

Abstract
Hippi functions as an adapter protein that mediates pro-apoptotic signaling from poly-glutamine-expanded huntingtin, an established cause of Huntington disease, to the extrinsic cell death pathway. To explore other functions of Hippi we generated Hippi knock-out mice. This deletion causes randomization of the embryo turning process and heart looping, which are hallmarks of defective left–right (LR) axis patterning. We report that motile monocilia normally present at the surface of the embryonic node, and proposed to initiate the break in LR symmetry, are absent on Hippi−/− embryos. Furthermore, defects in central nervous system development are observed. The Sonic hedgehog (Shh) pathway is downregulated in the neural tube in the absence of Hippi, which results in failure to establish ventral neural cell fate. Together, these findings demonstrate a dual role for Hippi in cilia assembly and Shh signaling during development, in addition to its proposed role in apoptosis signal transduction in the adult brain under pathogenically stressful conditions.
Keywords: Cilia, Hippi, Huntington disease, Left–right patterning, Sonic hedgehog
Introduction
During embryonic development, left–right (LR) asymmetry is the last of the three developmental axes to be specified, after dorso-ventral and anterior–posterior. The mechanism triggering the initial break in LR symmetry remains unclear. In Xenopus, asymmetrical phosphorylation of syndecan-2 and H+/K+-ATPase pump activity prior to gastrulation have been proposed as mechanisms initiating the break in LR symmetry (Kramer et al., 2002; Levin et al., 2002). Current models in mammals, however, suggest that the node plays a critical role in the early initiating events establishing LR patterning (reviewed by Hirokawa et al., 2006; Raya and Belmonte, 2006; Shiratori and Hamada, 2006). The node is a depression at the anterior end of the primitive streak in the early embryo where each cell features a motile monocilium. The beating of these cilia has been shown to produce a leftwards flow of fluid in the node region which is hypothesized to concentrate a morphogen on the left side of the embryo, triggering a signaling cascade (Nonaka et al., 1998). However, the exact identity of the morphogen has been elusive. An alternative model invokes mechanodetection of the directed nodal flow through non-motile sensory cilia in the nodal periphery (Brueckner, 2001; Tabin and Vogan, 2003). Ultimately, node function transfers asymmetrical signals to the lateral plate mesoderm (LPM) and consequently results in asymmetrical expression of the Nodal pathway genes involved in LR patterning (Nonaka et al., 1998). Genes expressed in the left LPM include Nodal, Lefty-2 and Pitx2. Axial (midline) structures of the embryo including the notochord, which is derived from the node, and the floorplate of the neural tube constitute a signaling barrier preventing transfer of information from left to right LPM, thus maintaining the integrity of the LR pattern (Danos and Yost, 1996; Meno et al., 1998).
During early vertebrate development, Sonic hedgehog (Shh) is first expressed in the node and the axial mesoderm, and later in the anterior definitive endoderm and the floorplate of the neural tube (Echelard et al., 1993). However, its role in LR patterning is complex. In the chick, Shh is asymmetrically expressed on the left side of the node (Hensen’s node). Exogenous expression of Shh on the right side induces Nodal expression and disrupt normal LR patterning, suggesting that Shh lies upstream of Nodal (Levin et al., 1995, 1997). In the mouse, however, Shh is not asymmetrically expressed, but in combination with Indian Hedgehog (Ihh), it regulates expression of Gdf1 and Nodal in the node (Zhang et al., 2001), both of which have been proposed as nodal flow morphogens. Recently, Shh, along with retinoic acid (RA), was found to be associated with nodal vesicular parcels (NVPs) (Tanaka et al., 2005). NVPs are membrane-sheathed particles that are secreted from cells within the node into the surrounding fluid in response to FGF signals. Evidence suggests that NVPs carrying Shh and RA are to be transported by the nodal flow and shatter near the left side of the node. This model proposes that both Shh and RA act coordinately as morphogens triggering the break of LR symmetry in mammals. At later stages, Shh is required for formation of the midline barrier through regulation of Lefty-1, an antagonist of Nodal (Tsukui et al., 1999). Thus, Shh is implicated both in establishing LR asymmetry in the node and maintaining LR patterning in the midline.
In addition to its role in midline barrier formation, notochord-derived Shh is required to induce the formation of the floorplate in the ventral neural tube (reviewed in Jessell and Sanes, 2000). Exogenous expression of Shh in the neural tube is sufficient to induce an ectopic floorplate while loss of Shh function results in embryo lacking a floorplate. Through activation of the transcription factor Gli2 in the overlying neural plate, Shh directly regulates expression of the forkhead transcription factor Hnf3β (also known as Foxa2, Ding et al., 1998; Matise et al., 1998; Sasaki et al., 1997) which in turn upregulates Shh expression in the floorplate (Jeong and Epstein, 2003). Moreover, Shh from the floorplate regulates dorso-ventral patterning in the neural tube including neural tube closure and cell fate specification (Ericson et al., 1995; Ybot-Gonzalez et al., 2002). The gradient of Shh signals along the neural tube dorso-ventral axis contributes to the differentiation of various neuronal cell types which populate the CNS (Jessell and Sanes, 2000). Shh is also involved in the development of many other tissues such as the limbs and somites (Echelard et al., 1993; Ingham and McMahon, 2001). In adult tissues, Shh is expressed in the brain, cerebellum, stomach and intestine (Traiffort et al., 1998, 2001).
Hippi (huntingtin interacting protein-1 protein interactor, also known as IFT57) was originally reported to contribute to the neuronal apoptotic cell death occurring in Huntington disease (HD) (Gervais et al., 2002). HD is a hereditary neurodegenerative disease characterized by chorea, personality changes and dementia resulting from cellular dysfunction that ultimately leads to cell death of cortical and striatal neurons (Vonsattel et al., 1985). Cell death occurs by an apoptotic mechanism as a result of polyglutamine (polyQ) expansion in the protein huntingtin (htt). This expansion abrogates the normal interaction of htt with the htt-interacting protein-1 (HIP1) and in turn favors increased interaction between HIP1 and Hippi (Gervais et al., 2002; Kalchman et al., 1997; Wanker et al., 1997). The HIP1-Hippi complex can activate caspase-8, one of the initiator proteases of apoptosis, and thus trigger cell death.
Hippi is orthologous to the Chlamydomonas intraflagellar transport protein IFT57 and has been shown to interact with an intraflagellar transport (IFT) protein complex in adult mouse ciliated cells (Baker et al., 2003). Its function in this latter context has not been studied. Studies in model organisms such as Drosophila melanogaster, Caenorhabditis elegans, and Chlamydomonas have demonstrated that the conserved intra-flagellar transport proteins are required for assembly, motility, and sensory function of flagella or cilia. Cilia lack the protein-synthetic machinery found in the cytoplasm, and accordingly, all structural components, and any other protein cargo to be transported to or from the tip of the cilium, are dependent on IFT (Scholey, 2003). IFT proteins form complexes incorporating anterograde motors of the kinesin family which drive transport of IFT particles to the axonemal tip and retrograde motors of the dynein family which drive transport towards the cell body. Ciliary maintenance reflects the dynamic equilibrium between these ongoing antagonistic processes.
