

Abstract
The protein C (PC) system conveys beneficial anticoagulant and cytoprotective effects in numerous in vivo disease models. The endothelial protein C receptor (EPCR) plays a central role in these pathways as cofactor for PC activation and by enhancing activated protein C (APC)-mediated protease-activated receptor (PAR) activation. During inflammatory disease, expression of EPCR on cell membranes is often diminished thereby limiting PC activation and APC’s effects on cells. Here a caveolae-targeting glycosylphosphatidylinositol (GPI)-anchored EPCR (EPCR-GPI) was engineered to restore EPCR’s bioavailability via “cell painting.” The painting efficiency of EPCR-GPI on EPCR-depleted endothelial cells was time- and dose-dependent. The EPCR-GPI bioavailability after painting was long lasting since EPCR surface levels reached 400% of wild-type cells after 2 hours and remained >200% for 24 hours. EPCR-GPI painting conveyed APC binding to EPCR-depleted endothelial cells where EPCR was lost due to shedding or shRNA. EPCR painting normalized PC activation on EPCR-depleted cells indicating that EPCR-GPI is functional active on painted cells. Caveolin-1 lipid rafts were enriched in EPCR after painting due to the GPI-anchor targeting caveolae. Accordingly, EPCR painting supported PAR1 and PAR3 cleavage by APC and augmented PAR1-dependent Akt phosphorylation by APC. Thus, EPCR-GPI painting achieved physiological relevant surface levels on endothelial cells, restored APC binding to EPCR-depleted cells, supported PC activation, and enhanced APC-mediated PAR cleavage and cytoprotective signaling. Therefore, EPCR-GPI provides a novel tool to restore the bioavailability and functionality of EPCR on EPCR-depleted and deficient cells.
Keywords: endothelial protein C receptor, glycosylphosphatidylinositol anchors, protease-activated receptors, protein C, vascular endothelium
INTRODUCTION
The protein C system conveys multiple important functions to maintain a regulated balance between hemostasis and host defense systems in response to vascular and inflammatory injury. The anticoagulant protein C pathway regulates coagulation, maintains blood fluidity within the vasculature, and prevents thrombosis, whereas the cytoprotective protein C pathway provides anti-inflammatory and cytoprotective activities to prevent vascular damage and stress (1–3). In vivo injury and disease models have attributed important beneficial protective effects to APC’s anticoagulant and cytoprotective activities when APC was generated endogenously or administered therapeutically (4–11). EPCR plays a central role in the protein C system as a cofactor for APC generation and by facilitating APC’s effects on cells (12–14).
Activation of PC by the thrombin-thrombomodulin complex is greatly augmented by binding of PC to EPCR and recruitment of PC to the endothelial cell surface (13, 15). After activation, APC can convey anticoagulant activity via proteolytic inactivation of the procoagulant cofactors Va and VIIIa, which effectively shuts down thrombin formation. EPCR is also critical for APC’s direct effects on cells mediated by PAR1 and PAR3 which, depending on cell type and cell stress, include anti-inflammatory and anti-apoptotic activities, alterations of gene expression profiles, and protection of the endothelial barrier function (12). According to the current paradigm, EPCR-dependent cytoprotective effects of APC occur when PAR1 and EPCR are co-localized in caveolin-1 enriched lipid rafts or caveolae, and APC-mediated cytoprotective cell signaling is initiated when EPCR-bound APC cleaves PAR1 at Arg46 or PAR3 at Arg41 (14, 16–19).
Functional EPCR is essential for optimal anticoagulant and cytoprotective effects of APC. However, EPCR’s functional bioavailability diminishes during prothrombotic and proinflammatory conditions by EPCR shedding and encryption (20–22). EPCR is particularly sensitive to shedding mediated by tumor necrosis factor (TNF)-alpha converting enzyme (TACE a.k.a. ADAM17) induced by inflammatory mediators (21). EPCR shedding results in increased levels of soluble EPCR in mice exposed to endotoxemia (23) and in plasma of patients with a prothrombotic and proinflammatory tendency (24–26). Alternatively, endothelial-derived secreted phospholipase A2 (sPLA2) can modify EPCR so that it loses the ability to bind APC (20). This EPCR encryption involves cleavage of the fatty-acid chain at the sn-2 position of the lipid embedded in the hydrophobic groove of EPCR by sPLA2. These studies suggest that under proinflammatory conditions cells become temporarily EPCR depleted due to shedding of EPCR from the cell membrane and/or EPCR encryption, and thus become refractory to EPCR-dependent functions such as PC activation and APC-mediated cytoprotective cell signaling. This is further supported by studies showing that EPCR inactivation in vivo, either genetically or induced by blocking antibodies, increases susceptibility to thrombotic and inflammatory disease (27). In addition, in vivo studies using genetically modified mice that differ in EPCR expression clearly demonstrate the important implication of EPCR availability for the efficacy of cytoprotective effects by APC. High EPCR expression reduces susceptibility for endotoxin-induced death in mice, while low EPCR expression increases endotoxin-induced death and aggravates the inflammatory response against endotoxin (28–30). Reduction in EPCR’s bioavailability has been implicated to contribute to the pathogenesis of various diseases and has been demonstrated for example at the site of microvascular thrombosis in tissues of children with meningococcemia sepsis (31), in intestinal tissue of patients with inflammatory bowel diseases (32), and in brains of severe malaria patients infected with Plasmodium falciparum (33). In summary, abundant in vitro and in vivo data indicates that functional depletion of EPCR is directly related to the efficacy of protein C activation and APC’s cytoprotective effects on cells, and that inflammation compromises EPCR-dependent anti-inflammatory mechanisms thereby fueling the vicious cycle of EPCR shedding (20, 27–30, 34, 35). Thus providing a rationale for approaches to restore functional EPCR on cells affected by EPCR shedding and encryption.
Improving EPCR’s bioavailability via cell painting with a membrane-anchored EPCR derivate is a novel unexplored area. Here we explored the potential of glycosylphosphatidylinositol (GPI)-anchored EPCR as a novel tool to restore the EPCR bioavailability and functionality on EPCR-depleted cells. As EPCR’s cofactor activity in the protein C system requires EPCR to locate in caveolin-enriched lipid rafts, the caveolae-targeting GPI-anchoring sequence originating from decay accelerating factor (DAF) was used (17, 18, 36). We show that GPI-anchored EPCR can be used to attain high surface EPCR levels, restore APC binding, improve PC activation, and augment PAR cleavage and APC-mediated cytoprotective signaling.
MATERIAL AND METHODS
Construction of EPCR-GPI
The downstream sequence from the pcDNA3.1(+) soluble EPCR intermediate construct with an AgeI cleavage site after Ser210 (37) was replaced with the glycosylphosphatidylinositol (GPI)-sequence from decay accelerating factor (DAF) (36, 38) using forward primer 5′-CCGGTCCCAAATAAAGGAAGTGGAACCACTTCAGGTACTACCCGTCTTCTATCTGGGCACACGTGTTTCACGTTGACAGGTTTGCTTGGGACGCTAGTAACCATGGGCTTGCTGACTTAG-3′ and reverse primer 5′-TCGACTAAGTCAGCAAGCCCATGGTTACTAGCGTCCCAAGCAAACCTGTCAACGTGAAACACGTGTGCCCAGATAGAAGACGGGTAGTACCTGAAGTGGTTCCACTTCCTTTATTTGGGA-3′. A BsrGI restriction site was introduced at the N-terminal sequence, followed by insertion of the His-tag using forward primer 5′-GTACCCGGTCATCATCACCATCACCATGC-3′ and reverse primer 5′-GTACGCATGGTGATGGTGATGATGACCGG-3′. The construct was sequenced and transfected into HEK-293 cells.
