

Abstract
Ultra-widefield fundus autofluorescence (UW-FAF) allows for the characterization of the peripheral retinal features of vitreoretinal diseases. The purpose of this study was to examine possible genotypic/phenotypic correlations of UW-FAF patterns in patients with a variety of retinal dystrophies and retinitis pigmentosa (RP). Seventeen patients were identified who had identified mutations in retinal dystrophy or RP genes and who also had undergone UW-FAF. Mutations with genes included RPGR, RHO, PRPF31, RDS/PRPH2, USH2A, CRB1, CEP290, and RPGRIP1. Variable UW-FAF patterns including ring hyperautofluorescence, double ring hyperautofluorescence, and peripheral hypoautofluorescence were identified. Further research is needed to better characterize this technology as an imaging biomarker for genotype association in retinal dystrophies and RP.
Inherited retinal dystrophies comprise a heterogeneous group of disorders with widely varied phenotypes. Retinitis Pigmentosa (RP) is most common and affects 1/3,000–1/5,000 people[1–3], and can be inherited as autosomal dominant, recessive, or X-linked.[4] To date, mutations in more than 50 genes are known to cause RP The most severe phenotype of infantile or early-onset childhood RP is termed Leber congenital amaurosis (LCA). Usher syndrome is characterized by hearing loss and progressive vision loss, due to retinitis pigmentosa. It accounts for up to 10–20% of RP cases.[5–9] Significant advances have been made in recent years in the molecular diagnosis of retinal dystrophies and there are a number of current trials of gene and other therapies for this group of disorders.
Fundus autofluorescence (FAF) imaging is an emerging technology that allows the functional evaluation of photoreceptors and retinal pigment epithelium (RPE).[10] The green light (532nm) widefield FAF imaging modality records signal mainly from RPE lipofuscin but also other fluorophores in the macula and periphery.[11] Reduced FAF is thought to result from photoreceptor loss in early life and is absent with RPE loss. Increased FAF occurs with increased lipofuscin in the RPE or loss of rod outer segments.[12] Ultra-widefield imaging (Optos 200 Tx) offers a 200-degree view of the retina on a single frame, which allows simultaneous FAF visualization in the macula and periphery.
We analyze ultra-widefield FAF (UW-FAF) in patients with retinal dystrophies and RP and attempt genotypic-phenotypic characterization of UW-FAF patterns.
We performed a retrospective, consecutive case series study after Institutional Review Board approval was obtained at the Cleveland Clinic, OH, USA. All procedures conformed to the tenets of the Declaration of Helsinki. Medical records of patients with RP and other retinal dystrophies at Cleveland Clinic were reviewed. Patients were included if they had UW-FAF and genotypic characterization of their retinal disorder. All patients underwent genetic counseling and were consented for molecular genetic testing. Molecular diagnostic testing was performed at CLIA approved laboratories.
All patients underwent visual acuity testing, slit lamp biomicroscopy and dilated fundus examination. All eyes underwent UW-FAF imaging was performed using the Optos 200 Tx. In AF mode the Optos 200 Tx uses a green (532nm) laser wavelengths to produce a digital, high-resolution image that is displayed on a monitor screen. UW-FAF characteristics were analyzed by two reviewers. The study included 34 eyes of 17 patients. The diagnoses and mutations (Table 1) included 3 patients with X-linked RP (RPGR mutations), 7 patients with dominant RP [RHO mutations (n = 4), PRPF31 mutation (n = 1), RDS/PRPH2 (n = 1), and EYS mutation (n = 1)], 3 patients with LCA [CRB1 mutation (n = 1), CEP290 mutation (n = 1), and RPGRIP1 mutation (n = 1)], and 4 patients with autosomal recessive Usher syndrome (all with USH2A mutations).
Table 1.
