

Abstract
Charcot-Marie-Tooth (CMT) disease is the most common hereditary peripheral neuropathy. More than 50 causative genes have been identified. The lack of genotype-phenotype correlations in many CMT patients make it difficult to decide which genes are affected. Recently, targeted next-generation sequencing (NGS) has been introduced as an alternative approach for diagnosis of genetic disorders. Here, we applied targeted NGS in combination with PMP22 duplication/deletion analysis to screen causative genes in 22 Chinese CMT families. The novel variants detected by targeted NGS were then further studied in cultured cells. Of the 22 unrelated patients, 8 had PMP22 duplication. The targeted NGS revealed 10 possible pathogenic variants in 11 patients, including 7 previously reported variants and 3 novel heterozygous variants (GJB1: p.Y157H; MFN2: p.G127S; YARS: p.V293M). Further classification of the novel variants according to American College of Medical Genetics and Genomics (ACMG) standards and guidelines and functional analysis in cultured cells indicated that p.Y157H in GJB1 was pathogenic, p.G127S in MFN2 was likely pathogenic, while p.V293M in YARS was likely benign. Our results suggest the potential for targeted NGS to make a more rapid and precise diagnosis in CMT patients. Moreover, the functional analysis is required when the novel variants are indistinct.
Keywords: Charcot-Marie-Tooth, molecular diagnosis, targeted next-generation sequencing, genetic variant, functional analysis
INTRODUCTION
Charcot-Marie-Tooth (CMT) is the most common inherited neuromuscular disorder with an incidence of 40 individuals in every 100 000 inhabitants [1]. The classical symptoms include slowly progressive distal muscle weakness, muscle atrophy and sensory loss of the lower and then upper limbs. As the motor and sensory peripheral nerves are affected, it is also called hereditary sensory and motor neuropathy. On the basis of electrophysiological results, CMT has been traditionally subdivided into two main groups: demyelinating type (CMT1) and axonal type (CMT2).
Thus far, more than 50 disease-causing genes have been identified to be associated with CMT (http://www.molgen.ua.ac.be/CMTMutations/; http://neuromuscular.wustl.edu/) of which the duplication/deletion of PMP22 is the most common cause of CMT1 [2]. The traditional strategy for molecular diagnosis of CMT is based on the clinical phenotype, inheritance pattern and electrophysiological examination. However, CMT is a highly heterogeneous disorder [3]. It may be inherited in more than one model of inheritance, and a single gene can result in multiple clinical phenotypes. Moreover, there are still many unknown causative genes of CMT waiting to be discovered [4]. It is impractical for a lab to investigate all the possible genes using conventional Sanger sequencing, which is time-consuming and expensive. Consequently, introducing a more comprehensive approach for molecular diagnosis of CMT is important. Targeted next-generation sequencing (NGS), a high-throughput DNA sequencing technology that performs parallel sequencing the genomic regions of interested [5], makes it possible.
Recently, targeted NGS has been successfully performed in the inherited neurologic diseases diagnosis [6–9]. It is an efficient and cost-effective tool for achieving a genetic diagnosis for inherited peripheral neuropathies [10–12]. However, up till now, there is still no literature to evaluate the efficiency of targeted NGS in Chinese CMT patients. Here, we applied targeted NGS in combination with PMP22 duplication/deletion analysis in a cohort of 22 Chinese CMT families. The novel sequence variants identified by targeted NGS were classified according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines [13], and further verified by the functional analysis in cultured cells.
RESULTS
PMP22 duplication/deletion analysis
The multiplex ligation-dependent probe amplification (MLPA) analysis was performed in all patients before the examination of targeted NGS. It showed that 8 out of 22 unrelated CMT patients had PMP22 duplication (Supplemental Table 1). The rest of the CMT patients were further screened for mutations with targeted NGS.
Identification of variants by targeted NGS and sanger sequencing
Targeted NGS was performed in the remaining 14 patients. Our gene panel included 44 genes (Table 1). The coverage of the fraction of target base can be found in Table 2. Over 99% of target bases had >10x coverage, 97.43% had >30x coverage and 94.24% had >50x coverage. The mean coverage of target bases ranged from 106.86 - 1287.75.
Table 1. Target genes included in the panel.