Stable EPCR knockdown in endothelial cells
A shRNA retroviral vector against EPCR’s 3′-untranslated region was constructed using forward primer 5′-GATCGTGGTTTGCTAAGAACCTAATTCGAAAATTAGGTTCTTAGCAAACCATTTTTTGAAGCT-3′ and reverse: primer 5′-AGCTAGCTTCAAAAAATGGTTTGCTAAGAACCTAATTTTCGAATTAGGTTCTTAGCAAACCAC-3′. Primers were ligated into fragment BamHI-HindIII from the pGFP-V-RS cloning vector (Origene). Vectors encoding shRNA against EPCR were produced in GP2-293 cells (Invitrogen) according to manufacturer’s protocol (HuSH-29; Origene). Viral supernatant was concentrated on an Amicon Ultra centrifugal filter with a 3K cut-off (Millipore). EA.hy926 cells were transduced with retroviral vectors in the presence of 10 μg/ml polybrene (Millipore) by spinoculating for 90 minutes at 1200 rpm. Complete medium supplemented with 0.5 μg/ml puromycin (Invitrogen) was added 24 hours after transduction. Stable knockdown of EPCR in the EA.hy926 EPCRKD cells was confirmed by Western blot.
Purified Proteins
Human protein C and APC was purified as described (10). Biotinylated APC was prepared by a 10-fold molar excess of biotinylated FPR-chloromethylketone (HTI) followed by dialysis against Tris buffered saline (TBS; 50 mM Tris, 150 mM NaCl, pH 7.4). Soluble EPCR was purified from HEK-293 cells as described (37). EPCR-GPI was purified from HEK-293 cells expressing N-terminal His-tagged EPCR-GPI. Cells were harvested with citric saline (15 mM sodium citrate, 135 mM KCl) and cell pellets were lysed with 0.3% saponin, 50 mM Tris pH 8.0 supplemented with EDTA-free protease inhibitor cocktail (Thermo Scientific) for 30 minutes on ice (39). Cell lysates were cleared by centrifugation for 30 minutes at 14,000g at 4°C, diluted 1:10 in TBS, and EPCR-GPI was purified using Ni-NTA Sepharose (Invitrogen). Bound EPCR-GPI was washed with TBS, and eluted in 0.5 ml fractions with 250 mM imidazole in TBS, followed by extensive dialysis against TBS.
Detection of soluble EPCR in medium from HEK-293 cells
To confirm the attachment of the GPI-anchor to EPCR, HEK-293 cells expressing EPCR-GPI were treated with 0–0.25 U/ml phosphatidylinositol-specific phospholipase C (PI-PLC)(Sigma-Aldrich) for 90 minutes at 37°C in serum-free medium media. Soluble EPCR in the supernatant was measured by sandwich-ELISA using goat anti-EPCR (R&D Systems) as capture antibody (1 μg/ml ), TBS-3%BSA as blocking, rabbit anti-EPCR (EPCR-III) as detecting antibody (1:1000), and HRP-labelled anti-rabbit antibodies (DAKO) for development (1:600).
Analysis of APC binding to EPCR-GPI
Soluble EPCR and EPCR-GPI were coated on Maxisorp 96-wells plates (Nunc) at 10 μg/ml in coating buffer (15 mM Na2CO3, 35 mM NaHCO3, 0.02% NaN3, pH 9.5). Plates were blocked with TBS, 3% BSA for 2 hours, and incubated with 0–200 nM biotinylated APC in HMM2 (Hank’s balanced salt solution containing 1.3 mM CaCl2, 0.6 mM MgCl2, and 0.1% (w/v) endotoxin-free BSA) for 1 hour at room temperature. Plates were washed, fixed with 4% paraformaldehyde (Thermo Scientific), blocked for 2 hours with TBS, 3% BSA, and developed with streptavidin-HRP (Thermo Scientific) diluted 1:4000 in TBS, 1.5% BSA.
Detection of EPCR shedding in the medium of EA.hy926 cells
Shedding of EPCR was detected by immunoprecipitation. Confluent EA.hy926 cells were labeled with 0.5 mg/ml Sulfo-NHS-LC-biotin (Thermo Scientific) in PBS for 10 minutes, washed, and incubated with 0.2 μM phorbol myristate acetate (PMA) for 1.5 hours at 37°C. Shed EPCR was immunoprecipitated from the medium with 10 μg rabbit anti-EPCR (EPCR-III) or 10 μg normal rabbit IgG (Santa Cruz). EPCR was eluted in non-reducing SDS sample buffer and analyzed on Western blot using goat anti-EPCR (R&D Systems) 1:2000 with IRDye 680 donkey anti-goat (Licor) and IRDye CW800 streptavidin. Fluorescent signals were quantified on the Odyssey Imaging System using Image Studio v2.0 and normalized to vehicle-treated controls.
Painting of EPCRKD cells with EPCR-GPI
Confluent EA.hy926 EPCRKD cells were painted with 0–80 μg/ml EPCR-GPI in serum-free DMEM high glucose (Invitrogen) supplemented with 25 mM Hepes (Invitrogen) for 1.5 hours at 37°C. Cells were washed with PBS before analysis. Wild type EA.hy926 cells were used as positive controls in all experiments.
Determination of EPCR cell surface levels
EPCR on the cell surface of EA.hy926 or EPCRKD EA.hy926 cells was determined with an on-cell Western. Cells were fixed with 4% methanol-free paraformaldehyde (Thermo Scientific) on ice, blocked with Odyssey Blocking buffer (Licor) supplemented with 3% bovine serum albumin (Sigma-Aldrich) for 2 hours, and incubated for 1 hour with 3 μg/ml goat anti-human EPCR antibody (R&D Systems). IRDye CW800 donkey anti-goat (Licor) was used 1:600 and DRAQ5 (Biostatus) diluted 1:100,000 for cell number normalization.
Fractionation of cellular membranes
Detergent-free lipid rafts of EA.hy926 EPCRKD cells painted with 20 μg/ml EPCR-GPI or vehicle-treated wild type EA.hy926 cells were prepared as described (40). Fractions were analyzed on Western blot using goat anti-EPCR (R&D Systems) 1:2000 with IRDye CW800 donkey anti-goat (Licor) and rabbit anti-cav-1 (N-20, Santa Cruz) 1:2000 with IRDye 680 donkey anti-rabbit (Licor).
Cellular APC binding assay
Binding of APC to the cell surface of EA.hy926 or EPCRKD EA.hy926 cells was studied by on-cell Western. Cells were incubated on ice with 200 nM biotinylated APC in HMM2 for 1 hour, fixed with 4% methanol-free paraformaldehyde (Thermo Scientific), and blocked for 2 hours with Odyssey Blocking buffer (Licor). Bound biotinylated APC was detected with IRDye 800CW streptavidin (Licor) diluted 1:4000 in blocking buffer and DRAQ5 (Biostatus) was used for cell number normalization. Plates were scanned using the Odyssey Imaging System, and analyzed with Image Studio Software v2.0 (Licor). Fluorescent signals were corrected for background and cell number, and normalized to vehicle-treated controls.
PC activation on cell surfaces
Painted or untreated EA.hy926 EPCRKD and wild type cells were incubated with 100 nM PC and 1 nM thrombin in HMM2 for 90 minutes at 37°C. Supernatant was supplemented with 25 U/ml hirudin and the amidolytic activity of APC was measured at 405nm with Pefachrome PCa (Pentapharm). Data were corrected for PC activation in the absence of cells.
