

Summary
Epigenetic reprogramming is a critical process of pathological gene induction during cardiac hypertrophy and remodeling. However, the underlying regulatory mechanism remains to be elucidated. Here we identified a heart-enriched long non-coding (lnc)RNA, named Cardiac Hypertrophy Associated Epigenetic Regulator (Chaer), necessary for the development of cardiac hypertrophy. Mechanistically, Chaer directly interacts with Polycomb Repressor Complex 2 (PRC2) catalytic subunit through a 66-mer motif, interferes with its targeting to genomic locus, and subsequently inhibits histone H3 lysine 27 methylation at hypertrophic genes. This interaction is transiently induced upon hormone or stress stimulation in an mTORC1 dependent manner, and is prerequisite for epigenetic reprogramming and induction of hypertrophic genes. Inhibition of Chaer in intact heart before, but not after, the onset of pressure overload significantly attenuates cardiac hypertrophy and dysfunction. Therefore, our study reveals that stress-induced pathological gene activation in heart requires a previously uncharacterized lncRNA-dependent epigenetic checkpoint.
Introduction
Cardiac hypertrophy, dysfunction and remodeling are common pathological features developed in diseased heart that are driven by transcriptome reprogramming1-4. Genome function is highly regulated at chromatin level by epigenetic modifications, among which histone lysine methylations are crucial events for the recruitment of key protein complexes regulating genome architecture, stability, and gene expression5. Methylation at histone H3 lysine 4 (H3K4) by trithorax group (TrxG)/mixed-lineage leukemia (MLL) complex represents a hall mark for gene activation, while methylation at H3 lys27 (K27) by polycomb repressive complex 2 (PRC2) leads to gene silence6. Significant changes of chromatin modifications have been demonstrated in diseased hearts7-9. It is clear that pathological reprogramming of cardiac transcriptome requires a well-orchestrated and stress-signal dependent regulation of different epigenetic modifications at the targeted promoters. However, the underlying molecular mechanism remains elusive.
Emerging evidence highlights the important roles of lncRNAs in heart diseases10-18. Global transcriptome analyses identify thousands of novel lncRNAs deregulated during heart development and pathogenesis with only a few being well studied19-23. Interestingly, a number of lncRNAs are involved in epigenetic regulations, termed as Epi-lncRNAs24, by directly interacting with epigenetic modifiers21,25-27. HOX antisense intergenic RNA (Hotair) binds to PRC2 and promotes H3K27 tri-methylation (H3K27me3) at the promoter region of target genes25,28. Meanwhile, it also binds to Lysine-specific demethylase 1 (LSD1) and suppresses histone H3K4 methyalion29. Two recently identified cardiac-specific lncRNAs, FOXF1 adjacent non-coding (Fendrr) and Braveheart (Bvht), play important roles in cardiac lineage commitment through interacting with PRC221,26. On the other hand, Myheart (Mhrt) controls cardiac hypertrophy via targeting BRM/SWI2-related gene 1 (BRG1)-mediated chromatin modification30. Moreover, H19 has been reported to function both as a sponge for let-7 family microRNAs during muscle differentiation31 and as a modifier of histone H3 methylation during embryonic development32, implicating divergent functions of lncRNAs in different spatio-temporal settings to coordinate epigenetic reprogramming in heart.
Here we identify a novel cardiac enriched lncRNA, named as cardiac hypertrophy associated epigenetic regulator (Chaer), which is both necessary and sufficient for cardiac hypertrophy and hypertrophic gene induction. Chaer directly binds to the enhancer of zeste homolog 2 (Ezh2) subunit of PRC2 via a 66-mer motif with predicted structural features shared between human and rodent, and negatively regulates PRC2 function on H3K27 methylation for hypertrophic genes. Unique among PRC2-interacting lncRNAs, Chaer-PRC2 interaction is transiently enhanced at the onset of hypertrophic stress in a mammalian target of rapamycin complex 1 (mTORC1) dependent manner. Both genetic and siRNA mediated inactivation of Chaer significantly blunted cardiac hypertrophy and pathological progression, while inhibiting Chaer in post-stressed heart had no effect. Therefore, Chaer defines a previously unrecognized lncRNA dependent early checkpoint for epigenetic switch necessary for hypertrophic gene expression, and serves as a critical molecular link between pathological signaling and hypertrophic gene induction in the stressed heart. The findings offer important insights into fundamental mechanisms of lncRNA function and epigenetic regulation, as well as a novel venue of therapy for cardiac hypertrophy and pathology.