To elucidate the normal and pathological functions of Hippi, we generated mice lacking its expression. We report here that Hippi has a role in LR axis patterning through its essential function in node cilia assembly. Moreover, we demonstrate that Hippi is also essential for Shh signaling in the neural tube. Hippi thus functions in essential steps in early embryonic development in addition to its role in HD pathogenesis.
Materials and methods
Generation of Hippi-null mice
pKO Scrambler NTKV-1905 (Stratagene) was used as the backbone vector to generate two Hippi-targeting constructs and the β-galactosidase cassette was retrieved from the pCMVβ vector (Clontech). Hippi homology regions were generated by PCR using 129S1/SvImJ liver genomic DNA as a template. Linearized constructs were electroporated in ES cells (mEMS128) derived from 129S1/SvImJ (JAX® 002448) mice (Festing et al., 1999; Simpson et al., 1997), a strain chosen from amongst the 129 substrains for its relative breeding success, black eyes, and suitability for behavior analysis (Hossain et al., 2004; Young et al., 2002). Positive clones were confirmed both by PCR and Southern blot. ES cells were injected into C57BL/6J blastocysts and resulting male chimeras were bread to C57BL/6J females to obtain Hippi+/− mice which were then intercrossed to obtain Hippi−/− mice on a mixed C57BL/6J-129S1/SvImJ genetic background. Embryos were considered 0.5 days post coitum at noon of the day of detection of the vaginal plug. Morphology and the number of somites was also used to stage embryos as appropriate. All experiments were performed in accordance with the UBC Committee on Animal Care guidelines.
Genotyping
For E7.5 to E8.0 embryos, cultured conceptus-derived trophoblast giant cells were lysed for genotyping. For E8.5 and older embryos, the yolk sac was collected and tail clips or ear punches were used for weaned mice. All samples were lysed in 100 mM Tris–HCl pH8.5, 200 mM NaCl, 5 mM EDTA, 0.2% SDS and 0.1 mg/ml proteinase K (Roche) for 2 to 16 h at 55 to 65°C. Proteinase K was inactivated by incubating samples for 20 min at 95°C. Samples were diluted 1:5 or 1:10 in PCR grade water and 1 μl was used in a 25 μl PCR reaction using the Qiagen PCR kit. Western blotting to confirm lack of Hippi protein in Hippi−/− embryos was performed as previously described (Houde et al., 2004) with the polyclonal affinity-purified anti-Hippi antibody (Gervais et al., 2002).
In situ hybridization
To prepare digoxigenin-labeled complementary RNA probes, standard transcription reactions were prepared using Roche cRNA transcription kit reagents. Probes were purified by centrifugation through a ProbeQuant G-50 microcolumn (Amersham) and then mixed 1:20 with whole-mount hybridization buffer (50% formamide, 5× SSC, 1% SDS, 50 μg/ml yeast tRNA, 50 μg/ml heparin) and stored at −20°C. Embryos were dissected in cold PBS and fixed in 4% paraformaldehyde in PBS at 4°C overnight. In situ hybridization was carried out following standard protocols. The probe to detect Hippi mRNA was obtained by BamHI digest of the I.M.A.G.E clone ID: 1243273. This fragment includes some of the 3′ end of the ORF and the 3′UTR totaling 686 nucleotides.
Embedding and sectioning
Stained embryos were refixed in 4% paraformaldehyde, 0.2% glutaraldehyde in PBS for 20 min. All steps were performed at 4°C. Embryos were washed 3 times for 15 min in PBS and incubated in 15% sucrose in PBS for 1 h, in 30% sucrose in PBS overnight and in OCT (Sakura Fineteck) for 1 h. Embryos were embedded in OCT in plastic molds on dry ice. 12- to 50-μm cryostat sections were mounted on poly-D-lysine coated slides with Vectashield water-based mounting media (Vector Laboratories).
Scanning electron microscopy
E7.75 to E8.0 embryos were dissected in room temperature PBS and transferred to SEM fix solution (2.5% glutaraldehyde in 0.1 M cacodylate pH 7.3) for 1 h at room temperature and then kept at 4°C for up to 36 h. Embryos were rinsed once and then kept in 0.1 M cacodylate pH 7.3 at 4°C for up to 7 days. Embryos were transferred to microporous capsules (Conemco) to facilitate transfer between the following solutions: 1% osmium in 0.1 M cacodylate pH 7.3, deionized water, 50%, 70%, 95% and 100% ethanol. One- or two-minute incubations were carried out in each solution using a Ted Pella microwave oven. Samples were then dried in a Balzers Union CPD 020 critical point dryer with 6 liquid exchanges. Samples were mounted and sputter coated with gold/palladium (Nanotech SEMPrepII Sputter Coater) for observation with a Hitachi S4700 scanning electron microscope.
Results
Targeted deletion of Hippi
To further investigate the in vivo function of Hippi, we generated Hippi-null mice. Two targeting constructs were engineered to generate mice lacking the expression of the Hippi protein following homologous recombination in ES cells. The first 95 codons of the Hippi open reading frame including its ATG start site were targeted for replacement with either a neomycin (neo) resistance cassette alone or with the β-galactosidase (β-gal) gene (Fig. 1A). Correctly targeted ES cell clones obtained with each gene-targeting construct were identified by PCR and Southern blotting and used to establish two independent lines of Hippi knock-out mice which showed identical phenotype.
Fig. 1.

Hippi targeted deletion. (A) Two targeting vectors were generated resulting in deletion of exon 1, intron A and part of exon 2 of the Hippi locus and insertion of either a neomycin resistance cassette alone (construct 1) or along with the β-galactosidase gene (construct 2). Black boxes are exons, dashed boxes are the inserted cassettes and capital letters indicate introns. (B) Western blot of protein extracts obtained from whole E9.5 embryos and probed with an affinity-purified anti-Hippi antibody showing Hippi expression in Hippi+/+ and Hippi+/−, but no expression in Hippi−/− extracts.
Hippi mutants, homozygous for the deletion, could not be detected in newborn mice from Hippi+/− intercrosses. Therefore, we analyzed embryos obtained from timed pregnancies. At E9.5, a Mendelian ratio of wildtype, heterozygous and homozygous embryos was observed (40 +/+:102 +/−:47 −/−). Hippi−/− embryos at E10.5 were often in a state of resorbtion and later stages were rarely observed suggesting that the Hippi mutant embryos die predominantly prior to E10.5. Western blot analysis of protein extracts from E9.5 embryos shows that whereas Hippi+/+ and Hippi+/− embryos both express the Hippi protein, the Hippi−/− embryos do not express any as detected with an affinity-purified antibody raised against the C-terminal end of Hippi (Gervais et al., 2002), a region not targeted by the construct (Fig. 1B).