PAR cleavage by EPCR-GPI-bound APC
APC-mediated PAR cleavage on HEK-293 cells expressing SEAP-PAR1 or SEAP-PAR3 with EPCR-GPI was measured as described (19, 37). To determine APC-mediated PAR cleavage on EPCR-GPI painted HEK-293 cells, cells were incubated with 50 nM APC in HMM2 for 3 hours (PAR3) or 400 nM APC in HMM2 for 1 hour (PAR1) at 37°C. SEAP released in the supernatant was determined using 1-step p-nitrophenyl phosphate (Thermo Scientific). Values were corrected for SEAP released in the absence of APC.
Cell signaling
Induction of Akt phosphorylation on EA.hy926 EPCRKD was performed as described with minor modifications (19). EA.hy926 EPCRKD cells were grown to confluence in 12-well dishes followed by incubation with 20 μg/mL EPCR-GPI in serum-free medium (SFM) for 2 hours. After incubation, cells were washed once with SFM and cells were starved over-night in 2% serum containing medium. After treatment with APC (50 nM) cell lysates were made with 100 μL of 1.5% NP-40 lysis buffer containing 1× protease inhibitor cocktail (Pierce) and 1× phosphatase inhibitor cocktail (Invitrogen). Lysates were centrifuged (30 min, 14000 rpm), mixed with SDS sample buffer (LI-COR) and separated on 4–12% SDS Nu-PAGE (Invitrogen). Proteins were transferred to Nitrocellulose membrane (Thermo Scientific), blocked with Odyssey Blocking Buffer (LI-COR) and incubated with primary antibodies (1/2000) against Akt and phospho-Ser473 Akt (Cell Signaling). Blots were developed with donkey anti-mouse IRDye 680 (total Akt) and donkey anti-rabbit IRDye 800CW (phospho-Akt) secondary antibodies (LI-COR) and scanned on the Odyssey Imager (LI-COR). Quantification of integrated fluorescence intensity (K counts) was done using Odyssey Application Software v3.0 (LI-COR).
Statistical analysis
Data was analyzed using the Student’s t-test or ANOVA with a multiple comparison test as appropriate. P-values <0.05 were considered statistically significant. Statistical analyses were performed using GraphPad Prism software, v5.04 (GraphPad).
RESULTS
Characterization of GPI-anchored EPCR
EPCR shedding may limit the efficacy of protein C’s anticoagulant and cytoprotective pathways. A GPI-anchored derivative of EPCR was engineered to restore EPCR’s bioavailability via “cell painting” with membrane-anchored EPCR. The GPI recognition sequence of decay accelerating factor (DAF) was fused to the C-terminus of soluble EPCR truncated at the beginning of the transmembrane sequence. The DAF hydrophobic domain is recognized by the GPI attachment machinery in the endoplasmic reticulum, which results in cleavage of the GPI recognition sequence and attachment of the GPI anchor to the newly created C-terminal serine (38)(Fig. 1).
Figure 1. Schematics of the EPCR-GPI anchor attachment.

The glycophosphatidylinositol (GPI) recognition sequence of decay accelerating factor (DAF) was fused to the C-terminus of soluble EPCR truncated at the beginning of the transmembrane sequence (Ser210). Two amino acids (PV) were introduced behind Ser210 for cloning purposes. Recognition of the DAF hydrophobic domain by the GPI attachment machinery in the endoplasmic reticulum (ER) results processing of the GPI recognition sequence, proteolytic cleavage of the hydrophobic region, and attachment of a glycophosphatidylinositol moiety to the newly created C-terminal serine.
Attachment of the GPI anchor to EPCR was confirmed by treating HEK-293 cells expressing EPCR-GPI with phosphatidylinositol-specific phospholipase C (PI-PLC). EPCR in the supernatant of EPCR-GPI expressing cells increased upon cleavage of the GPI-anchor by PI-PLC, whereas PI-PLC had no effect on the amount of EPCR in the supernatant of empty or wt-EPCR expressing cells (Fig. 2A). Accordingly, binding of APC to EPCR-GPI on the cell surface decreased upon PI-PLC treatment, while APC binding to wt-EPCR expressing cells was unaffected (Fig. 2B).
Figure 2. Functional analysis of cell surface expressed EPCR-GPI.

To confirm attachment of the GPI anchor to EPCR, HEK-293 cells expressing EPCR-GPI (square), wild type EPCR (circle) or no EPCR (diamond) were treated with PI-PLC. (A) EPCR released in the supernatant upon PI-PLC treatment was determined by ELISA and (B) APC binding on PI-PLC treated cells was analyzed by on-cell Western. To determine whether EPCR-GPI supports PAR cleavage, APC-mediated PAR cleavage on HEK-293 cells expressing SEAP-PAR1 (C), or SEAP-PAR3 (E) with wild type EPCR (circle), EPCR-GPI (square), E86A-EPCR (diamond), or no EPCR (X) was compared. (D, F) The role of EPCR-GPI in PAR1 (D) and PAR3 (F) cleavage was confirmed by incubating the cells with PI-PLC prior to addition of APC (50 nM). SEAP-PAR1 (D) and SEAP-PAR3 (F) cleavage by APC in the absence of PI-PLC is set to 100%. Total PAR1 (D) and total PAR3 (F) is the control for SEAP-PAR1 (D) or SEAP PAR3 (F) expression on the cell surface in the presence of increasing concentrations of PI-PLC, measured as the SEAP activity directly on the cell surface in the absence of APC. Shown are mean ± SEM of three independent experiments.
To validate EPCR-GPI’s functional equivalence to wt-EPCR, APC-mediated PAR cleavage on HEK-293 cells expressing EPCR-GPI with PAR1 was studied. Expression of EPCR-GPI enhanced APC-mediated PAR1 cleavage similar to expression of wt-EPCR, whereas expression of E86A-EPCR defective in APC binding did not (Fig 2C). In addition, the decrease in PAR1 cleavage upon PI-PLC treatment confirmed that EPCR-GPI was responsible for PAR cleavage (Fig. 2D). Similar results were obtained for PAR3 (Fig. 2E, F). This shows that EPCR-GPI supports APC-mediated PAR cleavage similar to wt-EPCR.
Purified EPCR-GPI binds APC with normal affinity
To facilitate purification, EPCR-GPI was expressed on HEK-293 cells with an N-terminal His-tag. EPCR-GPI was purified from HEK-293 lysate to >70% homogeneity (Fig. 3A). As expected, EPCR-GPI migrated slightly slower on SDS-PAGE (50 kDa) compared to soluble EPCR (ca. 46 kDa) because of the 11 additional amino acids and the GPI-anchor attached to the C-terminus of soluble EPCR (Fig. 3B). Most importantly, soluble EPCR and purified EPCR-GPI bound APC with a very similar apparent KD of 24±4 nM and 24±3 nM, respectively (Fig. 3C). Specificity of APC binding to EPCR-GPI was confirmed using the non-blocking (rcr-2) (41) control anti-EPCR antibody and blocking (rcr-252) (42) that reduced APC binding to EPCR-GPI >75% (Fig. 3D). This illustrates that EPCR-GPI can be readily purified from HEK-293 cells and remains functional after purification. Moreover, the N-terminal His-tag does not interfere with APC binding since APC binds equally well to His-tagged EPCR-GPI as to soluble EPCR. Finally, the ability of the GPI anchor to direct EPCR-GPI to lipid rafts was confirmed by fractionation of transfected HEK-293 cell membranes on a density gradient. EPCR-GPI localized almost exclusively in the buoyant lipid raft fractions, whereas wt-EPCR in transfected HEK-293 cells was predominantly associated with the denser non-lipid raft fractions (Fig. 3E).
Figure 3. Purification and APC binding to EPCR-GPI.