| Gene | Locus | Ref sequence | MIM | Exons | Gene | Locus | Ref sequence | MIM | Exons |
|---|---|---|---|---|---|---|---|---|---|
| AARS | 16q22 | NM_001605.2 | 601065 | 21 | KIF1B | 1p36.2 | NM_015074.3 | 605995 | 47 |
| ATL1 | 14q22.1 | NM_015915.4 | 606439 | 14 | KIF5A | 12q13.13 | NM_004984.2 | 602821 | 29 |
| ATL3 | 11q13.1 | NM_015459.4 | 609369 | 13 | LITAF | 16p13.13 | NM_004862.3 | 603795 | 4 |
| BSCL2 | 11q13 | NM_032667.6 | 606158 | 11 | LRSAM1 | 9q33.3 | NM_138361.5 | 610933 | 25 |
| DCTN1 | 2p13 | NM_004082.4 | 601143 | 32 | MARS | 12q13.3 | NM_004990.3 | 156560 | 21 |
| DHTKD1 | 10p14 | NM_018706.6 | 614984 | 17 | MFN2 | 1p36.22 | NM_014874.3 | 608507 | 19 |
| DNM2 | 19p13.2 | NM_001005361.2 | 602378 | 21 | MPZ | 1q23.3 | NM_000530.7 | 159440 | 6 |
| DNMT1 | 19p13.2 | NM_001130823.2 | 126375 | 41 | NAGLU | 17q21 | NM_000263.3 | 609701 | 6 |
| DYNC1H1 | 14q32 | NM_001376.4 | 600112 | 78 | NEFL | 8p21 | NM_006158.4 | 162280 | 4 |
| EGR2 | 10q21.1 | NM_000399.3 | 129010 | 2 | PDK3 | Xp22.11 | NM_001142386.2 | 300906 | 12 |
| GARS | 7p15 | NM_002047.3 | 600287 | 17 | PMP22 | 17p12 | NM_000304.3 | 601097 | 5 |
| GDAP1 | 8q21.11 | NM_018972.2 | 606598 | 6 | RAB7 | 3q21.3 | NM_004637.5 | 602298 | 6 |
| GJB1 | Xq13.1 | NM_001097642.2 | 304040 | 2 | SCN11A | 3p22.2 | NM_001287223.1 | 604385 | 26 |
| GNB4 | 3q26.33 | NM_021629.3 | 610863 | 10 | SEPT9 | 17q25 | NM_001113493.1 | 604061 | 11 |
| HARS | 5q31.3 | NM_001289094.1 | 142810 | 13 | SLC5A7 | 2q12 | NM_001305005.2 | 608761 | 9 |
| HOXD10 | 2q31.1 | NM_002148.3 | 142984 | 2 | SOX10 | 22q13.1 | NM_006941.3 | 602229 | 4 |
| HSPB1 | 7q11.23 | NM_001540.3 | 602195 | 3 | SPTLC1 | 9q22.2 | NM_001281303.1 | 605712 | 15 |
| HSPB3 | 5q11.2 | NM_006308.2 | 604624 | 1 | SPTLC2 | 14q24.3 | NM_004863.3 | 605713 | 12 |
| HSPB8 | 12q24.23 | NM_014365.2 | 608014 | 3 | TFG | 3q12.2 | NM_001195478.1 | 602498 | 8 |
| IFRD1 | 7q31.1 | NM_001550.3 | 603502 | 12 | TRPV4 | 12q24.1 | NM_021625.4 | 605427 | 16 |
| INF2 | 14q32.33 | NM_001031714.3 | 610982 | 22 | VCP | 9p13.3 | NM_007126.3 | 601023 | 17 |
| KARS | 16q23.1 | NM_001130089.1 | 601421 | 15 | YARS | 1p35.1 | NM_003680.3 | 608323 | 13 |
Abbreviations: MIM = Mendelian Inheritance in Man.
Table 2. The coverage of the fraction of target bases.
| % fraction of target bases | Mean coverage of target bases | |||||
|---|---|---|---|---|---|---|
| coverage > 10x | coverage > 20x | coverage > 30x | coverage > 40x | coverage > 50x | ||
| Mean | 99.36 | 98.53 | 97.43 | 96.08 | 94.24 | 649.39 |
| Range | 97.60 - 99.91 | 93.83 - 99.84 | 88.90 - 99.75 | 82.97 - 99.66 | 76.13 - 99.38 | 106.86 - 1287.75 |
On average, 105 variants were identified in each CMT patient. After being filtered and verified by Sanger sequencing, 10 possible pathogenic variants, including 7 previously reported ones (GJB1: p.V91M, p.R164Q, p.R164W, p.R183H; MFN2: p.R280H; HSPB8: p.K141N; BSCL2: p.S90L) and 3 novel ones (GJB1: p.Y157H; MFN2: p.G127S; YARS: p.V293M), were identified in 11 cases (2 cases carrying the p.R164W in GJB1, Table 3). No possible pathogenic variant was found in the remaining 3 patients (Supplemental Table 1).
Table 3. Clinical manifestations of 11 individuals with genetic variants detected by targeted NGS.
| Characteristics | Patients | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| case 2 | case 5 | case 6 | case 7 | case 9 | case 13 | case 15 | case 17 | case 18 | case 19 | case 20 | |
| Age at study, (years) | 17 | 43 | 16 | 43 | 23 | 39 | 56 | 15 | 26 | 32 | 20 |
| Sex | Female | Male | Male | Male | Female | Male | Male | Male | Male | Female | Male |
| Age at onset, (years) | 6 | 30 | 10 | 27 | 21 | 36 | 41 | 10 | 11 | 29 | 10 |
| Inheritance | D | D | D | D | D | D | D | D | D | D | D |
| Variants | MFN2 | MFN2 | GJB1 | YARS | GJB1 | HSPB8 | GJB1 | GJB1 | GJB1 | GJB1 | BSCL2 |
| Nucleotide | 379G>A | 839G>A | 491G>A | 877G>A | 469T>C | 423G>C | 490C>T | 548G>A | 490C>T | 271G>A | 269C>T |
| Amino acid | G127S | R280H | R164Q | V293M | Y157H | K141N | R164W | R183H | R164W | V91M | S90L |
| Novel or known | Novel | Known | Known | Novel | Novel | Known | Known | Known | Known | Known | Known |