Morphological defects in Hippi-null embryos
At E9.5, Hippi−/− embryos are visually abnormal and feature a spectrum of moderate to severe development defects including growth retardation, randomized turning, abnormal neural tube morphology and abnormal cardiac morphology (Fig. 2A for wildtype, B for moderate phenotype, C, D and E for severe phenotype). One of the most striking features of Hippi−/− embryos is that the turning process, by which the mouse embryo turns on itself to invert its germ layers and adopt the normal chordate fetal position, is delayed. In the most severe cases, turning is not initiated in Hippi−/− embryos at E9.5, a time at which turning should be completed. Wildtype embryos normally turn leftwards in this process, but when initiated, the turning direction is randomized in Hippi−/− embryos. Out of 40 E9.5 Hippi−/− embryos examined for their turning side, 11 turned leftwards, 11 towards their right side and 18 were severely distorted and turning direction could not be ascertained. In addition, various sizes of pericardial edema were observed on all Hippi−/− embryos (Figs. 2B, C and E) and heart looping sidedness was sometimes inverted (Fig. 2C, dotted line). Each one of these defects can have multiple explanations. However, the combination of these features suggests that the Hippi mutants have defects in left–right axis patterning.
Fig. 2.
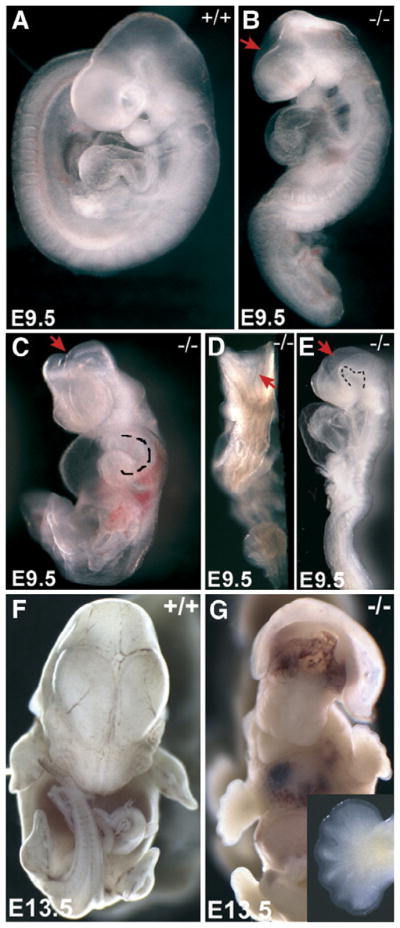
Hippi−/− embryos show severe development defects at mid-gestation. (A) Wildtype E9.5 embryo, lateral view. (B) Hippi−/− littermate, lateral view, same magnification as in panel A, showing an example of a mild phenotype with incomplete forebrain closure (arrow). (C–E) Hippi−/− embryos, ventral, dorsal and lateral views, same magnification as in panel A, showing examples of severe phenotypes. Note retardation in growth and turning, inverted heart looping (dotted line in panel C shows reverse right ventricule looping to the left side), pericardial edema, kinks in the spinal cord, retarded closure of the neural tube in the brain region (arrows), uneven closure of the spinal cord and abnormal cranial flexture (dotted line in panel E). (F) Wildtype E13.5 embryo, ventral view. (G) Ventral view of a Hippi−/− embryos at E13.5 exhibiting exencephaly, hypotelorism and abnormal maxillary processes. Insert shows forelimb with at least 6 digits.
Neural tube closure defects are observed in E9.5 Hippi−/− embryos, including opened brain (Figs. 2B, C, D and E, red arrows) and uneven “zipping” of the spinal cord (Fig. 2D). Closure of the neural tube at the brain level varies considerably between Hippi−/− embryos, but never completes and an unusual curvature of the floor of the cranial flexure is often observed (Fig. 2E, dotted line). These malformations at E9.5 suggest defects in neural tube development.
Interestingly, we were able to collect one mutant embryo which had survived until E13.5. That embryo featured exencephaly in the region above the presumed location of the midbrain, hypotelorism (narrowed eye spacing), failed fusion and reduced length of the maxillary processes (Fig. 2G) and polydactyly on both fore (Fig. 2G, insert) and hindlimbs (not shown) as compared to wildtype littermates (Fig. 2F).
Hippi expression pattern
To determine the correlation between Hippi expression and phenotypic abnormalities in Hippi−/− embryos, Hippi mRNA expression pattern was assessed by in situ hybridization. At E7.0, 7.25 and 7.5, Hippi expression is ubiquitous through the epiblast (Figs. 3A to D). Expression is detected in mesoderm and most strongly in ectoderm, but not in endoderm (Fig. 3C). The region of the node (arrows in Figs. 3A to D), a depression at the surface of the embryo proposed to have a role in left–right axis patterning, also expresses high levels of Hippi messenger. At E8.5, Hippi is widely expressed except in the heart (Fig. 3E). Stronger expression is observed in the anterior midline (arrow in Fig. 3F), the forebrain and the somites (Fig. 3F). Strong Hippi expression remains in the forebrain at E9.5 and E10.5 (Figs. 3G and I) and extends to all regions of the neural tube. At those stages, high expression is also found in the branchial arches and in the limb buds. Embryo section at E9.5 (Fig. 3H) also shows expression throughout the neural tube and the mesoderm (Fig. 3H, see n and m respectively), but not in the surface ectoderm (e). The strongest expression is observed on the luminal edge of the neural tube (Fig. 3H, arrowheads) and in the ventral foregut (arrows). These patterns of expression during early embryogenesis are in accordance with the potential role of Hippi in LR axis patterning and neural tube development.
Fig. 3.

Hippi expression pattern. Whole-mount in situ hybridization shows Hippi mRNA expression pattern. (A–D) From E7.0 to E7.5, Hippi is expressed most strongly in the ectoderm and in the region of the node (arrows). (E, F) At E8.5, expression is detected mainly in the forebrain and the midline (arrow). (G) At E9.5, the highest expression of Hippi is detected in the forebrain, branchial arches and limb buds but also throughout the neural tube. (H) Section though a E9.5 embryo. Note expression in the neuroepithelium (n), mesoderm (m), luminal neural tube (arrowheads) and ventral foregut (arrows) but not in the ectoderm (e). (I) Hippi expression pattern is maintained at E10.5.
Left–right axis patterning is defective in Hippi−/− embryos
The randomized turning process and abnormal heart looping observed in the Hippi−/− embryos may suggest a defect in LR axis patterning. LR axis formation is established shortly after node formation in early somite stage embryos. The earliest gene known to be asymmetrically expressed in the mouse embryo is Nodal. Nodal is a TGF-β family member that is initially expressed on both sides of the node in the mouse. At E8.0, expression in the node becomes enhanced on the left side and Nodal expression is induced on the left half of the lateral plate mesoderm (LPM). While in both Hippi+/+ and Hippi+/− embryos Nodal is strongly expressed in the left LPM (Figs. 4A and B), in Hippi−/− embryos Nodal expression is observed in both the left and the right LPM, indicating that LR patterning has been disrupted (Fig. 4C). Interestingly, expression of Nodal in the node region was reduced in Hippi mutants compared to wildtype and heterozygote embryos (compare Figs. 4A, B and C). In addition to Nodal, we examined two other genes, Lefty-2 and Pitx2 that are targets of Nodal signaling and are asymmetrically expressed in the LPM. Lefty-2 is a TGF-β family member expressed in the left LPM but not in the node (Fig. 4D). The transcription factor Pitx2 is expressed in the left LPM as well as symmetrically within the head mesenchyme (Fig. 4F). In Hippi−/− embryos, both Lefty-2 and Pitx2 are expressed symmetrically in the LPM (Figs. 4E and G), indicating the loss of LR asymmetry at this early stage, well before gross anatomical defects are observed.
Fig. 4.