EPCR-GPI expressed in HEK-293 cells was purified from cell lysates. (A) Coomassie stain and (B) Western blot of purified EPCR-GPI under non-reducing conditions. (C) APC binding to recombinant soluble EPCR (sEPCR; circle) and EPCR-GPI (square). Signals were corrected for aspecific binding of APC to wells without EPCR. Data was fitted to a non-linear regression curve for one-site specific binding to calculate the Bmax and apparent KD, followed by normalized of the data to the corresponding Bmax. (D) Specificity of APC binding to EPCR-GPI using the blocking (rcr-252) and non-blocking (rcr-2) control anti-EPCR antibodies (both used at 20 μg/ml). (C-D) Shown are mean ± SD of at least three independent experiments. (E) Representative experiment of cell membrane fractionation on a density gradient of EPCR-GPI or wt-EPCR transfected HEK-293 cells. TfR denotes the transferrin receptor.
Long-lasting cell paint with GPI-anchored EPCR
To develop a model system for cellular EPCR depletion such as induced by inflammatory mediators, a stable EPCR knockdown in immortalized endothelial cells (EA.hy926 cells) was engineered. EA.hy926 cells were transduced with a retroviral vector encoding shRNA against the 3′-UTR of EPCR. EPCR protein expression on EA.hy926 EPCRKD cells was reduced by 80% compared to wild type EA.hy926 cells, while EPCR expression was unchanged in cells expressing scramble shRNA (Fig. 4A). The 80% reduction in total EPCR expression corresponded well with a 75% decrease in surface EPCR staining (Fig. 4B).
Figure 4. Analysis of EPCR-GPI cell painting efficiency.

The painting efficiency for EPCR-GPI was determined on EA.hy926 EPCRKD cells. (A) Knockdown of EPCR was quantified by Western blot of cell lysates and normalized to wild type EA.hy926 cells (ctrl) and β-actin. A representative blot is shown in the inset. (B) Cell surface EPCR expression on EPCRKD cells was measured with an on-cell Western assay and normalized to wild type EA.hy926 cells (ctrl cells). (C) EPCR-GPI painted EPCRKD cells were analyzed for surface EPCR levels by on-cell Western. Painted EPCRKD cells (EPCR-GPI) and untreated wild type EA.hy926 cells (wt) were stained for EPCR (green) and cell number using Draq5 (red). Shown is a representative image of three independent experiments. Note that wt cells did not receive paint. (D) Quantification of EPCR cell surface levels after painting EPCRKD cells with EPCR-GPI. EPCR levels were corrected for background signal (no Ab) and normalized for cell number. Surface EPCR levels on wild type EA.hy926 cells were set at 100%. (E) EPCRKD cells were painted with 80 μg/ml EPCR-GPI for 0–180 minutes to determine the optimal time for painting. Loss of EPCR-GPI from the cell surface on (F) painted EPCRKD cells or (G) painted wild type EA.hy926 cells was analyzed in time. Cells were painted with 40 μg/ml EPCR-GPI for 90 minutes, washed in PBS (t=0) and fresh media was added to the cells to determine the stability of EPCR-GPI incorporation in the cell membrane in time. EPCR levels on untreated wild type cells were used as 100%. For all experiments mean ± SD of three independent experiments are shown. *** P < 0.001, **** P < 0.0001.
Painting EPCRKD cells with EPCR-GPI dose-dependently increased cell surface EPCR levels (Fig. 4C) with EPCR levels increasing up to 3-fold of normal endothelial levels (Fig. 4D). The painting efficiency for EPCR-GPI was time-dependent, and reached a plateau after 180 minutes (Fig. 4E). EPCR levels on painted EPCRKD cells stabilized at 2.5-fold the amount of EPCR on normal cells and remained on the cell surface for at least 24 hours after the medium was replaced (Fig. 4F). Interestingly, wild type EA.hy926 cells could also be painted with EPCR-GPI (Fig. 4G) indicating that EPCR-GPI can be employed to increase EPCR levels on normal cells. Thus, cell painting with GPI-anchored EPCR provides a long-lasting method to restore the bioavailability of EPCR on the endothelial cell surface.
EPCR-GPI colocalizes with caveolin-1 in lipid rafts
The localization of EPCR-GPI on the cell surface was examined in more detail as localization of EPCR in caveolin-1 enriched lipid rafts is required for APC-mediated cytoprotective signaling (12, 17, 43). Most endogenous EPCR and EPCR-GPI was detected in the heavier membrane fractions, although some could also be detected in caveolin-1 enriched fractions (Fig. 5A). About 20% of total EPCR-GPI on painted EPCRKD cells colocalized with caveolin-1 in lipid rafts, whereas 7% of total endogenous EPCR present in wild type EA.hy926 cells was detected in caveolin-enriched lipid rafts (Fig. 5B). Interestingly, the relative distribution of residual endogenous EPCR on EPCRKD cells was similar to endogenous EPCR on normal cells (Fig. 5A, quantification not shown), which suggests that a pronounced reduction in the absolute amount of EPCR does not influence the distribution of EPCR across the cellular membranes. Thus as predicted by the localization of DAF in lipid rafts (36), the DAF-derived GPI anchor localized EPCR-GPI to caveolin-enriched lipid rafts at least as efficient as endogenous EPCR.
Figure 5. Localization of painted EPCR-GPI in lipid rafts.

To determine the localization of painted EPCR-GPI on cells, cell membranes were fractionated on a density gradient. (A) Distribution of endogenous EPCR and painted EPCR-GPI in membrane fractions of wild type EA.hy926 cells or EA.hy926 EPCRKD cells painted with 20 μg/ml EPCR-GPI. Caveolin-1 (cav-1) was used as a marker for lipid rafts. Representative blots of three independent experiments are shown. (B) Quantification of endogenous EPCR or EPCR-GPI in caveolin-enriched lipid raft (fractions 1–8) for wild type EA.hy926 cells (EPCR) and painted EA.hy926 EPCRKD cells (EPCR-GPI). Shown are mean ± SD of three independent experiments. *** P < 0.001.
GPI-anchored EPCR enhances PC activation and APC binding on EPCR-depleted cells
EPCR-GPI’s ability to improve APC binding on EPCR-depleted cells was determined. APC binding to the cell surface of EA.hy926 EPCRKD cells was reduced by 90% (Fig. 6A), consistent with depletion of EPCR and loss of EPCR functional effects on these cells. EPCR-GPI painting dose-dependently restored APC binding to EPCRKD cells (Fig. 6B, C). Knockdown of EPCR also reduced PC activation on the endothelial cell surface by approximately 45% (Fig. 6D), but did not interfere with PC activation mediated by the thrombin-thrombomodulin complex independently of EPCR as evident by the residual PC activation on EPCRKD cells. EPCR-GPI painting improved PC activation on EPCRKD cells to roughly 100%, the level of PC activation observed on wild type EA.hy926 cells. Thus, GPI-anchored EPCR can support both APC binding on painted cells and enhance EPCR-mediated PC activation.
Figure 6. Improvement of APC binding and protein C activation by EPCR-GPI painting.

EPCR-GPI’s ability to support APC binding and PC activation on EPCRKD cells was determined. (A) APC binding to the surface of EA.hy926 EPCRKD cells was detected by on-cell Western and normalized to wild type EA.hy926 cells (ctrl cells). (B) Binding of APC to EPCRKD cells painted with 0–80 μg/ml EPCR-GPI for 90 minutes. Painted EPCRKD cells (paint) and untreated wild type EA.hy926 cells (wt) were stained for APC (green) and cell number (red). Shown is a representative image of three independent experiments. Note that wild type EA.hy926 cells did not receive paint. (C) Quantification of APC binding to painted EPCRKD cells. APC levels were corrected for background signal (no APC) and normalized for cell number. APC binding to untreated wild type EA.hy926 cells was set at 100%. (D) PC activation on EPCRKD cells painted with 80 μg/ml EPCR-GPI (EPCRKD+EPCR-GPI), untreated EPCRKD (EPCRKD), and wild type EA.hy926 cells (ctrl cells). PC activated by thrombin in the absence of cells was subtracted as background. For all experiments mean ± SD of three independent experiments are shown. *** P < 0.001, **** P < 0.0001.