Clinical Characteristics.
| Patient | Age | Gender | Diagnosis | Mutation | Best Corrected Visual Acuity Right - Left |
|---|---|---|---|---|---|
| P1 | 49 | Male | X-linked Retinitis Pigmentosa | RPGR | 20/125 - 20/150 |
| P2 | 19 | Male | X-linked Retinitis Pigmentosa | RPGR | 20/30 - 20/30 |
| P3 | 29 | Male | X-linked Retinitis Pigmentosa | RPGR | 20/30 - 20/30-1 |
| P4 | 50 | Female | Autosomal Dominant RP | RHO | 20/25 - 20/20 |
| P5 | 15 | Female | Autosomal Dominant RP | RHO | 20/125 - 20/100 |
| P6 | 43 | Female | Autosomal Dominant RP | RHO | 20/60 - 20/40 |
| P7 | 60 | Female | Autosomal Dominant RP | RHO | 20/30 - 20/30 |
| P8 | 47 | Female | Autosomal Recessive RP | USH2A | 20/20 - 20/40 |
| P9 | 37 | Female | Autosomal Recessive RP | USH2A | 20/25 - 20/25 |
| P10 | 46 | Male | Autosomal Recessive RP | USH2A | 20/20 - 20/20 |
| P11 | 28 | Male | Autosomal Recessive RP | USH2A | 20/20 - 20/15 |
| P12 | 65 | Female | RP/Usher Syndrome | PRPF31 | 20/600 - 20/400 |
| P13 | 72 | Male | Autosomal Recessive RP | RDS/PRPH2 | 20/30 - 20/100 |
| P14 | 24 | Male | Autosomal Recessive RP | EYS | 20/20 - 20/15 |
| P15 | 8 | Male | Leber Congenital Amaurosis | RPGRIP1 | 20/50 - 20/50 |
| P16 | 31 | Female | Leber Congenital Amaurosis | CRB1 | 20/200 - 20/200 |
| P17 | 15 | Male | Leber Congenital Amaurosis | CEP 290 | 20/40 - 20/50 |
Macular hyperautofluorescence was noted in all patients. Ring hyperautofluorescence was prominent in patients with RHO, USH2A, CEP290, RPGRIP1 and RPGR mutations.(Figure 1A) Patients with USH2A mutations demonstrated a second macular ring of hyperautofluorescence. (Figure 1B) Peripheral UW-FAF patterns in patients with RHO or RPGR mutations consisted of hyperautofluorescence with patchy areas of hypoautofluorescence. (Figure 1C) Patients with USH2A or RDS/PRPH2 mutations had a distinct pattern of diffuse peripheral hypoautofluorescence and a characteristic dark appearance compared to the rest of the cohort.(Figures 1D and 1E) The optic nerve appeared dark in most patients. However, patients with PRPF31 mutation or RHO mutations and optic nerve pallor appeared to have optic nerve hyperautofluorescence without optic nerve drusen. (Figure 1F)
Figure 1. Ultra-widefield Fundus Autofluorescence Patterns in Retinitis Pigmentosa and Other Retinal Dystrophies.
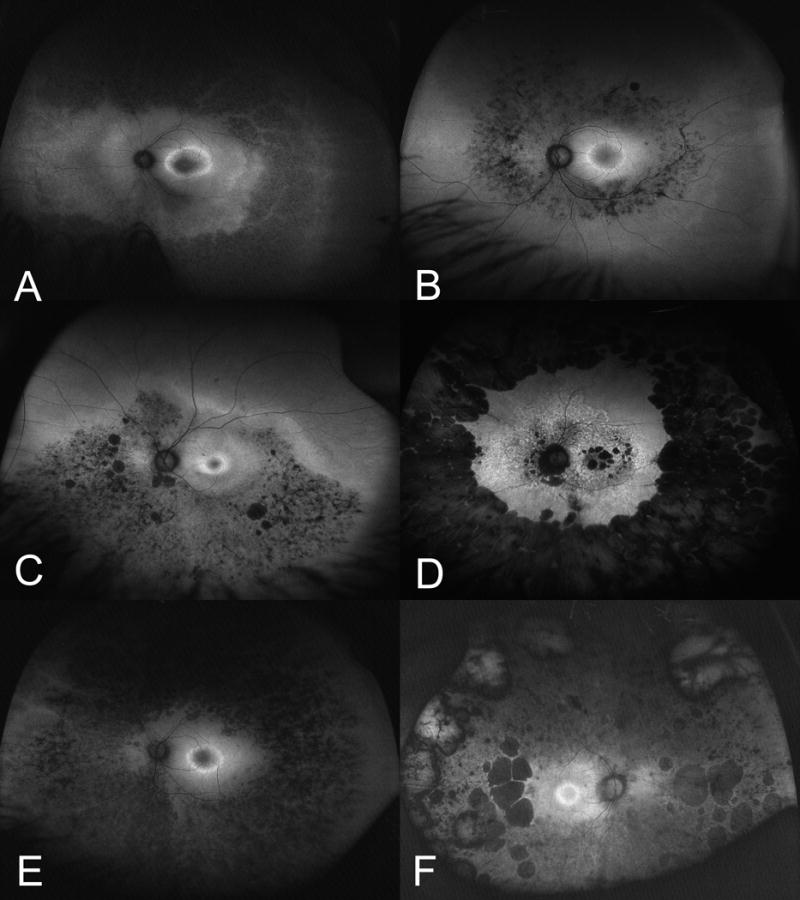
A. Macular ring hyperautofluorescence with as seen in patients with RHO, USH2A, CEP290, RPGRIP1 and RPGR mutations. B. Ultra-widefield fundus autofluorescence in retinitis pigmentosa with USH2A mutation demonstrates diffuse macular hyperautofluorescence and a ring of hyperautofluorescence. C. Ultra-widefield fundus autofluorescence in retinitis pigmentosa with RHO or RPGR with hyperautofluorescence with patchy areas of hypoautofluorescence. D. Ultra-widefield fundus autofluorescence in retinitis pigmentosa with RDS mutations had a distinct pattern of diffuse peripheral hypoautofluorescence. E. Ultra-widefield fundus autofluorescence in retinitis pigmentosa with USH2A mutation had diffuse peripheral hypoautofluorescence. F. Ultra-widefield fundus autofluorescence in retinitis pigmentosa with PRPF31 or RHO mutations with associated optic nerve pallor appeared to have optic nerve hyperautofluorescence.