Asymmetrical expression of LR axis patterning genes is lost in Hippi−/− embryos. Whole-mount in situ hybridization of E8.0 embryos. (A to C) Nodal is expressed bilaterally in the LPM in the 9 out of 9 Hippi−/− embryos observed, with downregulation around the node region. (D, E) Lefty-2 is expressed bilaterally in the 9 out of 9 Hippi−/− embryos observed and (F, G) Pitx2 is expressed bilaterally in the 9 out of 9 Hippi−/− embryos observed. (H, I) Brachyury is expressed in the midline in both wildtype and Hippi−/− embryos. (J, K) Lefty-1 is not expressed in the midline of the 5 out of 5 Hippi−/− embryos observed. (L, M) At E7.5–7.75, Hnf3β is expressed in the node and the midline in both wildtype and Hippi−/− embryos. For all genes, heterozygotes always stain as the wildtype embryos.
To further examine formation of the node and the notochord, the axial mesoderm that is derived from the node, we examined the expression of the T-box transcription factor Brachyury and the forkhead transcription factor Hnf3β. Hnf3β is expressed in the node and notochord and mice deficient in Hnf3β fail to form these structures (Ang and Rossant, 1994, Weinstein et al., 1994). Importantly, Hnf3β is expressed in early headfold stage (E7.75) Hippi−/− embryos, demonstrating that the node and notochord are specified in these embryos (Figs. 4L and M). Furthermore, Brachyury is expressed normally in both the wildtype and Hippi−/− embryos at E8.5 indicating that the node and notochord are maintained (Figs. 4H and I).
Due to the Nodal pathway bilateral gene expression in the LPM, we next examined expression of Lefty-1, a TGFβ family member that is expressed in the left prospective floorplate of the neural tube of embryos at 3–7 somites. Lefty-1 is thought to act as a midline barrier, preventing signals from diffusing from the left LPM to the right LPM (Meno et al., 1998). Lefty-1 expression was not observed in Hippi−/− embryos (Figs. 4J and K) suggesting that although the notochord is present, the midline may not function to prevent diffusion of signals from the left side to the right side of the embryo. This supports the observation that left LPM markers are bilaterally expressed in Hippi−/− embryos.
Hippi is essential for node cilia assembly
The break in LR asymmetry in the mouse is postulated to rely on the fluid flow towards the left side of the embryo generated by the beating monocilia of the node, although mechanistic details remain controversial (Nonaka et al., 1998). We thus investigated the integrity of the node structure in Hippi−/− embryos. Scanning electron microscopy of E8.0 embryos shows that Hippi−/− embryos exhibit a depression with similar location and gross morphology to the wildtype node (Figs. 5A and C). At higher magnification, however, we observed that monocilia present on Hippi+/+ embryos were absent from the surface of the Hippi−/− node cells (Figs. 5B and D). Thus, absence of nodal flow resulting from the loss of node cilia is a possible explanation for the severe LR patterning defects observed in Hippi−/− embryos.
Fig. 5.

Node monocilia are absent on Hippi−/− embryos. Scanning electron microscopy of the node region of E8.0 embryos. (A) 700× Magnification of the Hippi+/+ node. (B) 11,000× Magnification of the Hippi+/+ node showing monocilia. (C) 700× Magnification of the Hippi−/− node showing normal gross structure. (D) 11,000× Magnification of the Hippi−/− node. No cilia are present. Scale bars represent 25 μm in panels A and C and 1.7 μm in panels B and D.
Hippi is an essential component of the Sonic hedgehog pathway
In addition to LR patterning defects, Hippi−/− embryos fail to undergo correct neural tube closure. Interestingly, mutants for Smoothened (Smo) and Dispatched-1 (Disp1), two receptors of the Shh pathway, show some similar features to the E9.5 Hippi−/− embryo phenotype, both for LR patterning and neural tube closure defects (Caspary et al., 2002). In addition, exencephaly, hypotelorism and polydactyly similar to the phenotypes observed in the E13.5 Hippi−/− embryo are also associated with defective Shh signaling (Milenkovic et al., 1999; Wang et al., 2000). Therefore, we investigated whether the expression of genes reliant on an intact Shh signaling cascade are affected in the absence of Hippi expression.
Hnf3β and the Shh receptor Patched (Ptc) are both normally upregulated in the neural tube floorplate in response to Shh secretion from the underlying notochord (Goodrich et al., 1996; Roelink et al., 1995). In E8.5 and E9.5 Hippi−/− embryos, Hnf3β expression in the midline is downregulated compared to Hippi+/+ littermates (Figs. 6A and B, black arrowheads), although its expression in the developing gut (red arrowheads) seems unaffected. Moreover, Ptc expression in the neural tube at E8.5 and E9.5 is downregulated in Hippi−/− embryos (Fig. 6, compare C and E with D and F respectively). Neural tube expression of Shh in E9.5 Hippi−/− embryos is also downregulated (Figs. 6G and H) Collectively, these data suggest that the Shh signaling pathway is profoundly impaired in the absence of Hippi expression.
Fig. 6.

The Sonic hedgehog pathway is downregulated in the neural tube of Hippi−/− embryos. Whole-mount in situ hybridization for (A, B) Hnf3β (arrowheads: red for gut, black for neural tube) (C to F) Ptc and (G, H) Shh. All three genes are downregulated in the neural tube of Hippi−/− embryos. Panels A to D are E8.5 embryos, panels E to H are E9.5.
As Shh signaling contributes to determination of the dorso-ventral axis of the neural tube, we examined cell specification on cross sections of whole-mount in situ hybridized embryos. In E9.5 embryos, Shh is normally expressed in the notochord and induced secondarily by this notochord-derived signal within the floorplate, the most ventral region of the neural tube (Fig. 7A, arrow and arrowhead respectively). Strikingly, its expression is restricted to the notochord (Fig. 7B, arrow) in Hippi−/− embryos and is not detected in the ventral neuroepithelium (arrowhead). Correspondingly, in E9.5 embryos, Hnf3β is expressed the floorplate of wildtype embryos. However expression is not observed in that region in Hippi−/− embryos (Figs. 7C and D). The absence of Shh and Hnf3β in the neural tube suggests that the floorplate of the neural tube is not specified in the Hippi mutant embryos.
Fig. 7.

Neural tube ventralization is lost in Hippi−/− embryos. Whole-mount in situ hybridization sections with dotted lines highlighting neural tube borders. (A, B) Normal Shh expression in the floorplate (arrowheads) is lost in Hippi−/− embryos and remains only in the notochord (arrows). (C, D) Hnf3β expression is absent in Hippi−/− embryos floorplate (E, F) Pax6 lateral neural tube expression pattern observed in Hippi+/+ embryos extends to the ventral region (asterisk) in Hippi−/− tissue. (G, H) Isl-1 lateral ventral neural tube expression pattern (arrows) is lost in Hippi−/− embryos.
To confirm that the floorplate of the neural tube is not present in Hippi−/− embryos, we also examined the expression of Pax6 which is normally expressed in a discrete domain lying between dorsal and ventral cells in the lateral neural tube (Fig. 7E). In E9.5 Hippi−/− embryos, Pax6 expression extends to the ventral side of the neural tube (Fig. 7F, asterisks). Moreover, Isl1, a marker of motor neurons that are induced in the lateral ventral neural tube by the floorplate (Ericson et al., 1997) is absent in Hippi−/− embryos (Fig. 7G arrows and H). These results are consistent with a loss of ventral cell specification in the neural tube of Hippi−/− embryos and support an essential role of Hippi dorso-ventral axis patterning in the neural tube.