| Muscle weakness UL | Yes | Not | Not | Not | Not | Not | Yes | Yes | Yes | Yes | Not |
| Muscle weakness LL | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Muscle atrophy UL | Yes | Not | Not | Not | Not | Not | Not | Yes | Yes | Yes | Not |
| Muscle atrophy LL | Yes | Yes | Yes | Yes | Not | Yes | Not | Yes | Yes | Yes | Not |
| Ankle DTRs | Reduced | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Absent | Reduced |
| Median nerve, R/L | |||||||||||
| cMAP, mV | 1.1/NA | NA/NA | 0.2/1.7 | 0.6/1.4 | 5.8/3.7 | 6.7/NA | 0.9/NA | 12.1/8.1 | NA/NA | 1.2/2.7 | 1.5/NA |
| MNCV, m/s | 37.8/NA | NA/NA | 32.0/34.4 | 27.8/31.9 | 46.0/42.3 | 56.0/NA | 35.3/NA | 40.8/38.8 | NA/NA | 40.7/40.4 | 61.5/NA |
| Peroneal nerve, R/L | |||||||||||
| cMAP, mV | NP/NA | 1.2/5.3 | NP/NA | NP/NA | 0.1/NA | 1.4/NA | NP/NA | 1.9/2.2 | NA/NA | NA/NA | 3.0/2.2 |
| MNCV, m/s | NP/NA | 35.1/37.8 | NP/NA | NP/NA | 34.9/NA | 40.8/NA | NP/NA | 40.7/40.9 | NA/NA | NP/NA | 36.5/37.4 |
| Median nerve, R/L | |||||||||||
| SNAP, uv | 14/NA | NA/NA | 3.8/0.7 | NP/NP | 1.6/3.6 | 25/NA | 5.4/NA | 0.84/1.3 | NA/NA | 9.5/8.7 | 13.2/NA |
| SNCV, m/s | 50/NA | NA/NA | 39.4/34.6 | NP/NP | 44.4/43.1 | 56.5/NA | 37.8/NA | 36.6/40.7 | NA/NA | 44.8/44.8 | 50/NA |
| Sural nerve, R/L | |||||||||||
| SNAP, uv | 1.4/NP | NA/NA | NP/NP | NP/NP | NP/NP | NA/19 | 3.7/4.0 | 3.9/5.0 | NA/NA | 4.8/NA | 23.1/16.6 |
| SNCV, m/s | 26.3/NP | NA/NA | NP/NP | NP/NP | NP/NP | NA/46.8 | 36.2/38.3 | 36.8/36.5 | NA/NA | 36.7/NA | 41/40 |
| Laboratory test | |||||||||||
| CK, U/L | NA | NA | 290 | NA | 212 | 744 | NA | NA | NA | 200 | NA |
| LDH, U/L | NA | NA | 227 | NA | 242 | 255 | NA | NA | NA | 172 | NA |
Abbreviations: D = dominant; NA = not available; NP = not potential; MNCV = motor nerve conduction velocity; cMAP = compound muscle action potential; SNAP = sensory nerve action potential; SNCV = sensory nerve conduction velocity; R/L = right/left.
Normal values: median MNCV ≥ 49 m/s; median cMAP ≥ 4.5 mv; peroneal MNCV ≥ 40 m/s; peroneal cMAP ≥ 2.5 mv; median SNCV ≥ 50 m/s; median SMAP ≥ 5.0 uv; sural SNCV ≥ 40 m/s; sural SMAP ≥ 5.0 uv.
Reference range: CK < 174 U/L; LDH < 211 U/L.
Classification of novel variants and functional analysis
The three novel variants were absent in 1000Genomes and dbSNP database and were not present in 500 control subjects. The SIFT and PolyPhen-2 software programs were used to predict the functional disruption of proteins due to amino acid change.
The novel variant c.379G>A (p.G127S) in MFN2 affects the same amino acid as two previously reported mutations (p.G127D and p.G127V) [14, 15]. This variant was further identified in the proband's affected mother and younger brother who presented with a similar phenotype, while her unaffected father did not carry the same variant (Figure 1A and 1B). The amino acid change was predicted to be deleterious by SIFT (score: 0) and PolyPhen-2 (score: 1.0). Additionally, the variant site was conserved in different animal species (Figure 1C). According to ACMG standards and guidelines, this variant was classified as a likely pathogenic variant.
Figure 1. Pedigree, sequencing chromatograms and conservation analysis of novel variants detected by targeted NGS.
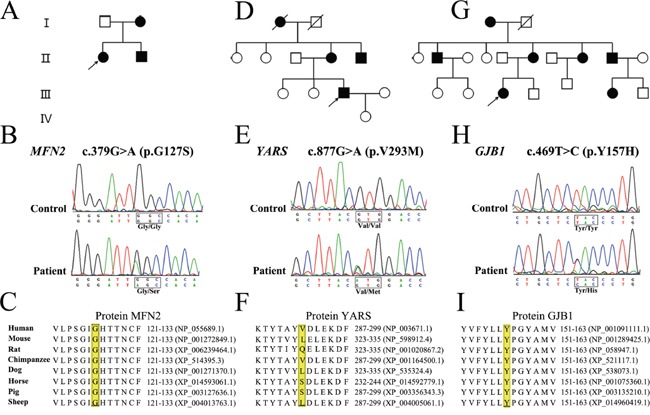
A-C. Pedigree of the proband with novel variant in MFN2, YARS and GJB1, respectively. D-F. Electropherograms showing the sequence variation in MFN2, YARS and GJB1, respectively. G-I. Conservation analysis. YARS, MFN2 and GJB1 protein sequence alignments from multiple, evolutionarily diverse species were depicted.
The novel variant c.877G>A (p.V293M) in YARS (Figure 1D and 1E) was predicted to be deleterious by the SIFT (score: 0.02), while benign in PolyPhen-2 (score: 0.049). The variant site was not conserved in different animal species (Figure 1F). The segregation analysis was not available as the blood samples were unobtainable from other family members. In contrast to all previously identified YARS mutations [16], this novel variant was located at the anticodon recognition region of YARS protein.