Results
Chaer is an lncRNA necessary for cardiac hypertrophy
From transcriptome analysis in pressure-overload-induced mouse failing heart, we identified nearly 150 long non-coding (lnc)RNAs being significantly deregulated22. One of these lncRNAs is a 2,737-nt transcript derived from a two-exon gene located on chromosome 5 of mouse genome showing highly enriched expression in heart (Fig. 1a,b), and its expression was progressively decreased in failing heart after trans-aortic constriction (TAC) (Supplementary Fig. 1a). Based on its function unveiled below, we named this lncRNA as Cardiac Hypertrophy Associated Epigenetic Regulator (Chaer). Translational analysis failed to find any significant open reading frames with translational propensity (Fig. 1a, lower). By in vitro translation assay in the presence of puromycin, it was confirmed that, similar as Hotair, no significant translation events could be detected for Chaer RNA (Fig. 1c). In addition, none of the predicted small peptides potentially derived from the putative small open-reading frames were detected in mass spectrum dataset from PFAM 27.033. Northern blot analysis identified conserved Chaer transcripts in mouse, rat and human hearts with a single band (2.7 kb for mouse and rat Chaer, roughly 2 kb for its human homolog; Supplementary Fig. 1b-d). Expression analysis in primary cardiomyocytes versus fibroblasts isolated from mouse hearts showed that Chaer was specifically expressed in cardiomyocytes but not in fibroblasts (Supplementary Fig. 1e). Subcellular fractionation assay detected Chaer predominantly in the nuclear fraction, comparable to Hotair (Supplementary Fig. 1f). This was further supported by imaging Chaer RNA with fluorescent in situ hybridization (FISH) in mouse heart tissue (Supplementary Fig. 2).
Figure 1. Chaer regulates cardiac hypertrophy.
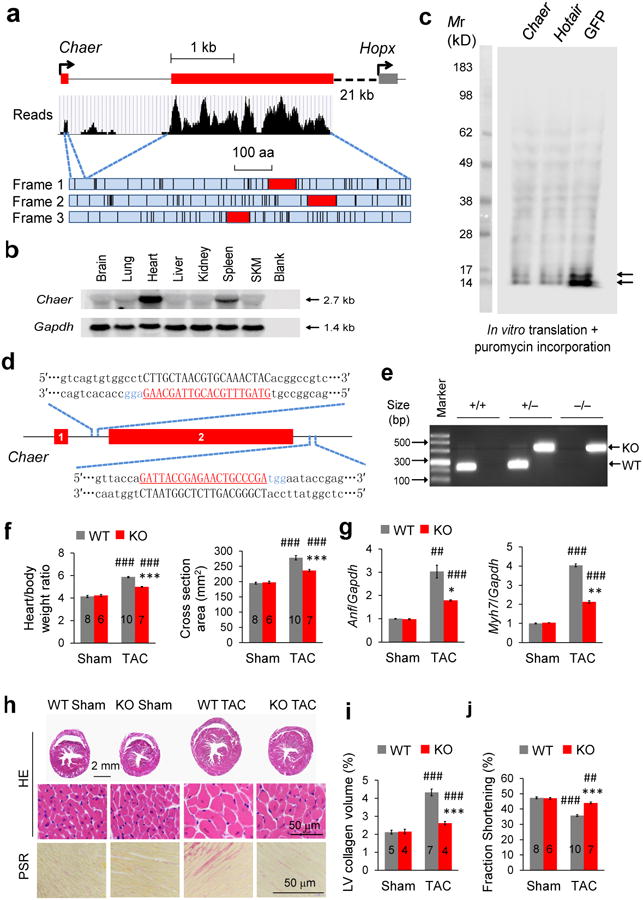
(a) RNA reads of mouse Chaer and genomic structure. Three reading frames are shown with stop codon labeled by black lines and the longest open reading frames labeled in red. (b) Northern blot analysis for Chaer in adult mouse tissues. Gapdh, Glyceraldehyde 3-phosphate dehydrogenase; SKM, skeletal muscle. (c) In vitro translation assay for Chaer, HOX transcript antisense RNA (Hotair) and GFP. (d) Schematic of Chaer knockout in mouse genome using CRISPR-cas9 system showing two guide RNA sequences used. (e) Wild-type (WT) and Chaer knockout (KO) alleles detected by genomic DNA PCR. (f) Effect of Chaer KO on heart weight and myofilament cross-section areas 4 weeks after trans-aortic constriction (TAC) surgery, Data were mean ± s.e.m. Sample numbers were labeled on bars. ***P < 0.001 versus WT; ###P < 0.001 versus sham (Students' t test). (g) Atrial natriuretic factor (Anf; left) and