EPCR-GPI restores APC binding to PMA-treated cells
To confirm that EPCR-GPI also restores APC binding on EPCR-depleted cells, EPCR shedding was induced with PMA, a compound well known for its ability to induce shedding (21). Soluble EPCR levels increased 6-fold upon PMA-induced EPCR shedding (Fig. 7A). PMA-induced shedding resulted in a 35% decrease in APC binding to the cell surface (Fig. 7B), whereas APC binding completely normalized when PMA-treated EA.hy926 cells were painted with EPCR-GPI (Fig. 7B). To determine the sensitivity of EPCR-GPI to shedding, HEK-293 cells transfected with EPCR-GPI or wt-EPCR were incubated with inducers of shedding (TNFα, PMA, or thrombin) (21, 34). Neither TNFα nor PMA induced EPCR shedding on EPCR-GPI cells whereas inflammatory mediator-induced shedding was evident from the appearance of sEPCR in the medium of wt-EPCR cells (Fig 7C). In contrast, PI-PLC induced shedding of EPCR-GPI, indicating that EPCR-GPI is relatively resistant to TNFα- or PMA-induced shedding. Thus, EPCR-GPI provides a novel tool to counteract cellular APC resistance resulting from EPCR shedding.
Figure 7. EPCR-GPI-mediated reconstitution of APC binding after PMA-induced EPCR shedding.
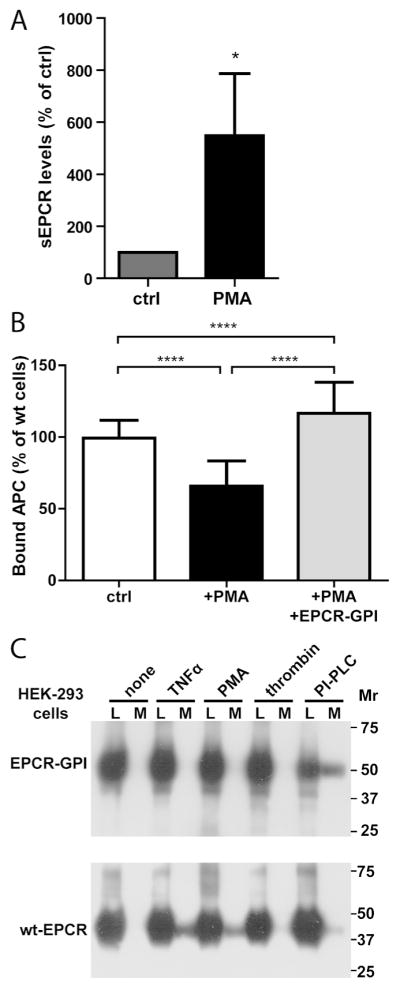
The ability of EPCR-GPI to restore APC binding after EPCR shedding was determined. (A) Shedding of EPCR was induced by treating wild type EA.hy926 cells with 0.2 μM PMA, and soluble EPCR (sEPCR) shed in the supernatant was measured by immunoprecipitation. Spontaneous shedding from untreated wild type cells was set to 100% (ctrl) (B) Reconstitution of APC binding by EPCR-GPI to the cell surface of PMA-treated cells was quantified using an on-cell Western assay. PMA-treated cells were incubation with 80 μg/ml EPCR-GPI (+PMA+EPCR-GPI) or left untreated (+PMA) before addition of APC. APC binding to non-treated wild type cells (ctrl) was used as 100%. Shown are mean ± SD of three independent experiments. * P < 0.05, **** P < 0.0001. (C) Sensitivity of EPCR-GPI to inflammatory mediator-induced shedding. HEK-293 cells transfected with EPCR-GPI or wt-EPCR were incubated with TNFα (10 ng/ml) for 12 hours or PMA (100 nM), thrombin (20 nM), or PI-PLC (0.5 U/ml) for 2 hours. Cell lysate (L) and conditioned medium (M) were analyzed for cellular and soluble shed EPCR by Western Blot using a goat anti-EPCR antibody. Shown is a representative example of two independent experiments.
EPCR-GPI supports APC-mediated PAR1 and PAR3 cleavage
To determine whether painted EPCR-GPI could stimulate PAR cleavage by APC, HEK-293 cells expressing PAR1 or PAR3 were painted with EPCR-GPI. APC-mediated PAR1 cleavage increased 3-fold after cells were painted with EPCR-GPI (Fig. 8A). In contrast, cleavage of PAR1 on untreated EPCR-negative cells was almost absent as only 2% of total PAR1 was cleaved. The increase in PAR cleavage due to cell painting with EPCR-GPI was identical on PAR3-expressing cells as PAR3 cleavage also increased 3-fold after painting (Fig. 8B). Accordingly, EPCR-GPI painting of EPCRKD EAhy926 cells augmented APC-mediated induction of PAR1-dependent cell signaling as evident by the phosphorylation of Akt (Fig. 8C). This shows that cell painting with EPCR-GPI not only restored EPCR’s bioavailability but also supported APC-mediated PAR1 and PAR3 cleavage and induction of cytoprotective cell signaling pathways.
Figure 8. Stimulation of PAR cleavage by EPCR-GPI painting.
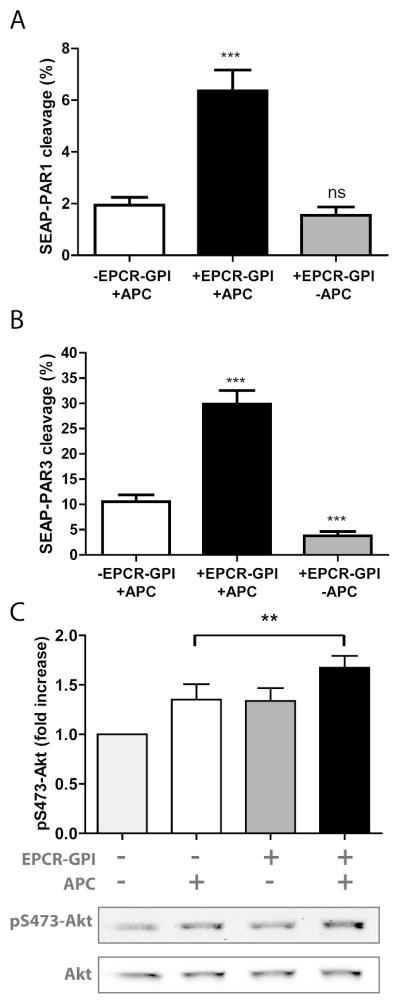
To determine whether EPCR-GPI painting supports APC-mediated PAR cleavage, N-terminal cleavage of PAR by APC was analyzed on HEK-293 cells expressing SEAP-PAR1 (A) or SEAP-PAR3 (B) that were painted with 80 μg/ml EPCR-GPI (+EPCR-GPI) or left untreated (-EPCR-GPI). APC-mediated PAR cleavage was expressed as the percentage of total PAR on the cell surface. (C) Induction of Akt phosphorylation at Ser473 by APC (50 nM) at 45 min on EPCRKD EA.hy926 cells in the presence and absence of EPCR-GPI painting (20 μg/ml). Shown are mean ± SD of a representative experiment of three independent experiments. * P < 0.05, ** P < 0.01, *** P < 0.001, ns = not significant.