The novel variant c.469T>C (p.Y157H) in GJB1 (Figure 1G and 1H) affected the same amino acid as the previously reported mutation (p.Y157C) [17]. The variant site was conserved in different animal species (Figure 1I) and was predicted to be deleterious by SIFT (score: 0) and PolyPhen-2 (score: 1.0). However, segregation analysis was not performed, as no other family members were available for further examination.
To determine the consequence of amino acid change in YARS or GJB1, functional analysis in cultured cells was further performed. The data revealed that HEK293 cells transfected with wild-type or mutant YARS had the comparable mRNA (Supplementary Figure 1) and protein level (Figure 2A). Furthermore, the p.V293M variant did not change the protein's distribution in HeLa cells (Figure 2B), HEK293 cells or SH-SY5Y neuroblastoma cell lines (data not shown). These studies suggested that the novel variant p.V293M in YARS was likely benign. With regard to GJB1, we found that the p.Y157H change did not affect the GJB1 mRNA (Supplementary Figure 1) or protein level (Figure 2C). The fluorescence study revealed that HeLa cells transfected with EGFP-GJB1-Wt formed intracellular granules, whereas cells expressed EGFP-GJB1-Y157H had diffuse intracellular staining (Figure 2D). These data indicated that p.Y157H variant affected intracellular distribution of GJB1, suggesting the pathogenicity of this novel variant.
Figure 2. Functional analysis in cultured cells.
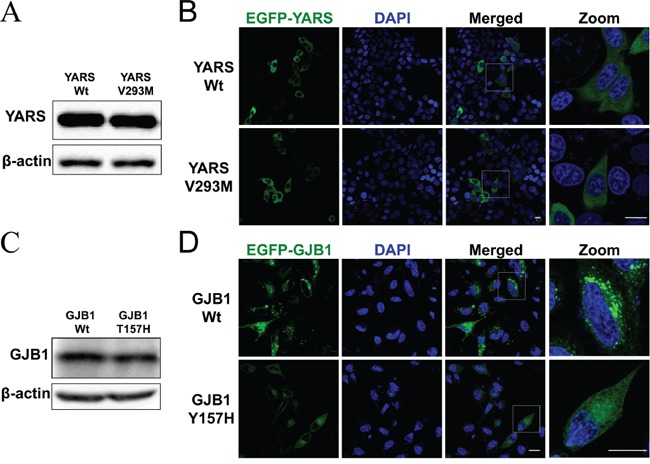
A. Western blot analysis of protein extracts obtained from HEK293 cells transfected with pFlag-YARS-Wt or pFlag-YARS-V293M vectors. The anti-Flag antibody was used to detect the interested band. B. Confocal images of HeLa cells transiently transfected with pEGFP-YARS-Wt or pEGFP-YARS-V293M vectors. Transfected cells were examined for green fluorescence 48 hours after transfection. Cell nuclei were stained with 4′-6-diamidino-2-phenylindole (DAPI; blue). Scale bar, 20 μm. C. Western blot analysis of GJB1 expression in HEK293 cells transfected with pEGFP-GJB1-Wt or pEGFP-GJB1-Y157H vector. The anti-GFP antibody was used to detected recombinant proteins. D. Confocal images of HeLa cells transiently transfected with pEGFP-GJB1-Wt or pEGFP-GJB1-Y157H constructs. Cells expressing wild type GJB1 displayed granules throughout the cytosol, whereas cells expressing the mutant GJB1 had diffuse intracellular staining. Scale bar, 20 μm.
Clinical features of CMT patients
The clinical features of recruited CMT patients were summarized in Supplemental Table 1. Among these patients, 17 were male and 5 were female, and ages ranged from 12 to 66. All these cases displayed a progressive phenotype. In the majority of CMT patients, the symptoms at disease onset were muscle weakness in the lower limbs. Motor deficits were more marked distally and more sever in the lower limbs.
The patient (case 2) carrying the likely pathogenic variant p.G127S in MFN2 was a 17-year-old female (Table 3). The symptoms and signs were began at age of 6 with gait disturbance. At the age of 15, she underwent orthopedic surgery. The clinical examinations revealed reduced deep tendon reflex, muscle weakness in the distal limbs and muscle atrophy in both hands. Conduction velocity of the median nerve was reduced, with a moderate decrease in distal amplitudes.
The patient (case 7) carrying the likely benign variant p.V293M in YARS was a 43-year-old male (Table 3). He displayed foot drop, distal muscle weakness and atrophy in the lower limbs for about 18 years. He did not feel numbness in the limbs. Neurological examinations revealed bilateral foot drop with pes cavus, muscle weakness and atrophy in the lower limbs, and areflexia in all limbs. He had horizontal nystagmus. The motor nerve conduction velocity and muscle compound action potential were markedly reduced in all limbs.
The patient (case 9) carrying the p.Y157H pathogenic variant in GJB1 was a 23-year-old female from a large family with several affected members (Table 3). Onset of the disease was at age 21 years, with distal weakness in legs and steppage gait. Neurological examinations revealed pes cavus, reduced muscle strength in the distal lower limbs and areflexia in all limbs. Electrophysiological studies showed reduced nerve conduction and compound muscle action potential in the peroneal nerve.
DISCUSSION
There are a number of different NGS technologies, including whole genome sequencing (WGS), whole exome sequencing (WES) and targeted NGS. All these technologies have already been successfully performed in CMT patients [18–20]. While it seems that WGS and WES are more attractive to identify underlying genetic causes than targeted NGS, one of the drawbacks of WES and WGS is that they generate a huge amount of unnecessary data. It is difficult and time-consuming to interpret those variants, especially when only one patient is available for sequencing. For targeted NGS, the panel covering a determined set of candidate genes can be designed to screen causative mutations. It offers some advantages owing to cost savings and the speed of data interpretation [5, 21]. Therefore, in the current study, we introduced targeted NGS to detect the causative genes in Chinese CMT patients. Since targeted NGS was relatively less effective to detect the copy number variants [12, 22], the PMP22 duplication/deletion was firstly analyzed.