β-myosin heavy chain (Myh7; right) expression. Data were mean ± s.e.m. n = 3. *P < 0.05, ***P < 0.001 versus WT; ##P < 0.01, ###P < 0.001 versus sham (Students' t test). (h) Hematoxylin and Eosin (H&E) staining and Picro Sirius Red (PSR) staining. (i,j) Left ventricular (LV) collagen volume (i) and fractional shortening (j). Data were mean ± s.e.m. Sample numbers were labeled on bars. *P < 0.05, **P < 0.01, ***P < 0.001 versus WT; ##P < 0.01, ###P < 0.001 versus sham (Students' t test).
To determine the functional role of Chaer, we employed a CRISPR-cas9 mediated genome editing approach to inactivate Chaer in C57BL6 mice by deleting the exon 2 which contains majority of the transcript (Fig. 1d). Genomic deletion and loss of Chaer transcript in the knockout (Chaer-KO) heart was validated by genomic DNA PCR (Fig. 1e) and Northern blot (Supplementary Fig. 1b). At basal level, we did not observe any morphological or functional phenotype in the Chaer-KO heart based on histology, echocardiogram parameters and marker gene expression (Fig. 1f-j). However, cardiac hypertrophy in response to pressure overload following TAC was significantly attenuated in the Chaer-KO mice (Fig. 1f,g). Moreover, pathological fibrosis after TAC was significantly blunted in the Chaer-KO heart (Fig. 1h,i), along with better preserved cardiac function (Fig. 1j, Supplementary Table 1 and Supplementary Fig. 3). These data demonstrate that Chaer plays an indispensable role in pathological hypertrophy and remodeling.
To further demonstrate how Chaer contributes to cardiac hypertrophy, we used a specific siRNA (siChaer) to achieve ∼70% knockdown of its expression in isolated neonatal rat ventricular myocytes (NRVMs). Chaer knockdown did not change myocyte morphology at basal level, but significantly suppressed hypertrophic growth induced by phenylephrine (PE, 50 μM; Fig. 2a and Supplementary Fig. 4a). In contrast, the NRVMs with Chaer over-expression showed significantly enlarged cell size compared with the control (Fig. 2b and Supplementary Fig. 4b). Consistently, PE induced hypertrophic genes, including atrial natriuretic factor (Anf), β-myosin heavy chain (Myh7) and skeletal muscle α-actin (Acta1) were markedly attenuated after Chaer knockdown (Supplementary Fig. 4c), whereas Chaer over-expression significantly increased their expression without stimulation except a trend for Anf (Supplementary Fig. 4d). In addition, the neighbor gene HOP homeobox (Hopx) was not affected by Chaer inactivation or over-expression, suggesting that Chaer functions independent of Hopx-mediated cis mechanism (Supplementary Fig. 4c,d). Both in vivo and in vitro data indicate that Chaer is necessary and sufficient for cardiomyocyte hypertrophy and pathological gene expression.