DISCUSSION
The protein C system conveys important beneficial effects in various models of sepsis, stroke, and other vascular-inflammatory diseases that require functional EPCR (6, 8, 9, 11). Since EPCR’s functions are directly related to its localization on the cell surface and its ability to recruit PC and APC to the cell surface, EPCR restoration approaches need to encompass localization on or in the cell membrane, something that is not accomplished by the soluble ectodomain alone. Although gene transfer approaches would allow EPCR to be expressed with its transmembrane domain, the acute and transient need to restore EPCR on the cell membrane for conditions such as sepsis and stroke make such an approach undesirable.
Numerous examples exist for targeting proteins and small molecules to the cell surface based on fusion with antibody fragments or binding domains of cell surface molecules. Accordingly, fusions of hemostatic proteins to cellular targets have been successfully employed to reconstitute these proteins on cell surfaces (44–46). Thrombomodulin fused to a single chain fragment of an antibody that targets red blood cells was successfully used to prevent thrombosis in animal models (44). In addition, tissue factor fused to annexin V to target sites of vascular injury had both procoagulant and anticoagulant properties depending on its concentration (45). Important considerations for EPCR are that functionality is not only determined by binding and recruiting (A)PC to the cell surface, but also by presenting PC to the thrombomodulin-thrombin complex and presenting APC to PAR1 in caveolin-1 rich microdomains. Efficient presentation of (A)PC to these macromolecular complexes by EPCR is unlikely to be compatible with binding of a bulky EPCR fusion protein to a cell surface protein. Therefore, this study pursued a membrane-anchored EPCR derivate that allowed for on-demand cell painting.
Cell painting has been successfully used to insert glycosylphosphatidylinositol (GPI)-anchored proteins into cell membranes to function as tumor homing ligand (47), cancer vaccine (48), and antigenic complex (49). GPI anchors on endogenous proteins are generally characterized by a C-terminal hydrophobic region of about 20 amino acids (38). Recognition of the hydrophobic region in the ER results in proteolytic processing at 10–12 residues from the N-terminal of the hydrophobic region, and attachment of a GPI anchor. Fusion of the minimal GPI recognition sequence to EPCR’s C-terminus, truncated at the beginning of its transmembrane domain (Ser210), allowed for genetic encoding of a lipid anchor at the cost of an 11 amino acid insertion. As EPCR’s cofactor activity in the protein C system requires EPCR to locate in caveolin-enriched lipid rafts, the caveolae-targeting GPI-anchoring sequence originating from decay accelerating factor (DAF) was used (17, 18, 36). As projected, EPCR-GPI painting resulted in co-localization of EPCR-GPI with caveolin-1 in lipid rafts at least as efficient as endogenous EPCR. Moreover, EPCR-GPI painting resulted in long lasting EPCR incorporation in the cell surface, while supporting EPCR’s important cofactor functions by improving PC activation and PAR cleavage.
An added benefit of the GPI anchor on EPCR is that it conveys resistance to shedding induced by PMA or TNFα. Presumably, the additional 11 amino acid spacer between EPCR and the GPI attachment and potentially a slightly altered orientation on the cell membrane prevents the interaction of TACE with the metalloproteinase-sensitive region in EPCR (residues Lys192-Lys200) (21). Thus, applications of EPCR-GPI painting may comprise situations where EPCR’s functional bioavailability is diminished due to shedding of EPCR by inflammatory mediators, such as during sepsis. Our data indicate that EPCR-GPI painting increased EPCR surface levels on both EPCR-depleted cells as well as on cells expressing normal levels of EPCR, suggesting that the presence of endogenous (encrypted) EPCR does not interfere with the incorporation of EPCR-GPI. In addition to on-demand delivery, EPCR-GPI cell painting also facilitates administration of engineered EPCR. Proof-of-principle studies for GPI-fused proteins targeting diseases include multiple examples of ex vivo cell painting (47).
EPCR-GPI cell painting would be well suited for ex vivo cell therapies aimed at improving survival and engraftment of transplants. Both APC and liposomal thrombomodulin improved the outcome of pancreatic islets transplantation for type I diabetes (50–53), and APC’s cytoprotective effects have been shown to reduce graft-related injury (54, 55). Furthermore, overexpression of EPCR on murine transplant tissue protected against transplantation-related thrombotic and inflammatory injury (56). Although the feasibility of EPCR over-expression on human tissues is unlikely, painting donor tissue with EPCR-GPI during transport or prior to transplantation could potentially improve cytoprotective effects of APC that were demonstrated to reduce graft-related injury (54, 55). Alternatively, EPCR-GPI painting of CD8+ dendritic cells may enhance efficacy of APC’s anti-sepsis therapy, since these cells were shown to convey susceptibility to APC-mediated mortality reduction in experimental murine sepsis models (4). EPCR-GPI painting of erythrocytes and making use of their unique ability to transfer GPI-anchored proteins to the endothelium in vivo as was shown for DAF (CD55) may provide a specific targeting approach of EPCR-GPI for endothelial cells (57, 58) presenting opportunities for EPCR-GPI to promote vascular integrity, such as when endogenous EPCR is rendered inactive during severe malaria by Plasmodium falciparum infected erythrocytes (59).
In summary, controlling the EPCR content in cellular membranes using EPCR-GPI provides a novel tool for alterations of the efficacy of PC activation and APC’s direct effects on cells. However, additional in vivo experiments are necessary to provide proof for the utility of EPCR-based cell painting in diseases states.
WHAT IS KNOWN ABOUT THIS TOPIC?
APC conveys beneficial anticoagulant and cytoprotective effects in numerous in vivo disease models.
EPCR is required for efficient PC activation and for APC-mediated PAR activation.
During inflammatory disease, expression of EPCR on cell membranes is often diminished thereby limiting PC activation and APC’s effects on cells.
WHAT DOES THIS PAPER ADD
A caveolae-targeting glycosylphosphatidylinositol (GPI)-anchored EPCR (EPCR-GPI) restored EPCR’s bioavailability via “cell painting”
EPCR-GPI cell painting improved the efficacy of the protein C system without a need for gene transfer.
EPCR-GPI painting achieved physiological relevant surface levels on endothelial cells, restored APC binding to EPCR-depleted cells, supported PC activation, and enhanced APC-mediated PAR cleavage and cytoprotective signaling.
Acknowledgments
This work was supported by an American Heart Association Western States Affiliate postdoctoral fellowship (E.A.M.B.) and National Institutes of Health (NHLBI) grant HL104165 (L.O.M.).
The anti-EPCR antibodies (rcr-2 and rcr-252) were generously made available by Dr. Kenji Fukudome (Saga Medical School, Saga, Japan) and Dr. Ramon Montes (University of Navarra, Pamplona, Spain). We gratefully acknowledge Ms. Madeleine Mathias for excellent experimental support.
Footnotes
AUTHORSHIP CONTRIBUTIONS
E.A.M.B., F.S., and L.O.M. designed and performed experiments, and the analyzed data. E.A.M.B. and L.O.M. wrote the paper. All authors contributed to final editing of the manuscript.
DISCLOSURE OF CONFLICT OF INTEREST
The authors have nothing to disclose.