Thus far, targeted NGS approach has been applied to CMT patients [11, 12, 18]. Just like these studies, our results demonstrated that targeted NGS is an effective method to make a molecular diagnosis in CMT patients. However, to compare with those studies, there are some differences in our study. Firstly, apart from the PMP22 duplication/deletion analysis, the other common causative genes, such as GJB1 and MFN2, had not been detected before the examination of targeted NGS. The published studies have revealed that over 90% of the CMT patients carried mutations in PMP22, MPZ, GJB1 and MFN2 [4, 23]. In our study, 8 cases had mutations in GJB1 and MFN2. Secondly, a specific dominant gene panel was designed in our study. Therefore, we recruited CMT families with dominant inheritance pattern. For targeted NGS, the most significant difficulty is how to analyze and interpret the possible pathogenic variants. To overcome this obstacle, a more logical approach is to design a smaller panel that covers major subtypes. The disease subtype-specific NGS panel costs less than the complete CMT gene panel, and the number of irrelevant variants is reduced [5]. In our study, variants identified in the dominant CMT gene panel were fewer than the complete CMT gene panel [11]. Lastly, we made a molecular diagnosis in 10 patients using targeted NGS (case 7 carrying a likely benign variant). The diagnostic success rate is higher than the previously reported in the literatures [10–12].
After being interpreted, 10 possible pathogenic variants were found in our study. The clinical characteristics of these CMT patients with known variants are comparable to the reported data [15, 24–28]. In case 2, a likely pathogenic novel variant in MFN2 was found. MFN2 is localized in the outer mitochondrial membrane which contains two hydrophobic heptad repeat domains and a GTPase domain. The novel variant was located at the GTPase domain. An intact GTPase domain is indispensable for the function of mitochondrial fusion [29]. In case 9, a novel pathogenic variant in GJB1 was identified. Functional analysis revealed that wild-type GJB1 formed puncta staining in the mammalian cells, while this phenomenon was disappeared when the cells expressed GJB1 mutant (p.Y157H). The result was consistent with the previously reported GJB1 mutants (p.M34K, p.N205I and p.Y211X). Many GJB1 mutants showed abnormal trafficking when expressed in mammalian cells [30].
In case 7, the novel variant in YARS was detected. YARS, encoding tyrosyl-tRNA synthetase (TyrRS), is an aminoacyl-tRNA synthetase involved in dominant-intermediate CMT [16, 31]. TyrRS contains three functional domains: an N-terminal catalytic domain, a central anticodon recognition domain and a C-terminal EMAP II-like domain. So far, all previously identified pathogenic TyrRS mutations, including p.G41R, p.E196K and p.153-156delVKQV, were all located at the catalytic domain of the protein. These mutations affect the protein's localization in sprouting neurites of neuroblastoma cells [16]. A missense p.K265N variant located at the anticodon recognition domain of TyrRS has been reported. However, it was demonstrated to be a benign polymorphism [32]. The novel variant we found was also located at the anticodon recognition domain of TyrRS. Further functional analysis revealed that this variant did not change the protein's expression or intracellular localization. Therefore, this novel variant in YARS was classified as likely benign.
Even after targeted NGS, 4 cases (including case 7) still did not have a molecular diagnosis. There may be several reasons for this. First of all, as the filtering method was too strict, it's possible that some truly pathogenic variants were recognized as benign. Secondly, in addition to the copy number variations (CNVs) of PMP22, we did not detect any further CNVs in other causative genes. Although CNVs outside of PMP22 locus are rare, it is still important to test them in CMT cases negative for the PMP22 duplication [33–35]. Finally, there are still many other CMT causative-genes that remain to be identified. It has been estimated that 50% of CMT patients may carry mutations in unknown disease genes [4].
In summary, we have combined targeted NGS and PMP22 duplication/deletion analysis as a diagnostic strategy for CMT patients. Besides 7 previously reported variants, we found 3 novel variants after targeted NGS analysis. Further classification of sequence variants according to ACMG standards and guidelines and functional analysis in cultured cells indicated that p.Y157H in GJB1 was pathogenic, p.G127S in MFN2 was likely pathogenic, while p.V293M in YARS was likely benign. Although the number of cases was small, our results still revealed an increase in diagnostic success rate.
MATERIALS AND METHODS
Patients
Patients were recruited from the Department of Neurology, Huashan Hospital, and Second Affiliated Hospital of Zhejiang University School of Medicine from December 2007 to June 2015. Patients were considered to suffer from CMT if they had a sensorimotor peripheral neuropathy (according to their medical history, neurological examination and neurophysiological testing) and a family history of similar characteristics. The clinical diagnostic strategy was performed as described in the previously reported literature [23]. In total, 22 Chinese CMT families with dominant inheritance pattern were enrolled. Clinical evaluations were carried out by at least two senior neurologists. Routine blood biochemical tests and electrophysiological tests were performed. Five hundred unrelated aged individuals (≥65 years) without history of CMT were selected as a control group. Written informed consents were obtained from all the participants. This study was approved by the ethics board of Huashan Hospital and Second Affiliated Hospital.
PMP22 duplication/deletion analysis
All the CMT patients were tested for the PMP22 duplication/deletion using MLPA as reported previously [36]. MLPA was performed using the MLPA kit (MRC Holland, Netherlands), according to the manufacture's protocol.