Figure 2. Chaer negatively regulates H3K27me3 via interacting with the catalytic subunit of PRC2.
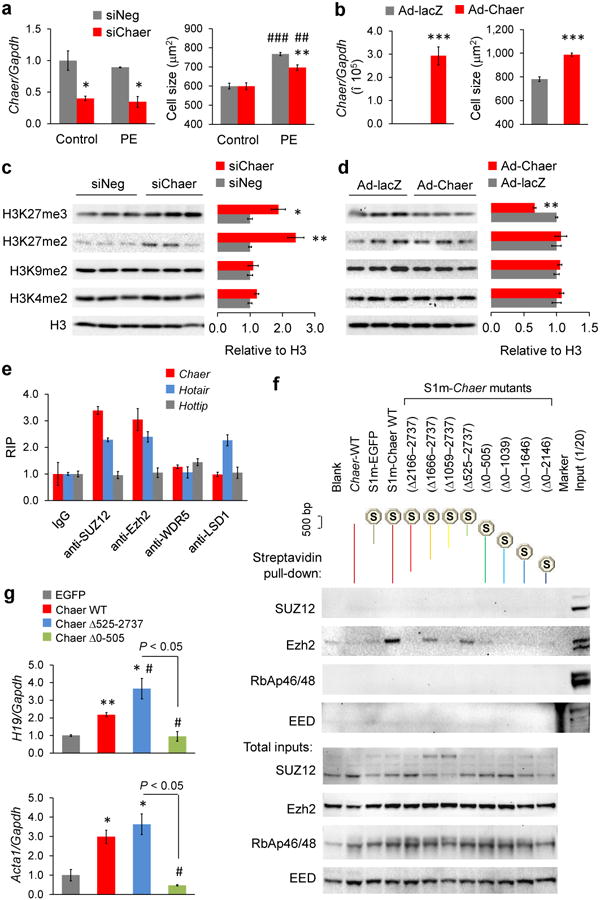
(a) Chaer expression (left) and phenylephrine (PE, 50 μM)-induced hypertrophy after Chaer knockdown (siChaer) in NRVMs (right). siNeg, negative control siRNA. Data were mean ± s.e.m. n = 3. *P < 0.05, **P < 0.01 versus siNeg; ##P < 0.01, ###P < 0.001 versus control (Students' t test). (b) Chaer expression (left) and NRVM cell size (right) following Adv-Chaer infection. Data were mean ± s.e.m. n = 3 ***P < 0.001 (Students' t test). (c,d) Effects of Chaer knock-down (c) or over-expression (d) on H3 methylation as indicated. Data shown in mean ± s.e.m. n = 3. *P < 0.05, *P < 0.01 versus siNeg or Ad-lacZ controls (Students' t test). (e) RNA immune-precipitation (RIP) analysis using antibodies as indicated followed by qRT-PCR for Chaer, Hotair and Hottip (HOXA transcript at the distal tip). Values were normalized to corresponding normal IgG groups. Data were mean ± s.d. from triplicates with one repeat. (f) Tagged RNA streptavidin pull-down assay. Wild-type (WT) Chaer and truncated Chaer mutants as indicated were tagged with a modified S1 motif (S1m), and transfected into MEFs for 48 h followed by streptavidin-beads pull-down. PRC2 components including SUZ12, Ezh2, Retinoblastoma-associated proteins 46 and 48 (RbAp46/48) and embryonic ectoderm development (EED) were detected by immunoblotting in the pull-down products (upper) and total inputs (lower). (g) qRT-PCR for H19 (upper) and Acta1 (lower) expression in MEFs expressing EGFP, Chaer WT, Chaer 0-524 fragment (ChaerΔ525-2737) or Chaer 506-2737 fragment (ChaerΔ0-505). Data were mean ± s.e.m. n = 3.*P < 0.05, **P < 0.01 versus EGFP; #P < 0.05 versus Chaer WT (Students' t test).