References
- 1.Dahlback B, Villoutreix BO. Regulation of blood coagulation by the protein C anticoagulant pathway. Novel insights into structure-function relationships and molecular recognition. Arterioscler Thromb Vasc Biol. 2005;25(7):1311–20. doi: 10.1161/01.ATV.0000168421.13467.82. [DOI] [PubMed] [Google Scholar]
- 2.Rezaie AR. Regulation of the protein C anticoagulant and antiinflammatory pathways. Curr Med Chem. 2010;17(19):2059–69. doi: 10.2174/092986710791233706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bouwens EA, Stavenuiter F, Mosnier LO. Mechanisms of anticoagulant and cytoprotective actions of the protein C pathway. J Thromb Haemost. 2013;11(Suppl 1):242–53. doi: 10.1111/jth.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kerschen EJ, Hernandez I, Zogg M, et al. Activated protein C targets CD8+ dendritic cells to reduce the mortality of endotoxemia in mice. J Clin Invest. 2010;120(9):3167–78. doi: 10.1172/JCI42629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geiger H, Pawar SA, Kerschen EJ, et al. Pharmacological targeting of the thrombomodulin-activated protein C pathway mitigates radiation toxicity. Nat Med. 2012;18(7):1123–9. doi: 10.1038/nm.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Danese S, Vetrano S, Zhang L, et al. The protein C pathway in tissue inflammation and injury: pathogenic role and therapeutic implications. Blood. 2010;115(6):1121–30. doi: 10.1182/blood-2009-09-201616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu J, Ji Y, Zhang X, et al. Endogenous activated protein C signaling is critical to protection of mice from lipopolysaccaride induced septic shock. J Thromb Haemost. 2009;7(5):851–6. doi: 10.1111/j.1538-7836.2009.03333.x. [DOI] [PubMed] [Google Scholar]
- 8.Zlokovic BV, Griffin JH. Cytoprotective protein C pathways and implications for stroke and neurological disorders. Trends Neurosci. 2011;34(4):198–209. doi: 10.1016/j.tins.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esmon CT. Protein C anticoagulant system-anti-inflammatory effects. Semin Immunopathol. 2012;34(1):127–32. doi: 10.1007/s00281-011-0284-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mosnier LO, Yang XV, Griffin JH. Activated protein C mutant with minimal anticoagulant activity, normal cytoprotective activity, and preservation of thrombin activable fibrinolysis inhibitor-dependent cytoprotective functions. J Biol Chem. 2007;282(45):33022–33. doi: 10.1074/jbc.M705824200. [DOI] [PubMed] [Google Scholar]
- 11.Bock F, Shahzad K, Vergnolle N, et al. Activated protein C based therapeutic strategies in chronic diseases. Thromb Haemost. 2014;111(4):610–7. doi: 10.1160/TH13-11-0967. [DOI] [PubMed] [Google Scholar]
- 12.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109(8):3161–72. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 13.Stearns-Kurosawa DJ, Kurosawa S, Mollica JS, et al. The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proc Natl Acad Sci U S A. 1996;93(19):10212–6. doi: 10.1073/pnas.93.19.10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Riewald M, Petrovan RJ, Donner A, et al. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296(5574):1880–2. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- 15.Taylor FB, Jr, Peer GT, Lockhart MS, et al. Endothelial cell protein C receptor plays an important role in protein C activation in vivo. Blood. 2001;97(6):1685–8. doi: 10.1182/blood.v97.6.1685. [DOI] [PubMed] [Google Scholar]
- 16.Burnier L, Mosnier LO. Novel mechanisms for activated protein C cytoprotective activities involving non-canonical activation of protease-activated receptor 3. Blood. 2013;122(5):807–16. doi: 10.1182/blood-2013-03-488957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rezaie AR. The occupancy of endothelial protein C receptor by its ligand modulates the PAR-1 dependent signaling specificity of coagulation proteases. IUBMB Life. 2011;63(6):390–6. doi: 10.1002/iub.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Russo A, Soh UJ, Trejo J. Proteases Display Biased Agonism at Protease-Activated Receptors: Location matters! Mol Interv. 2009;9(2):87–96. doi: 10.1124/mi.9.2.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mosnier LO, Sinha RK, Burnier L, et al. Biased agonism of protease-activated receptor 1 by activated protein C caused by non-canonical cleavage at Arg46. Blood. 2012;120(26):5237–46. doi: 10.1182/blood-2012-08-452169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopez-Sagaseta J, Puy C, Tamayo I, et al. sPLA2-V inhibits EPCR anticoagulant and antiapoptotic properties by accommodating lysophosphatidylcholine or PAF in the hydrophobic groove. Blood. 2012;119(12):2914–21. doi: 10.1182/blood-2011-05-353409. [DOI] [PubMed] [Google Scholar]
- 21.Qu D, Wang Y, Esmon NL, et al. Regulated endothelial protein C receptor shedding is mediated by tumor necrosis factor-alpha converting enzyme/ADAM17. J Thromb Haemost. 2007;5(2):395–402. doi: 10.1111/j.1538-7836.2007.02347.x. [DOI] [PubMed] [Google Scholar]
- 22.Bouwens EA, Mosnier LO. EPCR encryption induces cellular APC resistance. Blood. 2012;119(12):2703–5. doi: 10.1182/blood-2012-01-400762. [DOI] [PubMed] [Google Scholar]
- 23.Gu JM, Katsuura Y, Ferrell GL, et al. Endotoxin and thrombin elevate rodent endothelial cell protein C receptor mRNA levels and increase receptor shedding in vivo. Blood. 2000;95(5):1687–93. [PubMed] [Google Scholar]
- 24.Uitte De Willige S, van Marion V, Rosendaal FR, et al. Haplotypes of the EPCR gene, plasma sEPCR levels and the risk of deep venous thrombosis. J Thromb Haemost. 2004;2(8):1305–10. doi: 10.1046/j.1538-7836.2004.00855.x. [DOI] [PubMed] [Google Scholar]
- 25.Biguzzi E, Franchi F, Bucciarelli P, et al. Endothelial protein C receptor plasma levels increase in chronic liver disease, while thrombomodulin plasma levels increase only in hepatocellular carcinoma. Thromb Res. 2007;120(2):289–93. doi: 10.1016/j.thromres.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 26.Kurosawa S, Stearns-Kurosawa DJ, Carson CW, et al. Plasma Levels of Endothelial Cell Protein C Receptor Are Elevated in Patients With Sepsis and Systemic Lupus Erythematosus: Lack of Correlation With Thrombomodulin Suggests Involvement of Different Pathological Processes. Blood. 1998;91(2):725–7. [PubMed] [Google Scholar]
- 27.Esmon CT. The endothelial cell protein C receptor. Thromb Haemost. 2000;83(5):639–43. [PubMed] [Google Scholar]
- 28.Taylor FB, Jr, Stearns-Kurosawa DJ, Kurosawa S, et al. The endothelial cell protein C receptor aids in host defense against Escherichia coli sepsis. Blood. 2000;95(5):1680–6. [PubMed] [Google Scholar]
- 29.Kerschen EJ, Fernandez JA, Cooley BC, et al. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. J Exp Med. 2007;204(10):2439–48. doi: 10.1084/jem.20070404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.von Drygalski A, Furlan-Freguia C, Ruf W, et al. Organ-Specific Protection Against Lipopolysaccharide-Induced Vascular Leak Is Dependent on the Endothelial Protein C Receptor. Arterioscler Thromb Vasc Biol. 2013;33(4):769–76. doi: 10.1161/ATVBAHA.112.301082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Faust SN, Levin M, Harrison OB, et al. Dysfunction of endothelial protein C activation in severe meningococcal sepsis. N Engl J Med. 2001;345(6):408–16. doi: 10.1056/NEJM200108093450603. [DOI] [PubMed] [Google Scholar]