Targeted NGS
Genomic DNA was extracted from peripheral EDTA-treated blood using Blood Genomic Extraction Kit (Qiagen, Germany). A dominant gene panel was designed to cover 44 genes known to be associated with dominant CMT and other inherited peripheral neuropathies (Table 1). Genetic features can be found in the online databases including Neuromuscular Disease Center (http://neuromuscular.wustl.edu/time/hmsn.html) and Inherited Peripheral Neuropathies Mutation Database (http://www.molgen.ua.ac.be/cmtmutations/). All of the exons and the 20 flanking base pairs of the splice junctions surrounding the exons of targeted genes were included. The samples were captured by NimbleGen SeqCap EZ products (Roche, Switzerland). Deep sequencing was further done on an Illumina HiSeq2000 platform (Genergy Biotechnology Co Ltd, Shanghai, China). Each read was aligned to the hg19 reference genome (http://hgdownload.cse.ucsc.edu/) using the Burrows-Wheeler Aligner (BWA, version 0.7.12-r1039) [37]. The variants were detected using Genome Analysis Toolkit (GATK, version 3.1-1-g07a4bf8) following the GATK best practices [38]. All the variants were annotated by the ANNOVAR (Version 2014-11-12). Variants were further filtered, as described in our previously publication [39]. Two software programs, SIFT (http://sift.jcvi.org/) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), were used to predict the possible protein functional change caused by the variant.
Sanger sequencing
Sanger sequencing was used to validate all the potential variants using standard protocols. PCR was performed to amplify the fragments covering the variant sites. The PCR products were purified and further directly sequenced on ABI 3730 DNA Sequencer. The sequencing results were aligned to human reference genome published in Ensembl (http://www.ensembl.org/).
Functional studies in cultured cells
The cDNA encoding human GJB1 was cloned into HindIII/KpnI site of pEGFP-C2 vector. The cDNA encoding human YARS was cloned into HindIII/KpnI site of pFLAG-CMV-4 vector and pEGFP-C2 vector. All mutant constructs of GJB1 and YARS were created by PCR mutagenesis and verified by Sanger sequencing.
HEK293 and HeLa cells were cultured in DMEM supplemented 10% fetal bovine serum in a 5% CO2 incubator. To explore whether the novel variant affected mRNA and protein expression, HEK293 cells were transfected with wild-type, mutant expressing or empty vectors. Transient transfection was performed using the Lipofectamine 2000 according to the manufacture's protocol (Invitrogen, USA). Forty-eight hours after transfection, cells were lysed and harvested. The mRNA expression level of YARS and GJB1 was detected using RT-PCR analysis. The following primers were used: GJB1 Forward: GCGTGAACCGGCATTCTACT; GJB1 Reverse: TTGGTCATAGCAAACGCTGTT; YARS Forward: CTGCACCTTATCACCCGGAAC; YARS Reverse: TCCGCAAACAGAATTGTTACCT; GAPDH Forward: ACTCCACGACGTACTCAG; GAPDH Reverse: CATGTTCCAATATGATTCCACC. The protein samples were resolved by SDS-PAGE, transferred to nitrocellulose membrane and blotted with the desired antibodies. The antibodies against Flag (1:5 000; Abmart, China), GFP (1:5 000; Santa Cruz, USA) and β-actin (1:5 000; Sigma-Aldrich, USA) were used.
To further define the intracellular distribution of wild-type and mutant protein in mammalian cell lines, HeLa cells transiently over-expressing EGFP-YARS-Wt, EGFP-YARS-V293M, EGFP-GJB1-Wt, EGFP-GJB1-Y157H or the empty vector were directly observed under a confocal microscope (Leica, Germany).
SUPPLEMENTARY FIGURE AND TABLE
Acknowledgments
The authors sincerely thank the participants for their help and willingness to participate in this study, and also thank Nellie Nelson and Xiaowen Wang for their help on the language. This work was supported by a grant from the National Natural Science Foundation of China to Zhi-Ying Wu (81125009).
Footnotes
CONFLICTS OF INTEREST
All authors declare no conflicts of interest.
