5587-99; PMID:24271645; http://dx.doi.org/ 10.1007/s00894-013-2044-0 [DOI] [PubMed] [Google Scholar]
- [59].Hsiung Y, Jannatipour M, Rose A, McMahon J, Duncan D, Nitiss JL. Functional expression of human topoisomerase II alpha in yeast: mutations at amino acids 450 or 803 of topoisomerase II alpha result in enzymes that can confer resistance to anti-topoisomerase II agents. Cancer Res 1996; 56(1):91-9; PMID:8548781 [PubMed] [Google Scholar]
- [60].Espinosa M, Cantu D, Herrera N, Lopez CM, De la Garza JG, Maldonado V, Melendez-Zajgla J. Inhibitors of apoptosis proteins in human cervical cancer. BMC Cancer 2006; 6:45; PMID:16504151; http://dx.doi.org/ 10.1186/1471-2407-6-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Chen BS, Tsai KW, Li CW. Using nonlinear stochastic evolutionary game strategy to model an evolutionary biological network of organ carcinogenesis under a natural selection scheme. Evol Bioinform 2015; 11:155-78; PMID:26244004; http://dx.doi.org/ 10.4137/EBO.S26195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Woo YH, Li WH. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nat Commun 2012; 3:1004; PMID:22893128; http://dx.doi.org/ 10.1038/ncomms1982 [DOI] [PubMed] [Google Scholar]
- [63].Sohr S, Engeland K. RHAMM is differentially expressed in the cell cycle and downregulated by the tumor suppressor p53. Cell Cycle 2008; 7(21):3448-60; PMID:18971636; http://dx.doi.org/ 10.4161/cc.7.21.7014 [DOI] [PubMed] [Google Scholar]
- [64].Chhabra R. Cervical cancer stem cells: opportunities and challenges. J Cancer Res Clin Oncol 2015; 141(11):1889-97; PMID:25563493; http://dx.doi.org/ 10.1007/s00432-014-1905-y [DOI] [PubMed] [Google Scholar]
- [65].Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC, Lempicki RA. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol 2007; 8(9):R183; PMID:17784955; http://dx.doi.org/ 10.1186/gb-2007-8-9-r183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wehbe H, Henson R, Meng FY, Mize-Berge J, Patel T. Interleukin-6 contributes to growth in cholangiocarcinoma cells by aberrant promoter methylation and gene expression. Cancer Res 2006; 66(21):10517-24; PMID:17079474; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-2130 [DOI] [PubMed] [Google Scholar]
- [67].Baba A, Ohtake F, Okuno Y, Yokota K, Okada M, Imai Y, Ni M, Meyer CA, Igarashi K, Kanno J, et al.. PKA-dependent regulation of the histone lysine demethylase complex PHF2-ARID5B. Nat Cell Biol 2011; 13(6):668-75; PMID:21532585; http://dx.doi.org/ 10.1038/ncb2228 [DOI] [PubMed] [Google Scholar]
- [68].Sethi G, Pathak HB, Zhang H, Zhou Y, Einarson MB, Vathipadiekal V, Gunewardena S, Birrer MJ, Godwin AK. An RNA interference lethality screen of the human druggable genome to identify molecular vulnerabilities in epithelial ovarian cancer. Plos One 2012; 7(10):e47086; PMID:23056589; http://dx.doi.org/ 10.1371/journal.pone.0047086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Cheng Y, Yan Z, Liu Y, Liang C, Xia H, Feng J, Zheng G, Luo H. Analysis of DNA methylation patterns associated with the gastric cancer genome. Oncol Lett 2014; 7(4):1021-6; PMID:24944662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Cinquetti R, Badi I, Campione M, Bortoletto E, Chiesa G, Parolini C, Camesasca C, Russo A, Taramelli R, Acquati F. Transcriptional deregulation and a missense mutation define ANKRD1 as a candidate gene for total anomalous pulmonary venous return. Hum Mutat 2008; 29(4):468-74; PMID:18273862; http://dx.doi.org/ 10.1002/humu.20711 [DOI] [PubMed] [Google Scholar]
- [71].Niittymaki I, Gylfe A, Laine L, Laakso M, Lehtonen HJ, Kondelin J, Tolvanen J, Nousiainen K, Pouwels J, Järvinen H, et al.. High frequency of TTK mutations in microsatellite-unstable colorectal cancer and evaluation of their effect on spindle assembly checkpoint. Carcinogenesis 2011; 32(3):305-11; PMID:21163887 [DOI] [PubMed] [Google Scholar]
- [72].Wangpermtam P, Sanguansin S, Petmitr S, Punyarit P, Weerapradist W. Genetic alteration in oral squamous cell carcinoma detected by arbitrarily primed polymerase chain reaction. Asian Pac J Cancer Prev 2011; 12(8):2081-5; PMID:22292655 [PubMed] [Google Scholar]
- [73].Cancer Genome Atlas Research N, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, et al.. Integrated genomic characterization of endometrial carcinoma. Nature 2013; 497(7447):67-73; PMID:23636398; http://dx.doi.org/ 10.1038/nature12113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kaina B. DNA damage-triggered apoptosis: critical role of DNA repair, double-strand breaks, cell proliferation and signaling. Biochem Pharmacol 2003; 66(8):1547-54; PMID:14555233; http://dx.doi.org/ 10.1016/S0006-2952(03)00510-0 [DOI] [PubMed] [Google Scholar]
- [75].Borze I, Juvonen E, Ninomiya S, Jee KJ, Elonen E, Knuutila S. High-resolution oligonucleotide array comparative genomic hybridization study and methylation status of the RPS14 gene in de novo myelodysplastic syndromes. Cancer Genet Cytogenet 2010; 197(2):166-73; PMID:20193850 [DOI] [PubMed] [Google Scholar]