Chaer negatively regulates PRC2 during hypertrophy
To investigate the molecular mechanism underlying Chaer function in cardiomyocytes, we performed RNA sequencing analysis for cardiomyocytes with or without Chaer knockdown under basal and hypertrophic stimulation. Clustering analysis unveiled that 48.3% of the genes changed by PE treatment (>1.5 folds) were significantly reversed by Chaer knockdown (Supplementary Fig. 5a, left; Supplementary Fig. 5b, upper), whereas 68.8% of the genes affected by Chaer deficiency (>1.5 folds) were oppositely regulated by PE treatment (Supplementary Fig. 5a, right; Supplementary Fig. 5b, lower). Cardiomyopathy-related and cell cycle-related genes were enriched in the negatively correlated genes between Chaer deficiency and PE treatment (Supplementary Tables 2 and 3). This observation suggests a broad impact of Chaer-mediated regulation on transcriptome reprogramming during hypertrophy. Interestingly, 27 of the top 50 down-regulated genes following Chaer knockdown were found to be clustered in 11 conserved transcription loci (Supplementary Table 4) including the imprinted gene H19 (Supplementary Fig. 5c-e), implicating a potential involvement of chromatin remodeling. Histone methylation has been implicated in transcriptome reprogramming during cardiac hypertrophy and heart failure7-9. Indeed, we observed a dynamic change in global histone methylations at the H3K9 and the H3K27 sites but not at the H3K4 site following PE treatment in NRVMs (Supplementary Fig. 5f). Chaer deficiency in cardiomyocytes specifically increased di- and tri-methylation at H3K27 without affecting the levels of di-methylation at H3K4 or H3K9 sites (Fig. 2c), while Chaer over-expression specifically reduced H3K27 tri-methylation without detectable impact on other histone methylations (Fig. 2d). These data imply a specific but negative regulation of H3K27 methylation by Chaer in cardiomyocytes.
Di- and tri-methylation at H3K27 are catalyzed by the histone methyltransferase PRC2, which is a well-known molecular target of several regulatory lncRNAs25,34. Using RNA immuno-precipitation (RIP) assay, we detected a remarkable enrichment of Chaer in the interactome with PRC2 components SUZ12 and Ezh2 but not with TrxG/MLL component WDR5 or LSD1. As a control, Hotair was detected interacting with both PRC2 and LSD1 as previously reported29 (Fig. 2e). Interestingly, Chaer knockdown in cardiomyocytes significantly augmented the levels of interaction between PRC2 and Hotair or Fendrr21 without changing their total expression levels (Supplementary Fig. 6a,b). Conversely, Chaer over-expression in mouse embryonic fibroblasts (MEFs), in which no endogenous Chaer was expressed, markedly reduced interactions of Hotair or Fendrr with PRC2, even though their total expression levels were increased (Supplementary Fig. 6c,d). To further demonstrate the functional impact of Chaer-PRC2 interaction, we examined the effect of Chaer on H19 expression, a well-established target suppressed by lncRNA-PRC2 complex. In contrast to the inhibitory effect of Hotair, Chaer expression significantly induced H19 expression in MEFs, while over-expression of Hotair competed and blocked the Chaer-induced H19 expression (Supplementary Fig. 6e). All these data suggest that Chaer interaction antagonizes other PRC2 binding lncRNAs and relieves its suppressive function to target genes, an effect distinct from other known PRC2-binding lncRNAs.