REFERENCES
- 1.Pareyson D, Marchesi C. Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. The Lancet Neurology. 2009;8:654–667. doi: 10.1016/S1474-4422(09)70110-3. [DOI] [PubMed] [Google Scholar]
- 2.Murphy SM, Laura M, Fawcett K, Pandraud A, Liu YT, Davidson GL, Rossor AM, Polke JM, Castleman V, Manji H, Lunn MP, Bull K, Ramdharry G, et al. Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. Journal of neurology, neurosurgery, and psychiatry. 2012;83:706–710. doi: 10.1136/jnnp-2012-302451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ostern R, Fagerheim T, Hjellnes H, Nygard B, Mellgren SI, Nilssen O. Segregation analysis in families with Charcot-Marie-Tooth disease allows reclassification of putative disease causing mutations. BMC medical genetics. 2014;15:12. doi: 10.1186/1471-2350-15-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ekins S, Litterman NK, Arnold RJ, Burgess RW, Freundlich JS, Gray SJ, Higgins JJ, Langley B, Willis DE, Notterpek L, Pleasure D, Sereda MW, Moore A. A brief review of recent Charcot-Marie-Tooth research and priorities. F1000Research. 2015;4:53. doi: 10.12688/f1000research.6160.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verma A. Next-generation sequencing and genetic diagnosis of Charcot-Marie-Tooth disease. Annals of Indian Academy of Neurology. 2014;17:383–386. doi: 10.4103/0972-2327.144004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nemeth AH, Kwasniewska AC, Lise S, Parolin Schnekenberg R, Becker EB, Bera KD, Shanks ME, Gregory L, Buck D, Zameel Cader M, Talbot K, de Silva R, Fletcher N, et al. Next generation sequencing for molecular diagnosis of neurological disorders using ataxias as a model. Brain. 2013;136:3106–3118. doi: 10.1093/brain/awt236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sevy A, Cerino M, Gorokhova S, Dionnet E, Mathieu Y, Verschueren A, Franques J, Maues de Paula A, Figarella-Branger D, Lagarde A, Desvignes JP, Beroud C, Attarian S, et al. Improving molecular diagnosis of distal myopathies by targeted next-generation sequencing. Journal of neurology, neurosurgery, and psychiatry. 2016;87:340–342. doi: 10.1136/jnnp-2014-309663. [DOI] [PubMed] [Google Scholar]
- 8.Dai Y, Wei X, Zhao Y, Ren H, Lan Z, Yang Y, Chen L, Cui L. A comprehensive genetic diagnosis of Chinese muscular dystrophy and congenital myopathy patients by targeted next-generation sequencing. Neuromuscular disorders. 2015;25:617–624. doi: 10.1016/j.nmd.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 9.Chae JH, Vasta V, Cho A, Lim BC, Zhang Q, Eun SH, Hahn SH. Utility of next generation sequencing in genetic diagnosis of early onset neuromuscular disorders. Journal of medical genetics. 2015;52:208–216. doi: 10.1136/jmedgenet-2014-102819. [DOI] [PubMed] [Google Scholar]
- 10.Antoniadi T, Buxton C, Dennis G, Forrester N, Smith D, Lunt P, Burton-Jones S. Application of targeted multi-gene panel testing for the diagnosis of inherited peripheral neuropathy provides a high diagnostic yield with unexpected phenotype-genotype variability. BMC medical genetics. 2015;16:84. doi: 10.1186/s12881-015-0224-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoyer H, Braathen GJ, Busk OL, Holla OL, Svendsen M, Hilmarsen HT, Strand L, Skjelbred CF, Russell MB. Genetic diagnosis of Charcot-Marie-Tooth disease in a population by next-generation sequencing. BioMed research international. 2014;2014:210401. doi: 10.1155/2014/210401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ylikallio E, Johari M, Konovalova S, Moilanen JS, Kiuru-Enari S, Auranen M, Pajunen L, Tyynismaa H. Targeted next-generation sequencing reveals further genetic heterogeneity in axonal Charcot-Marie-Tooth neuropathy and a mutation in HSPB1. European journal of human genetics. 2014;22:522–527. doi: 10.1038/ejhg.2013.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engelfried K, Vorgerd M, Hagedorn M, Haas G, Gilles J, Epplen JT, Meins M. Charcot-Marie-Tooth neuropathy type 2A: novel mutations in the mitofusin 2 gene (MFN2) BMC medical genetics. 2006;7:53. doi: 10.1186/1471-2350-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chung KW, Kim SB, Park KD, Choi KG, Lee JH, Eun HW, Suh JS, Hwang JH, Kim WK, Seo BC, Kim SH, Son IH, Kim SM, et al. Early onset severe and late-onset mild Charcot-Marie-Tooth disease with mitofusin 2 (MFN2) mutations. Brain. 2006;129:2103–2118. doi: 10.1093/brain/awl174. [DOI] [PubMed] [Google Scholar]
- 16.Jordanova A, Irobi J, Thomas FP, Van Dijck P, Meerschaert K, Dewil M, Dierick I, Jacobs A, De Vriendt E, Guergueltcheva V, Rao CV, Tournev I, Gondim FA, et al. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nature genetics. 2006;38:197–202. doi: 10.1038/ng1727. [DOI] [PubMed] [Google Scholar]
- 17.Bone LJ, Deschenes SM, Balice-Gordon RJ, Fischbeck KH, Scherer SS. Connexin32 and X-linked Charcot-Marie-Tooth disease. Neurobiology of disease. 1997;4:221–230. doi: 10.1006/nbdi.1997.0152. [DOI] [PubMed] [Google Scholar]
- 18.Lupo V, Garcia-Garcia F, Sancho P, Tello C, Garcia-Romero M, Villarreal L, Alberti A, Sivera R, Dopazo J, Pascual-Pascual SI, Marquez-Infante C, Casasnovas C, Sevilla T, et al. Assessment of Targeted Next-Generation Sequencing as a Tool for the Diagnosis of Charcot-Marie-Tooth Disease and Hereditary Motor Neuropathy. The Journal of molecular diagnostics. 2016;18:225–234. doi: 10.1016/j.jmoldx.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 19.Choi BO, Koo SK, Park MH, Rhee H, Yang SJ, Choi KG, Jung SC, Kim HS, Hyun YS, Nakhro K, Lee HJ, Woo HM, Chung KW. Exome sequencing is an efficient tool for genetic screening of Charcot-Marie-Tooth disease. Human mutation. 2012;33:1610–1615. doi: 10.1002/humu.22143. [DOI] [PubMed] [Google Scholar]