Discussion
Hippi is essential for cilia formation in the node
An increasing number of human and mouse abnormal phenotypes are attributable to mutations or disorders in genes leading to defective ciliary assembly or function. These include defects in ciliated tissues such as the trachea or cells such as spermatozoa, but also are implicated in correct development of the kidneys, pancreas, and liver, sensory cilia-containing organs such as the retinal photoreceptors, hair cells of the inner ear, and neurons of the olfactory system (Scholey, 2003).
During mouse embryogenesis, cilia are first observed in the node at E7.5. Here, we demonstrate that the earliest morphological defect observed in the Hippi mutants is the loss of cilia in the node, indicating that Hippi has an essential function in cilia formation. This is in accordance with previous studies showing that mutants for the closest Hippi homolog in C. elegans and in zebrafish have defective sensory and motile cilia (Haycraft et al., 2003; Kramer-Zucker et al., 2005; Tsujikawa and Malicki, 2004). Null mutations of other intraflagellar transport proteins also result in abnormal or complete absence of monocilia at the surface of the embryo node; the dynein motor subunit md2lic (Rana et al., 2004), Ift172 (wimple) (Huangfu et al., 2003), Ift88 (polaris/tg737/orpk) (Murcia et al., 2000), and the kinesin motor subunits Kif3A (Takeda et al., 1999) and Kif3B (Nonaka et al., 1998). Interestingly, in adult mouse ciliated cells, Hippi forms a complex with Ift88, Kif3A and Kif3B (Baker et al., 2003). Our study is thus further supporting the role of Hippi in IFT in mammalian cells.
Hippi mutant embryos exhibit left–right patterning defects
Currently, the only known function of cilia in the node is in the determination of the left–right axis in the embryo. Although the exact mechanism underlying this specification remains unclear, the nodal flow generated by the beating monocilia of the node results in asymmetrical gene expression in the LPM of the embryo (Nonaka et al., 1998). In keeping with the absence of cilia observed in the Hippi mutants, these embryos display morphological defects in LR patterning at E9.5, including randomized turning direction and abnormal heart looping. Moreover, genes that are normally expressed in the left LPM such as Nodal, Lefty-2, and Pitx2, are symmetrically expressed on both sides of the embryo in Hippi mutants, demonstrating that Hippi plays an essential role in LR patterning. Other IFT protein mutants such as Ift88 and Ift172 also show LR patterning defects and bilateral expression of LPM markers (Huangfu et al., 2003; Murcia et al., 2000). In addition, the majority of embryos with mutations in Dnchc2 or md2lic, which have stunted or abnormal cilia, also show bilateral expression (Huangfu and Anderson, 2005; Rana et al., 2004).
Bilateral expression of lateral plate markers may also be due to diffusion of signals from the left to the right LPM of the embryo. Lefty-1 has been proposed to restrict the expression of Nodal and Lefty-2 to the left LPM. In Hippi−/− embryos, Lefty-1 is not expressed in the prospective floorplate of the neural tube, suggesting that the midline is not functioning as a barrier. Interestingly, Lefty-1−/− embryos survive later (some newborns are found) (Meno et al., 1998) than Hippi mutants. As well, mutants mentioned above with immotile or stunted cilia which cannot generate nodal flow show less severe phenotypes than Hippi mutants. The Hippi−/− phenotype is thus likely to be a consequence of additive effects on both nodal flow and midline barrier establishment rather than on only one of these phenomena.
Hippi mutants display defects in Shh signaling
Hippi−/− embryos display several phenotypic abnormalities observed in embryos carrying mutations in components of the Shh signaling pathway, including left–right and dorsal–ventral patterning defects. LR patterning defects have been observed in Shh and Smo mutant embryos. Similar to the Hippi mutant, embryos lacking Shh express Pitx2 in both the left and right LPM. Interestingly, Ihh mutant embryos displays normal expression of Pitx2 in the left LPM. However, combined Ihh/Shh compound mutants and Smo mutants display a complete absence of LPM markers. This suggests that Hippi mutants are more closely related to Shh mutants (Zhang et al., 2001).
Extensive studies have demonstrated that Shh arising from the floorplate of the neural tube provides a morphogenic signal that directs the normal specification of cells in the ventral regions of the neural tube (Wijgerde et al., 2002). Furthermore, formation of the floorplate is dependent on Shh signals derived from the notochord. In Hippi−/− embryos, we observed a loss of dorsal–ventral patterning in the neural tube as indicated by the loss of Isl1 expressing motor neurons in the ventral lateral region, and expansion of the Pax6 expression domain into the ventral neural tube. Furthermore, we observed an absence of floorplate expression of Hnf3β, Lefty-1 and Shh suggesting that the floorplate is not specified in these mutants. Significantly, expression of Hnf3β and Lefty-1 has been shown to be regulated by Shh in the floorplate (Sasaki et al., 1997; Tsukui et al., 1999). Furthermore, expression of the Shh receptor, Ptc, another direct transcriptional target for Shh signaling (Goodrich et al., 1996), is downregulated in the Hippi−/− embryos. Together theses results suggest that Hippi functions in the Shh signaling pathway.
A connection between cilia and Shh signaling has only recently begun to emerge. Similar to Hippi mutants, Ift88, Ift172 and their associated kinesin and dynein motors mutants exhibit downregulated Shh pathway activity (Huangfu et al., 2003; Liu et al., 2005; Rana et al., 2004; Takeda et al., 1999). Although the mechanism is unclear, Ift88 and Ift172 were implicated in regulating the proteolytic cleavage of the transcription factor Gli3 to produce the repressor form of the protein, suggesting that IFTs may play a role in regulating both the positive and negative functions of Gli proteins (Huangfu and Anderson, 2005; Liu et al., 2005). Intriguingly, recent experiments have demonstrated that Shh-dependent localization of the Shh co-receptor Smo to cilia is essential for Smo function (Corbit et al., 2005). Moreover, Smo cilia localization is dependent on IFT as the Dnchc2 retrograde motor component deletion abrogates it (May et al., 2005). It is unlikely, however, that Hippi deletion completely blocks Smo function as Smo−/− embryos have a phenotype overlapping but distinct from Hippi mutants, including non-looping of the heart tube, closure of the neural tube accompanied by holoprosencephaly and absence of Nodal pathway genes expression (Zhang et al., 2001). Hippi may however partially restrict Smo localization, or impinge negatively on downstream Smo targets including the Gli proteins which have both activating and repressing effects on Shh signaling (Altaba, 1999; Sasaki et al., 1999).