Molecular basis of Chaer mediated PRC2 regulation
To further characterize the Chaer-PRC2 interaction at molecular level, we conjugated a modified streptavidin-binding S1 RNA tag (S1m)35 with full-length or truncated fragments of Chaer as shown in Supplementary Fig. 7. All constructs were expressed in MEFs at similar levels (Supplementary Fig. 7) followed by protein pull-down using streptavidin-conjugated beads. Compared with negative controls including blank, untagged wild-type Chaer and tagged EGFP, the tagged full-length Chaer specifically pulled down Ezh2 but not SUZ12, RbAp46/48 or EED components of the PRC2 complex (Fig. 2f). Based on the interaction with different truncated mutants, several regions including nt2166-2737 and nt1059-1666 were identified to be necessary for its interaction with Ezh2. However, only one 524-nt region (Chaer Δ525-2737) near the 5′ end of Chaer was found to be both necessary and sufficient for Ezh2 binding (Fig. 2f). Remarkably, expressing this fragment alone was sufficient to increase the expression of H19 and a muscle specific gene Acta1 in MEFs to the same level as using the full-length Chaer, while expressing a similar sized fragment without the Ezh2 binding fragment (Chaer Δ0-505) showed no effect (Fig. 2g,h).
Within the 5′ 524-nt fragment, we identified a 66-nt motif with a similar predicted secondary structure as the 56-mer motif in Fendrr and the 89-mer Ezh2-EED interacting motif in Hotair36. This motif has shared features in predicted structure among mouse, rat and human Chaer homologs (Fig. 3a and Supplementary Fig. 8a-c), including paired SNPs in the predicted stem structures between mouse and rat homologs (Fig. 3a). To demonstrate the direct interaction between this motif and PRC2, we employed the RNA electrophoresis mobility shift assay (EMSA) and detected a specific complex between the Chaer 66-mer and the recombinant Ezh2 (Fig. 3b). This interaction was effectively competed away by either Chaer or Hotair Ezh2-binding motifs, supporting the notion of a shared binding entity between these two lncRNAs (Fig. 3b). However, the Ezh2-binding motif from Chaer showed lower binding affinity comparing to the motif from Hotair (Fig. 3c). Moreover, binding propensity analysis revealed a similar interaction profile in the established Ezh2 RNA-binding region between Chaer and Hotair (Fig. 3d, Supplementary Fig. 8d). These data suggest that Chaer possesses a potential Ezh2 binding motif with shared features with other known PRC2-binding lncRNAs like Hotair.
Figure 3. Characterization of Chaer motif for PRC2 interaction.
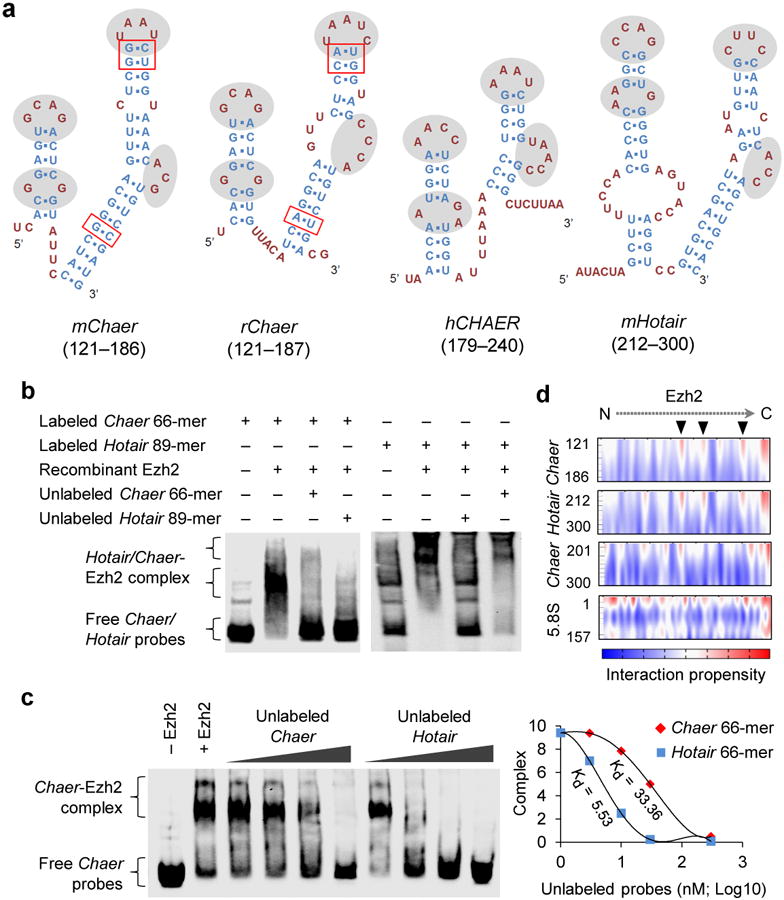