FAF imaging is considered an indirect indicator of retinal function as it mainly results from lipofuscin and lipofuscin accumulation and is suggestive of oxidative stress and increased metabolic activity.[13,14] A common finding in patients with RP is hyperautofluorescence in the macula that indicates RPE stress. A hyperautofluorescent ring in the macula is commonly found in RP and was frequently observed in this study.[15] One hypothesis is that this represents a circumscribed area of increased photoreceptor phagocytosis that results in accumulation of lipofuscin. In this study, all eyes exhibited hyperautofluorescence in the macula even in absence of a ring. However, we observed specific patterns of UW-FAF associated with select mutations. It is not clear what the second ring of hyperautofluoresence represents but it was more evident in patients with USH2A mutations. Longitudinal changes in the hyperautofluorescent ring have been described in RP including constriction of the ring thought to be related to disease progression.[16]
The benefit of UW-FAF is that it allows for panretinal assessment of disease characteristics. Most of the RP patients in this study demonstrate variable hyperautofluorescence and hypoautofluorescence in the periphery. However, patients with autosomal recessive Usher syndrome (USH2A mutations) had distinctly dark UW-FAF in the periphery. The prominence of this finding suggests RPE death or atrophy in these areas. Typically, the optic nerve has a dark appearance on FAF imaging in healthy patients. Most of the patients with RP have also unremarkable FAF features to the optic nerve. However, patients with significant waxy pallor exhibited hyperautofluorescence at the disc.
The present study has several limitations. It is retrospective and does not provide data on time-dependent longitudinal changes. The number of patients with mutations in the same gene limits quantitative and statistical analysis and further characterization of genotype-phenotype correlation. Prospective large-scale assessment of longitudinal changes on UW-FAF is needed to better characterize UW-FAF as a potential imaging biomarker for inherited retinal diseases with different underlying genetic alterations. This study suggests the UW-FAF findings in patients with retinal dystrophies is heterogeneous and may have distinctive features in different genotypes of inherited retinal diseases.
Acknowledgments
We thank Meghan Marino for assisting with identifying patients who underwent genetic testing at Cole Eye institute. Grant Support: NIH/NEI K23-EY022947-01A1 (JPE); Ohio Department of Development TECH-13-059 (JPE); Foundation Fighting Blindness (EIT, JPE). Research to Prevent Blindness (Cole Eye Institutional)
Footnotes
Financial Disclosures: GT: None; EIT: Sanofi (C), Retrophin (C), Sparks Therapeutics (C); JPE: Bioptigen (P, C), Synergetics (P), Leica (C), Zeiss (C), Thrombogenics (C, R); Genentech (R); Regeneron (R); Santen (C).