In the Shh pathway, Ptc, the trans-membrane receptor of Shh, normally inhibits Smo signaling. The Shh pathway is activated when Shh binds to Ptc, which releases Smo inhibition, allowing the upregulation of Smo downstream targets, including Ptc itself, the Gli genes and Hnf3β. Smo and Ptc are found both at the cell surface and in intracellular vesicles. In vertebrates, Ptc and Smo are internalized together following Shh binding and whereas the Shh/Ptc complex is targeted to the lysosome for degradation, Smo remains in endosomes where it is free to mediate its downstream effects (Incardona et al., 2002). In contrast, in Drosophila, Ptc normally causes Smo to be localized to internal vesicles and Hedgehog stimulation triggers Smo relocalization and stabilization at the plasma membrane (Zhu et al., 2003) where cilia are found. In both cases, the subcellular localization of Ptc and Smo is crucial for proper signaling. Moreover, the recycling endosome-associated protein Rab23 regulates subcellular localization of some Shh pathway components lying between Smo and the Gli proteins (Eggenschwiler et al., 2006) again supporting an important role for intracellular trafficking in Shh signaling.
Interestingly, Hippi and some kinesin and dynein subunits involved in IFT also localize to cytoplasmic structures, raising the possibility that at least part of the machinery used for intraflagellar transport could also be involved in cytoplasmic transport (Gervais et al., 2002; Grissom et al., 2002; Vaisberg et al., 1996; Yamazaki et al., 1995). Cytoplasmic localization and function of IFT proteins have been little studied, but preliminary results indicate a role for Hippi in clathrin-coated vesicle trafficking (Saleh, M., personal communication). Subcellular spatial regulation of Shh signaling remains an important and challenging aspect of this pathway to elucidate.
Could impaired Shh signaling contribute to HD pathology?
In Huntington disease, Hippi function is altered by increased interaction with HIP-1, which triggers apoptosis by activating caspase-8 (Gervais et al., 2002). Other mechanisms, including neurotrophic deprivation and transcription downregulation, have also been reported to contribute to neuronal cell death in HD (Li and Li, 2004). Interestingly, Shh, Ptc and Smo are broadly expressed in the adult rat brain in several regions including the striatum and the cortex (Traiffort et al., 1998, 2001) the primary sites of neurodegeneration in HD and several studies implicate Shh in mediating survival and proliferative effects in adult brain. For example, Shh has been shown to be a trophic factor for post-mitotic dopaminergic and GABAergic neurons (Miao et al., 1997), to promote neurogenesis from stem cells in adult mice (Palma et al., 2005), and to improve behavior in a Parkinson’s disease rat model (Tsuboi and Shults, 2002). Further studies are needed to determine if Hippi abnormal interaction with the HIP-1 trafficking molecule in HD results in sequestering Hippi away from its normal function, which could possibly affect the Shh pathway and its pro-survival signals, providing an additional mechanism in the pathogenesis of HD.
In summary, Hippi appears to have a dual role in vivo. During embryonic development it is critical for LR axis patterning owing to its essential role as an IFT protein, and to neural development due to its critical function in Shh signaling. In the adult brain, Hippi facilitates neurodegeneration in HD through a novel apoptotic pathway. The question remains wether Hippi may also contribute to HD through perturbation of Shh survival signaling.
Acknowledgments
We would like to thank Nagat Bissada, Lisa Bertram, Tara Davidson and Lonna Miller for the animal work. Thanks also to Karin Schuster Gossler of the Cell biology Service at The Jackson Laboratory for ES cell derivation. We also acknowledge Elaine Humphrey and the BioImaging Facility at the University of British Columbia in Vancouver, Canada, where the scanning electron microscopy was performed, as well as Andrew McMahon, Brigid Hogan, Hiroshi Hamada, Sumihare Noji, Peter Guss and Philip Beachy for providing in situ hybridization probes. C.H. was supported by the Canadian Institutes of Health Research (CIHR). R.J.D. is supported by the Michael Smith Foundation for Health Research (MSFHR). E.M.S. is a Canada Research Chair in Genetics and Behaviour and PAH is a Canadian Institutes of Health Research (CIHR) New Investigator and a MSFHR Scholar. This work was funded in part by grants to PAH from the National Cancer Institute of Canada with funds from the Terry Fox Foundation and the CIHR Rx and D–HRF Research Program.
References
- Altaba A. Gli proteins encode context-dependent positive and negative functions: implications for development and disease. Development. 1999;126:3205–3216. doi: 10.1242/dev.126.14.3205. [DOI] [PubMed] [Google Scholar]
- Ang SL, Rossant J. HNF-3 beta is essential for node and notochord formation in mouse development. Cell. 1994;78:561–574. doi: 10.1016/0092-8674(94)90522-3. [DOI] [PubMed] [Google Scholar]
- Baker SA, Freeman K, Luby-Phelps K, Pazour GJ, Besharse JC. IFT20 links kinesin II with a mammalian intraflagellar transport complex that is conserved in motile flagella and sensory cilia. J Biol Chem. 2003;278:34211–34218. doi: 10.1074/jbc.M300156200. [DOI] [PubMed] [Google Scholar]
- Brueckner M. Cilia propel the embryo in the right direction. Am J Med Genet. 2001;101:339–344. doi: 10.1002/1096-8628(20010715)101:4<339::aid-ajmg1442>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Caspary T, Garcia-Garcia MJ, Huangfu D, Eggenschwiler JT, Wyler MR, Rakeman AS, Alcorn HL, Anderson KV. Mouse Dispatched homolog1 is required for long-range, but not juxtacrine, Hh signaling. Curr Biol. 2002;12:1628–1632. doi: 10.1016/s0960-9822(02)01147-8. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- Danos MC, Yost HJ. Role of notochord in specification of cardiac left–right orientation in zebrafish and Xenopus. Dev Biol. 1996;177:96–103. doi: 10.1006/dbio.1996.0148. [DOI] [PubMed] [Google Scholar]
- Ding Q, Motoyama J, Gasca S, Mo R, Sasaki H, Rossant J, Hui CC. Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development. 1998;125:2533–2543. doi: 10.1242/dev.125.14.2533. [DOI] [PubMed] [Google Scholar]
- Echelard Y, Epstein DJ, St Jacques B, Shen L, Mohler J, McMahon JA, McMahon AP. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75:1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- Eggenschwiler JT, Bulgakov OV, Qin J, Li T, Anderson KV. Mouse Rab23 regulates hedgehog signaling from smoothened to Gli proteins. Dev Biol. 2006;290:1–12. doi: 10.1016/j.ydbio.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Ericson J, Muhr J, Placzek M, Lints T, Jessell TM, Edlund T. Sonic hedgehog induces the differentiation of ventral forebrain neurons: a common signal for ventral patterning within the neural tube. Cell. 1995;81:747–756. doi: 10.1016/0092-8674(95)90536-7. [DOI] [PubMed] [Google Scholar]