- 32.Faioni EM, Ferrero S, Fontana G, et al. Expression of endothelial protein C receptor and thrombomodulin in the intestinal tissue of patients with inflammatory bowel disease. Crit Care Med. 2004;32(5 Suppl):S266–S70. doi: 10.1097/01.ccm.0000128032.85396.83. [DOI] [PubMed] [Google Scholar]
- 33.Moxon CA, Wassmer SC, Milner DA, Jr, et al. Loss of endothelial protein C receptors links coagulation and inflammation to parasite sequestration in cerebral malaria in African children. Blood. 2013;122(5):842–51. doi: 10.1182/blood-2013-03-490219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukudome K, Esmon CT. Identification, cloning, and regulation of a novel endothelial cell protein C/activated protein C receptor. J Biol Chem. 1994;269(42):26486–91. [PubMed] [Google Scholar]
- 35.Xu J, Qu D, Esmon NL, et al. Metalloproteolytic release of endothelial cell protein C receptor. J Biol Chem. 2000;275(8):6038–44. doi: 10.1074/jbc.275.8.6038. [DOI] [PubMed] [Google Scholar]
- 36.Legler DF, Doucey MA, Schneider P, et al. Differential insertion of GPI-anchored GFPs into lipid rafts of live cells. FASEB J. 2005;19(1):73–5. doi: 10.1096/fj.03-1338fje. [DOI] [PubMed] [Google Scholar]
- 37.Mosnier LO, Zampolli A, Kerschen EJ, et al. Hyper-antithrombotic, non-cytoprotective Glu149Ala-activated protein C mutant. Blood. 2009;113(23):5970–8. doi: 10.1182/blood-2008-10-183327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moran P, Caras IW. Fusion of sequence elements from non-anchored proteins to generate a fully functional signal for glycophosphatidylinositol membrane anchor attachment. J Cell Biol. 1991;115(6):1595–600. doi: 10.1083/jcb.115.6.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bumgarner GW, Zampell JC, Nagarajan S, et al. Modified cell ELISA to determine the solubilization of cell surface proteins: Applications in GPI-anchored protein purification. J Biochem Biophys Methods. 2005;64(2):99–109. doi: 10.1016/j.jbbm.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 40.Macdonald JL, Pike LJ. A simplified method for the preparation of detergent-free lipid rafts. J Lipid Res. 2005;46(5):1061–7. doi: 10.1194/jlr.D400041-JLR200. [DOI] [PubMed] [Google Scholar]
- 41.Lopez-Sagaseta J, Montes R, Puy C, et al. Binding of factor VIIa to the endothelial cell protein C receptor reduces its coagulant activity. J Thromb Haemost. 2007;5(9):1817–24. doi: 10.1111/j.1538-7836.2007.02648.x. [DOI] [PubMed] [Google Scholar]
- 42.Fukudome K, Ye X, Tsuneyoshi N, et al. Activation mechanism of anticoagulant protein C in large blood vessels involving the endothelial cell protein C receptor. J Exp Med. 1998;187(7):1029–35. doi: 10.1084/jem.187.7.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Russo A, Soh UJ, Paing MM, et al. Caveolae are required for protease-selective signaling by protease-activated receptor-1. Proc Natl Acad Sci U S A. 2009;106(15):6393–7. doi: 10.1073/pnas.0810687106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zaitsev S, Kowalska MA, Neyman M, et al. Targeting recombinant thrombomodulin fusion protein to red blood cells provides multifaceted thromboprophylaxis. Blood. 2012;119(20):4779–85. doi: 10.1182/blood-2011-12-398149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang X, Ding WQ, Vaught JL, et al. A soluble tissue factor-annexin V chimeric protein has both procoagulant and anticoagulant properties. Blood. 2006;107(3):980–6. doi: 10.1182/blood-2005-07-2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carnemolla R, Greineder CF, Chacko AM, et al. PECAM targeted oxidant-resistant mutant thrombomodulin fusion protein with enhanced potency in vitro and vivo. J Pharmacol Exp Ther. 2013;347(2):339–45. doi: 10.1124/jpet.113.205104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Legler DF, Johnson-Leger C, Wiedle G, et al. The alpha v beta 3 integrin as a tumor homing ligand for lymphocytes. Eur J Immunol. 2004;34(6):1608–16. doi: 10.1002/eji.200424938. [DOI] [PubMed] [Google Scholar]
- 48.McHugh RS, Ahmed SN, Wang YC, et al. Construction, purification, and functional incorporation on tumor cells of glycolipid-anchored human B7-1 (CD80) Proc Natl Acad Sci U S A. 1995;92(17):8059–63. doi: 10.1073/pnas.92.17.8059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang JH, Getty RR, Chisari FV, et al. Protein transfer of preformed MHC-peptide complexes sensitizes target cells to T cell cytolysis. Immunity. 1994;1(7):607–13. doi: 10.1016/1074-7613(94)90050-7. [DOI] [PubMed] [Google Scholar]
- 50.Contreras JL, Eckstein C, Smyth CA, et al. Activated protein C preserves functional islet mass after intraportal transplantation: a novel link between endothelial cell activation, thrombosis, inflammation, and islet cell death. Diabetes. 2004;53(11):2804–14. doi: 10.2337/diabetes.53.11.2804. [DOI] [PubMed] [Google Scholar]
- 51.Xue M, Dervish S, Harrison LC, et al. Activated Protein C Inhibits Pancreatic Islet Inflammation, Stimulates T Regulatory Cells, and Prevents Diabetes in Non-obese Diabetic (NOD) Mice. J Biol Chem. 2012;287(20):16356–64. doi: 10.1074/jbc.M111.325951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen H, Teramura Y, Iwata H. Co-immobilization of urokinase and thrombomodulin on islet surfaces by poly(ethylene glycol)-conjugated phospholipid. J Control Release. 2011;150(2):229–34. doi: 10.1016/j.jconrel.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 53.Cui W, Angsana J, Wen J, et al. Liposomal Formulations of Thrombomodulin Increase Engraftment After Intraportal Islet Transplantation. Cell Transplant. 2010;19:1359–67. doi: 10.3727/096368910X513964. [DOI] [PubMed] [Google Scholar]
- 54.Ilmakunnas M, Pesonen EJ, Hockerstedt K, et al. Graft protein C entrapment is associated with reduced phagocyte activation during reperfusion in human liver transplantation. Crit Care Med. 2006;34(2):426–32. doi: 10.1097/01.ccm.0000198108.38349.28. [DOI] [PubMed] [Google Scholar]
- 55.Kuriyama N, Isaji S, Hamada T, et al. The cytoprotective effects of addition of activated protein C into preservation solution on small-for-size grafts in rats. Liver Transpl. 2009;16(1):1–11. doi: 10.1002/lt.21923. [DOI] [PubMed] [Google Scholar]
- 56.Lee KF, Lu B, Roussel JC, et al. Protective Effects of Transgenic Human Endothelial Protein C Receptor Expression in Murine Models of Transplantation. Am J Transplant. 2012;12(9):2363–72. doi: 10.1111/j.1600-6143.2012.04122.x. [DOI] [PubMed] [Google Scholar]
- 57.Kooyman DL, Byrne GW, McClellan S, et al. In vivo transfer of GPI-linked complement restriction factors from erythrocytes to the endothelium. Science. 1995;269(5220):89–92. doi: 10.1126/science.7541557. [DOI] [PubMed] [Google Scholar]
- 58.Kooyman DL, Byrne GW, Logan JS. Glycosyl phosphatidylinositol anchor. Exp Nephrol. 1998;6(2):148–51. doi: 10.1159/000020516. [DOI] [PubMed] [Google Scholar]
- 59.Petersen JEV, Bouwens EA, Tamayo I, et al. Protein C system defects inflicted by the malaria parasite protein PfEMP1 can be overcome by a soluble EPCR variant. Thromb Haemost. 2015 doi: 10.1160/TH15-01-0018. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]