References
- 1.Haim M. Epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol Scand. 2002;(Suppl. 80):1–34. doi: 10.1046/j.1395-3907.2002.00001.x. [DOI] [PubMed] [Google Scholar]
- 2.Hartong DT1, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006 Nov 18;368(9549):1795–809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 3.Heckenlively JR. Retinitis Pigmentosa. Philadelphia: Lippincott; 1988. [Google Scholar]
- 4.Kaplan J, Bonneau D, Frezal J, Munnich A, Dufier JL. Clinical and genetic heterogeneity in retinitis pigmentosa. Hum Genet. 1990;85:635–642. doi: 10.1007/BF00193589. [DOI] [PubMed] [Google Scholar]
- 5.Gu S, Thompson DA, Srikumari CRS, Lorenz B, Finckh U, Nicoletti A, Murthy KR, Rathmann M, Kumaramanickavel G, Denton MJ, Gal A. Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nature Genet. 1997;17:194–197. doi: 10.1038/ng1097-194. [DOI] [PubMed] [Google Scholar]
- 6.Heher KL, Traboulsi EI, Maumenee IH. The natural history of Leber’s congenital amaurosis: age-related findings in 35 patients. Ophthalmology. 1992;99:241–245. doi: 10.1016/s0161-6420(92)31985-2. [DOI] [PubMed] [Google Scholar]
- 7.Fazzi E, Signorini SG, Scelsa B, Bova SM, Lanzi G. Leber’s congenital amaurosis: an update. Eur J Paediatr Neurol. 2003;7:13–22. doi: 10.1016/s1090-3798(02)00135-6. [DOI] [PubMed] [Google Scholar]
- 8.Foxman SG, Heckenlively JR, Bateman JB, Wirtschafter JD. Classification of congenital and early onset retinitis pigmentosa. Arch Ophthalmol. 1985;103:1502–1506. doi: 10.1001/archopht.1985.01050100078023. [DOI] [PubMed] [Google Scholar]
- 9.Fishman GA, Kumar A, Joseph ME, Torok N, Anderson RJ. Usher’s syndrome. Ophthalmic and neuro-otologic findings suggesting genetic heterogeneity. Arch Ophthalmol. 1983 Sep;101(9):1367–74. doi: 10.1001/archopht.1983.01040020369005. [DOI] [PubMed] [Google Scholar]
- 10.Delori FC, Dorey CK, Staurenghi G, Arend O, Goger DG, Weiter JJ. In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Invest Ophthalmol Vis Sci. 1995;36(3):718–729. [PubMed] [Google Scholar]
- 11.Eldred GE, Katz ML. Fluorophores of the human retinal pigment epithelium: separation and spectral characterization. Exp Eye Res. 1988;47:71–86. doi: 10.1016/0014-4835(88)90025-5. [DOI] [PubMed] [Google Scholar]
- 12.Lorenz B, Wabbels B, Wegscheider E, Hamel CP, Drexler W, Preising MN. Lack of fundus autofluorescence to 488 nanometers from childhood on in patients with early onset severe retinal dystrophy associated with mutations in RPE65. Ophthalmology. 2004;111:1585–1594. doi: 10.1016/j.ophtha.2004.01.033. [DOI] [PubMed] [Google Scholar]
- 13.Lois N, Forrester J. Fundus Autofluorescence. Lippincott Williams and Wilkins; Philadelphia, PA: 2009. [Google Scholar]
- 14.Delori FC1, Goger DG, Dorey CK. Age-related accumulation and spatial distribution of lipofuscin in RPE of normal subjects. Invest Ophthalmol Vis Sci. 2001 Jul;42(8):1855–66. [PubMed] [Google Scholar]
- 15.Lima LH, Cella W, Greenstein VC, Wang NK, Busuioc M, Smith RT, Yannuzzi LA, Tsang SH. Structural assessment of hyperautofluorescent ring in patients with retinitis pigmentosa. Retina. 2009 Jul-Aug;29(7):1025–31. doi: 10.1097/IAE.0b013e3181ac2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lima LH, Burke T, Greenstein VC, Chou CL, Cella W, Yannuzzi LA, Tsang SH. Progressive constriction of the hyperautofluorescent ring in retinitis pigmentosa. Am J Ophthalmol. 2012 Apr;153(4):718–27. doi: 10.1016/j.ajo.2011.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]