- Ericson J, Rashbass P, Schedl A, Brenner-Morton S, Kawakami A, van Heyningen V, Jessell TM, Briscoe J. Pax6 controls progenitor cell identity and neuronal fate in response to graded Shh signaling. Cell. 1997;90:169–180. doi: 10.1016/s0092-8674(00)80323-2. [DOI] [PubMed] [Google Scholar]
- Festing MF, Simpson EM, Davisson MT, Mobraaten LE. Revised nomenclature for strain 129 mice. Mamm Genome. 1999;10:836. doi: 10.1007/s003359901099. [DOI] [PubMed] [Google Scholar]
- Gervais FG, Singaraja R, Xanthoudakis S, Gutekunst CA, Leavitt BR, Metzler M, Hackam AS, Tam J, Vaillancourt JP, Houtzager V, et al. Recruitment and activation of caspase-8 by the Huntingtin-interacting protein Hip-1 and a novel partner Hippi. Nat Cell Biol. 2002;4:95–105. doi: 10.1038/ncb735. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Johnson RL, Milenkovic L, McMahon JA, Scott MP. Conservation of the hedgehog/patched signaling pathway from flies to mice: induction of a mouse patched gene by Hedgehog. Genes Dev. 1996;10:301–312. doi: 10.1101/gad.10.3.301. [DOI] [PubMed] [Google Scholar]
- Grissom PM, Vaisberg EA, McIntosh JR. Identification of a novel light intermediate chain (D2LIC) for mammalian cytoplasmic dynein 2. Mol Biol Cell. 2002;13:817–829. doi: 10.1091/mbc.01-08-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft CJ, Schafer JC, Zhang Q, Taulman PD, Yoder BK. Identification of CHE-13, a novel intraflagellar transport protein required for cilia formation. Exp Cell Res. 2003;284:251–263. doi: 10.1016/s0014-4827(02)00089-7. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Tanaka Y, Okada Y, Takeda S. Nodal flow and the generation of left–right asymmetry. Cell. 2006;125:33–45. doi: 10.1016/j.cell.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Hossain SM, Wong BK, Simpson EM. The dark phase improves genetic discrimination for some high throughput mouse behavioral phenotyping. Genes Brain Behav. 2004;3:167–177. doi: 10.1111/j.1601-183x.2004.00069.x. [DOI] [PubMed] [Google Scholar]
- Houde C, Banks KG, Coulombe N, Rasper D, Grimm E, Roy S, Simpson EM, Nicholson DW. Caspase-7 expanded function and intrinsic expression level underlies strain-specific brain phenotype of caspase-3-null mice. J Neurosci. 2004;24:9977–9984. doi: 10.1523/JNEUROSCI.3356-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. From the cover: cilia and hedgehog responsiveness in the mouse. Proc Natl Acad Sci U S A. 2005;102:11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–3087. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- Incardona JP, Gruenberg J, Roelink H. Sonic hedgehog induces the segregation of patched and smoothened in endosomes. Curr Biol. 2002;12:983–995. doi: 10.1016/s0960-9822(02)00895-3. [DOI] [PubMed] [Google Scholar]
- Jeong Y, Epstein DJ. Distinct regulators of Shh transcription in the floor plate and notochord indicate separate origins for these tissues in the mouse node. Development. 2003;130:3891–3902. doi: 10.1242/dev.00590. [DOI] [PubMed] [Google Scholar]
- Jessell TM, Sanes JR. Development. The decade of the developing brain. Curr Opin Neurobiol. 2000;10:599–611. doi: 10.1016/s0959-4388(00)00136-7. [DOI] [PubMed] [Google Scholar]
- Kalchman MA, Koide HB, McCutcheon K, Graham RK, Nichol K, Nishiyama K, Kazemi-Esfarjani P, Lynn FC, Wellington C, Metzler M, et al. HIP1, a human homologue of S. cerevisiae Sla2p, interacts with membrane-associated huntingtin in the brain. Nat Genet. 1997;16:44–53. doi: 10.1038/ng0597-44. [DOI] [PubMed] [Google Scholar]
- Kramer KL, Barnette JE, Yost HJ. PKCgamma regulates syndecan-2 inside-out signaling during Xenopus left–right development. Cell. 2002;111:981–990. doi: 10.1016/s0092-8674(02)01200-x. [DOI] [PubMed] [Google Scholar]
- Kramer-Zucker AG, Olale F, Haycraft CJ, Yoder BK, Schier AF, Drummond IA. Cilia-driven fluid flow in the zebrafish pronephros, brain and Kupffer’s vesicle is required for normal organogenesis. Development. 2005;132:1907–1921. doi: 10.1242/dev.01772. [DOI] [PubMed] [Google Scholar]
- Levin M, Johnson RL, Stern CD, Kuehn M, Tabin C. A molecular pathway determining left–right asymmetry in chick embryogenesis. Cell. 1995;82:803–814. doi: 10.1016/0092-8674(95)90477-8. [DOI] [PubMed] [Google Scholar]
- Levin M, Pagan S, Roberts DJ, Cooke J, Kuehn MR, Tabin CJ. Left/right patterning signals and the independent regulation of different aspects of situs in the chick embryo. Dev Biol. 1997;189:57–67. doi: 10.1006/dbio.1997.8662. [DOI] [PubMed] [Google Scholar]
- Levin M, Thorlin T, Robinson KR, Nogi T, Mercola M. Asymmetries in H+/K+-ATPase and cell membrane potentials comprise a very early step in left–right patterning. Cell. 2002;111:77–89. doi: 10.1016/s0092-8674(02)00939-x. [DOI] [PubMed] [Google Scholar]
- Li SH, Li XJ. Huntingtin–protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004;20:146–154. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132:3103–3111. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- Matise MP, Epstein DJ, Park HL, Platt KA, Joyner AL. Gli2 is required for induction of floor plate and adjacent cells, but not most ventral neurons in the mouse central nervous system. Development. 1998;125:2759–2770. doi: 10.1242/dev.125.15.2759. [DOI] [PubMed] [Google Scholar]
- May SR, Ashique AM, Karlen M, Wang B, Shen Y, Zarbalis K, Reiter J, Ericson J, Peterson AS. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev Biol. 2005;287:378–389. doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- Meno C, Shimono A, Saijoh Y, Yashiro K, Mochida K, Ohishi S, Noji S, Kondoh H, Hamada H. Lefty-1 is required for left–right determination as a regulator of lefty-2 and nodal. Cell. 1998;94:287–297. doi: 10.1016/s0092-8674(00)81472-5. [DOI] [PubMed] [Google Scholar]
- Miao N, Wang M, Ott JA, D’Alessandro JS, Woolf TM, Bumcrot DA, Mahanthappa NK, Pang K. Sonic hedgehog promotes the survival of specific CNS neuron populations and protects these cells from toxic insult In vitro. J Neurosci. 1997;17:5891–5899. doi: 10.1523/JNEUROSCI.17-15-05891.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenkovic L, Goodrich LV, Higgins KM, Scott MP. Mouse patched1 controls body size determination and limb patterning. Development. 1999;126:4431–4440. doi: 10.1242/dev.126.20.4431. [DOI] [PubMed] [Google Scholar]
- Murcia NS, Richards WG, Yoder BK, Mucenski ML, Dunlap JR, Woychik RP. The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left–right axis determination. Development. 2000;127:2347–2355. doi: 10.1242/dev.127.11.2347. [DOI] [PubMed] [Google Scholar]
- Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y, Kido M, Hirokawa N. Randomization of left–right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95:829–837. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- Palma V, Lim DA, Dahmane N, Sanchez P, Brionne TC, Herzberg CD, Gitton Y, Carleton A, Alvarez-Buylla A, Altaba A. Sonic hedgehog controls stem cell behavior in the postnatal and adult brain. Development. 2005;132:335–344. doi: 10.1242/dev.01567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana AA, Barbera JP, Rodriguez TA, Lynch D, Hirst E, Smith JC, Beddington RS. Targeted deletion of the novel cytoplasmic dynein mD2LIC disrupts the embryonic organiser, formation of the body axes and specification of ventral cell fates. Development. 2004;131:4999–5007. doi: 10.1242/dev.01389. [DOI] [PubMed] [Google Scholar]
- Raya A, Belmonte JC. Left–right asymmetry in the vertebrate embryo: from early information to higher-level integration. Nat Rev, Genet. 2006;7:283–293. doi: 10.1038/nrg1830. [DOI] [PubMed] [Google Scholar]
- Roelink H, Porter JA, Chiang C, Tanabe Y, Chang DT, Beachy PA, Jessell TM. Floor plate and motor neuron induction by different concentrations of the amino-terminal cleavage product of sonic hedgehog autoproteolysis. Cell. 1995;81:445–455. doi: 10.1016/0092-8674(95)90397-6. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Hui C, Nakafuku M, Kondoh H. A binding site for Gli proteins is essential for HNF-3beta floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development. 1997;124:1313–1322. doi: 10.1242/dev.124.7.1313. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Nishizaki Y, Hui C, Nakafuku M, Kondoh H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development. 1999;126:3915–3924. doi: 10.1242/dev.126.17.3915. [DOI] [PubMed] [Google Scholar]
- Scholey JM. Intraflagellar transport. Annu Rev Cell Dev Biol. 2003;19:423–443. doi: 10.1146/annurev.cellbio.19.111401.091318. [DOI] [PubMed] [Google Scholar]
- Shiratori H, Hamada H. The left–right axis in the mouse: from origin to morphology. Development. 2006;133:2095–2104. doi: 10.1242/dev.02384. [DOI] [PubMed] [Google Scholar]
- Simpson EM, Linder CC, Sargent EE, Davisson MT, Mobraaten LE, Sharp JJ. Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nat Genet. 1997;16:19–27. doi: 10.1038/ng0597-19. [DOI] [PubMed] [Google Scholar]
- Tabin CJ, Vogan KJ. A two-cilia model for vertebrate left–right axis specification. Genes Dev. 2003;17:1–6. doi: 10.1101/gad.1053803. [DOI] [PubMed] [Google Scholar]
- Takeda S, Yonekawa Y, Tanaka Y, Okada Y, Nonaka S, Hirokawa N. Left–right asymmetry and kinesin superfamily protein KIF3A: new insights in determination of laterality and mesoderm induction by kif3A−/− mice analysis. J Cell Biol. 1999;145:825–836. doi: 10.1083/jcb.145.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Okada Y, Hirokawa N. FGF-induced vesicular release of Sonic hedgehog and retinoic acid in leftward nodal flow is critical for left–right determination. Nature. 2005;435:172–177. doi: 10.1038/nature03494. [DOI] [PubMed] [Google Scholar]
- Traiffort E, Charytoniuk DA, Faure H, Ruat M. Regional distribution of Sonic Hedgehog, patched, and smoothened mRNA in the adult rat brain. J Neurochem. 1998;70:1327–1330. doi: 10.1046/j.1471-4159.1998.70031327.x. [DOI] [PubMed] [Google Scholar]
- Traiffort E, Moya KL, Faure H, Hassig R, Ruat M. High expression and anterograde axonal transport of aminoterminal sonic hedgehog in the adult hamster brain. Eur J Neurosci. 2001;14:839–850. doi: 10.1046/j.0953-816x.2001.01708.x. [DOI] [PubMed] [Google Scholar]
- Tsuboi K, Shults CW. Intrastriatal injection of sonic hedgehog reduces behavioral impairment in a rat model of Parkinson’s disease. Exp Neurol. 2002;173:95–104. doi: 10.1006/exnr.2001.7825. [DOI] [PubMed] [Google Scholar]
- Tsujikawa M, Malicki J. Intraflagellar transport genes are essential for differentiation and survival of vertebrate sensory neurons. Neuron. 2004;42:703–716. doi: 10.1016/s0896-6273(04)00268-5. [DOI] [PubMed] [Google Scholar]
- Tsukui T, Capdevila J, Tamura K, Ruiz-Lozano P, Rodriguez-Esteban C, Yonei-Tamura S, Magallon J, Chandraratna RA, Chien K, Blumberg B, et al. Multiple left–right asymmetry defects in Shh(−/−) mutant mice unveil a convergence of the shh and retinoic acid pathways in the control of Lefty-1. Proc Natl Acad Sci U S A. 1999;96:11376–11381. doi: 10.1073/pnas.96.20.11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisberg EA, Grissom PM, McIntosh JR. Mammalian cells express three distinct dynein heavy chains that are localized to different cytoplasmic organelles. J Cell Biol. 1996;133:831–842. doi: 10.1083/jcb.133.4.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100:423–434. doi: 10.1016/s0092-8674(00)80678-9. [DOI] [PubMed] [Google Scholar]
- Wanker EE, Rovira C, Scherzinger E, Hasenbank R, Walter S, Tait D, Colicelli J, Lehrach H. HIP-I: a huntingtin interacting protein isolated by the yeast two-hybrid system. Hum Mol Genet. 1997;6:487–495. doi: 10.1093/hmg/6.3.487. [DOI] [PubMed] [Google Scholar]
- Weinstein DC, Altaba A, Chen WS, Hoodless P, Prezioso VR, Jessell TM, Darnell JE., Jr The winged-helix transcription factor HNF-3 beta is required for notochord development in the mouse embryo. Cell. 1994;78:575–588. doi: 10.1016/0092-8674(94)90523-1. [DOI] [PubMed] [Google Scholar]
- Wijgerde M, McMahon JA, Rule M, McMahon AP. A direct requirement for Hedgehog signaling for normal specification of all ventral progenitor domains in the presumptive mammalian spinal cord. Genes Dev. 2002;16:2849–2864. doi: 10.1101/gad.1025702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki H, Nakata T, Okada Y, Hirokawa N. KIF3A/B: a heterodimeric kinesin superfamily protein that works as a microtubule plus end-directed motor for membrane organelle transport. J Cell Biol. 1995;130:1387–1399. doi: 10.1083/jcb.130.6.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ybot-Gonzalez P, Cogram P, Gerrelli D, Copp AJ. Sonic hedgehog and the molecular regulation of mouse neural tube closure. Development. 2002;129:2507–2517. doi: 10.1242/dev.129.10.2507. [DOI] [PubMed] [Google Scholar]
- Young KA, Berry ML, Mahaffey CL, Saionz JR, Hawes NL, Chang B, Zheng QY, Smith RS, Bronson RT, Nelson RJ, et al. Fierce: a new mouse deletion of Nr2e1; violent behaviour and ocular abnormalities are background-dependent. Behav Brain Res. 2002;132:145–158. doi: 10.1016/s0166-4328(01)00413-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XM, Ramalho-Santos M, McMahon AP. Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R asymmetry by the mouse node. Cell. 2001;105:781–792. [PubMed] [Google Scholar]
- Zhu AJ, Zheng L, Suyama K, Scott MP. Altered localization of Drosophila Smoothened protein activates Hedgehog signal transduction. Genes Dev. 2003;17:1240–1252. doi: 10.1101/gad.1080803. [DOI] [PMC free article] [PubMed] [Google Scholar]