- [76].Vizoso M, Puig M, Carmona FJ, Maqueda M, Velásquez A, Gómez A, Labernadie A, Lugo R, Gabasa M, Rigat-Brugarolas LG, et al.. Aberrant DNA methylation in non-small cell lung cancer-associated fibroblasts. Carcinogenesis 2015; 36(12):1453-63; PMID:26449251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Yong WH, Shabihkhani M, Telesca D, Yang S, Tso JL, Menjivar JC, Wei B, Lucey GM, Mareninov S, Chen Z, et al.. Ribosomal proteins RPS11 and RPS20, two stress-response markers of glioblastoma stem cells, are novel predictors of poor prognosis in glioblastoma patients. PLoS One 2015; 10(10):e0141334; PMID:26506620; http://dx.doi.org/ 10.1371/journal.pone.0141334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Asselin-Labat ML, Vaillant F, Sheridan JM, Pal B, Wu D, Simpson ER, Yasuda H, Smyth GK, Martin TJ, Lindeman GJ, et al.. Control of mammary stem cell function by steroid hormone signalling. Nature 2010; 465(7299):798-802; PMID:20383121; http://dx.doi.org/ 10.1038/nature09027 [DOI] [PubMed] [Google Scholar]
- [79].Sauter CN, McDermid RL, Weinberg AL, Greco TL, Xu X, Murdoch FE, Fritsch MK. Differentiation of murine embryonic stem cells induces progesterone receptor gene expression. Exp Cell Res 2005; 311(2):251-64; PMID:16223481; http://dx.doi.org/ 10.1016/j.yexcr.2005.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Gallego MJ, Porayette P, Kaltcheva MM, Bowen RL, Vadakkadath Meethal S, Atwood CS. The pregnancy hormones human chorionic gonadotropin and progesterone induce human embryonic stem cell proliferation and differentiation into neuroectodermal rosettes. Stem Cell Res Ther 2010; 1:28; PMID:20836886; http://dx.doi.org/ 10.1186/scrt28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Wang ES, Reyes NA, Melton C, Huskey NE, Momcilovic O, Goga A, Blelloch R, Oakes SA. Fas-activated mitochondrial apoptosis culls stalled embryonic stem cells to promote differentiation. Curr Biol 2015; 25(23):3110-8; PMID:26585277; http://dx.doi.org/ 10.1016/j.cub.2015.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Fang J, Zhang T, Liu Y, Li Y, Zhou S, Song D, Zhao Y, Feng R, Zhang X, Li L, et al.. PAX6 downregulates miR-124 expression to promote cell migration during embryonic stem cell differentiation. Stem Cells Dev 2014; 23(19):2297-310; PMID:24773074; http://dx.doi.org/ 10.1089/scd.2013.0410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Li J, Bei YH, Liu Q, Lv D, Xu T, He Y, Chen P, Xiao J. MicroRNA-221 is required for proliferation of mouse embryonic stem cells via P57 targeting. Stem Cell Rev Rep 2015; 11(1):39-49; PMID:25086570; http://dx.doi.org/ 10.1007/s12015-014-9543-y [DOI] [PubMed] [Google Scholar]
- [84].Sato T, Liu XD, Basma H, Togo S, Sugiura H, Nelson A, Nakanishi M, Kanaji N, Wang X, Kim M, et al.. IL-4 induces differentiation of human embryonic stem cells into fibrogenic fibroblast-like cells. J Allergy Clin Immunol 2011; 127(6):1595-603; PMID:21388667; http://dx.doi.org/ 10.1016/j.jaci.2011.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Tarantino C, Paolella G, Cozzuto L, Minopoli G, Pastore L, Parisi S, Russo T. miRNA 34a, 100, and 137 modulate differentiation of mouse embryonic stem cells. FASEB J 2010; 24(9):3255-63; PMID:20439489; http://dx.doi.org/ 10.1096/fj.09-152207 [DOI] [PubMed] [Google Scholar]
- [86].Maitra A, Arking DE, Shivapurkar N, Ikeda M, Stastny V, Kassauei K, Sui G, Cutler DJ, Liu Y, Brimble SN, et al.. Genomic alterations in cultured human embryonic stem cells. Nat Genet 2005; 37(10):1099-103; PMID:16142235; http://dx.doi.org/ 10.1038/ng1631 [DOI] [PubMed] [Google Scholar]
- [87].Farinelli SE, Greene LA. Cell cycle blockers mimosine, ciclopirox, and deferoxamine prevent the death of PC12 cells and postmitotic sympathetic neurons after removal of trophic support. J Neurosci 1996; 16(3):1150-62; PMID:8558244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Krude T. Mimosine arrests proliferating human cells before onset of DNA replication in a dose-dependent manner. Exp Cell Res 1999; 247(1):148-59; PMID:10047457; http://dx.doi.org/ 10.1006/excr.1998.4342 [DOI] [PubMed] [Google Scholar]
- [89].Vidya Priyadarsini R, Senthil Murugan R, Maitreyi S, Ramalingam K, Karunagaran D, Nagini S. The flavonoid quercetin induces cell cycle arrest and mitochondria-mediated apoptosis in human cervical cancer (HeLa) cells through p53 induction and NF-kappaB inhibition. Eur J Pharmacol 2010; 649(1–3):84-91; PMID:20858478; http://dx.doi.org/ 10.1016/j.ejphar.2010.09.020 [DOI] [PubMed] [Google Scholar]
- [90].Chen J, Gu W, Yang L, Chen C, Shao R, Xu K, Xu ZP. Nanotechnology in the management of cervical cancer. Rev Med Virol 2015; 25(Suppl 1):72-83; PMID:25752817 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.