- 20.Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D, Chen DC, Nazareth L, Bainbridge M, Dinh H, Jing C, Wheeler DA, McGuire AL, Zhang F, Stankiewicz P, et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. The New England journal of medicine. 2010;362:1181–1191. doi: 10.1056/NEJMoa0908094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lynch DS, Koutsis G, Tucci A, Panas M, Baklou M, Breza M, Karadima G, Houlden H. Hereditary spastic paraplegia in Greece: characterisation of a previously unexplored population using next-generation sequencing. European journal of human genetics. 2015 doi: 10.1038/ejhg.2015.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sathirapongsasuti JF, Lee H, Horst BA, Brunner G, Cochran AJ, Binder S, Quackenbush J, Nelson SF. Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics. 2011;27:2648–2654. doi: 10.1093/bioinformatics/btr462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Annals of neurology. 2011;69:22–33. doi: 10.1002/ana.22166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oterino A, Monton FI, Cabrera VM, Pinto F, Gonzalez A, Lavilla NR. Arginine-164-tryptophan substitution in connexin32 associated with X linked dominant Charcot-Marie-Tooth disease. Journal of medical genetics. 1996;33:413–415. doi: 10.1136/jmg.33.5.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dubourg O, Tardieu S, Birouk N, Gouider R, Leger JM, Maisonobe T, Brice A, Bouche P, LeGuern E. Clinical, electrophysiological and molecular genetic characteristics of 93 patients with X-linked Charcot-Marie-Tooth disease. Brain. 2001;124:1958–1967. doi: 10.1093/brain/124.10.1958. [DOI] [PubMed] [Google Scholar]
- 26.Irobi J, Van Impe K, Seeman P, Jordanova A, Dierick I, Verpoorten N, Michalik A, De Vriendt E, Jacobs A, Van Gerwen V, Vennekens K, Mazanec R, Tournev I, et al. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nature genetics. 2004;36:597–601. doi: 10.1038/ng1328. [DOI] [PubMed] [Google Scholar]
- 27.Kochanski A, Kabzinska D. Molecular genetic analysis of the GJB1 gene: a study of six mutations. Journal of applied genetics. 2004;45:95–100. [PubMed] [Google Scholar]
- 28.Bissar-Tadmouri N, Parman Y, Boutrand L, Deymeer F, Serdaroglu P, Vandenberghe A, Battaloglu E. Mutational analysis and genotype/phenotype correlation in Turkish Charcot-Marie-Tooth Type 1 and HNPP patients. Clinical genetics. 2000;58:396–402. doi: 10.1034/j.1399-0004.2000.580511.x. [DOI] [PubMed] [Google Scholar]
- 29.Cartoni R, Martinou JC. Role of mitofusin 2 mutations in the physiopathology of Charcot-Marie-Tooth disease type 2A. Experimental neurology. 2009;218:268–273. doi: 10.1016/j.expneurol.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 30.Yum SW, Kleopa KA, Shumas S, Scherer SS. Diverse trafficking abnormalities of connexin32 mutants causing CMTX. Neurobiology of disease. 2002;11:43–52. doi: 10.1006/nbdi.2002.0545. [DOI] [PubMed] [Google Scholar]
- 31.Storkebaum E, Leitao-Goncalves R, Godenschwege T, Nangle L, Mejia M, Bosmans I, Ooms T, Jacobs A, Van Dijck P, Yang XL, Schimmel P, Norga K, Timmerman V, et al. Dominant mutations in the tyrosyl-tRNA synthetase gene recapitulate in Drosophila features of human Charcot-Marie-Tooth neuropathy. Proceedings of the National Academy of Sciences of the United States of America; 2009; pp. 11782–11787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leitao-Goncalves R, Ermanoska B, Jacobs A, De Vriendt E, Timmerman V, Lupski JR, Callaerts P, Jordanova A. Drosophila as a platform to predict the pathogenicity of novel aminoacyl-tRNA synthetase mutations in CMT. Amino acids. 2012;42:1661–1668. doi: 10.1007/s00726-011-0868-4. [DOI] [PubMed] [Google Scholar]
- 33.Huang J, Wu X, Montenegro G, Price J, Wang G, Vance JM, Shy ME, Zuchner S. Copy number variations are a rare cause of non-CMT1A Charcot-Marie-Tooth disease. Journal of neurology. 2010;257:735–741. doi: 10.1007/s00415-009-5401-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okamoto Y, Goksungur MT, Pehlivan D, Beck CR, Gonzaga-Jauregui C, Muzny DM, Atik MM, Carvalho CM, Matur Z, Bayraktar S, Boone PM, Akyuz K, Gibbs RA, et al. Exonic duplication CNV of NDRG1 associated with autosomal-recessive HMSN-Lom/CMT4D. Genetics in medicine. 2014;16:386–394. doi: 10.1038/gim.2013.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pehlivan D, Beck CR, Okamoto Y, Harel T, Akdemir ZH, Jhangiani SN, Withers MA, Goksungur MT, Carvalho CM, Czesnik D, Gonzaga-Jauregui C, Wiszniewski W, Muzny DM, et al. The role of combined SNV and CNV burden in patients with distal symmetric polyneuropathy. Genetics in medicine. 2015 doi: 10.1038/gim.2015.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Slater H, Bruno D, Ren H, La P, Burgess T, Hills L, Nouri S, Schouten J, Choo KH. Improved testing for CMT1A and HNPP using multiplex ligation-dependent probe amplification (MLPA) with rapid DNA preparations: comparison with the interphase FISH method. Human mutation. 2004;24:164–171. doi: 10.1002/humu.20072. [DOI] [PubMed] [Google Scholar]
- 37.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu ZJ, Li HF, Tan GH, Tao QQ, Ni W, Cheng XW, Xiong ZQ, Wu ZY. Identify mutation in amyotrophic lateral sclerosis cases using HaloPlex target enrichment system. Neurobiology of aging. 2014;35(2881):e2811–2885. doi: 10.1016/j.neurobiolaging.2014.07.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.