(a) Predicted secondary structures of a 66-mer motif in mouse mChaer, a 67-mer motif in rat rChaer, a 62-mer motif in human hCHAER and an 89-mer motif in mouse mHotair. The unpaired loops with similar pattern were highlighted by gray background, and the paired single nucleotide variations in stems between mouse and rat Chaer were highlighted with red boxes. (b) Validation of the direct binding between the 66-mer mChaer motif and recombinant Ezh2 by RNA electrophoretic mobility shift assay (EMSA). (c) RNA EMSA using labeled Chaer and unlabeled Chaer or Hotair (left). Dissociation constants were calculated by the concentration of unlabeled Chaer or Hotair causing 50% dissociation of the Ezh2-Chaer complex (right). (d) Interaction propensity for Ezh2 binding with Chaer 66-mer motif, Hotair 89-mer motif, Chaer 201-300-nt fragment and 5.8 S rRNA predicted by CatRAPID. Arrowheads highlights the predicted RNA-binding sites of Ezh2.
Regulation of Chaer-PRC2 interaction by hypertrophic signal
Although Chaer negatively regulates PRC2 function through direct interaction in cells, it did not directly change the enzymatic activity of PRC2 in vitro (Fig. 4a). However, chromatin immuno-precipitation (ChIP) assay with anti-Ezh2 antibody revealed a reduced PRC2 binding to the H19 promoter in MEFs with Chaer over-expression (Supplementary Fig. 9a), suggesting that Chaer affected PRC2-mediated epigenetic modification by altering its binding at specific gene loci. In support of the Chaer-mediated regulation of PRC2 function during cardiac hypertrophy, RIP analysis with anti-Ezh2 and anti-SUZ12 antibodies detected a robust but transient enhancement of Chaer-PRC2 interaction within the first 8 h of PE treatment in NRVMs, which was inversely correlated with the Hotair-PRC2 interaction profile (Fig. 4b). This early transient interaction proceeded, in terms of timing, a progressive decrease of global H3K27me3 levels (Supplementary Fig. 9b) followed by induction of hypertrophic genes (Supplementary Fig. 9c). Remarkably, pre-emptive inactivation of Chaer expression significantly blocked PE-induced global H3K27me3 reduction in cardiomyocytes (Supplementary Fig. 9d). Consistent with our observation in MEFs, PRC2 ChIP analysis showed that PE treatment in NRVMs decreased PRC2 binding to the promoter regions of hypertrophic genes Anf, Myh7 and Acta1 (Fig. 4c), which could be reversed by Chaer inactivation (Fig. 4d). Over-expressing Chaer in NRVM was also sufficient to reduce PRC2 targeting to these genes (Fig. 4e). Consequently, Chaer knockdown reversed the PE triggered reduction of H3K27me3 levels at the hypertrophic genes (Fig. 4f,g), while Chaer over-expression was sufficient to significantly decrease the H3K27me3 levels (Fig. 4h). Finally, to demonstrate that Chaer-mediated gene regulation was dependent on PRC2 activity, we tested the effect of Ezh2 inhibitor GSK126 (1 μM) in PE-treated NRVMs (Fig. 4i). As expected, PRC2 inhibition indeed abolished the effect of Chaer knockdown on hypertrophic gene expression (significant for Myh7 and Acta1, and a trend for Anf) in PE-treated NRVMs (Fig. 4j). These data establish that a transiently enhanced interaction between Chaer and PRC2 at the onset of hypertrophic stimulation is both prerequisite and sufficient to release pathological gene suppression by H3K27 tri-methylation in cardiomyocytes.
Figure 4. Transiently enhanced Chaer-PRC2 interaction regulates hypertrophic gene induction and targeted H3K27me3 modification.
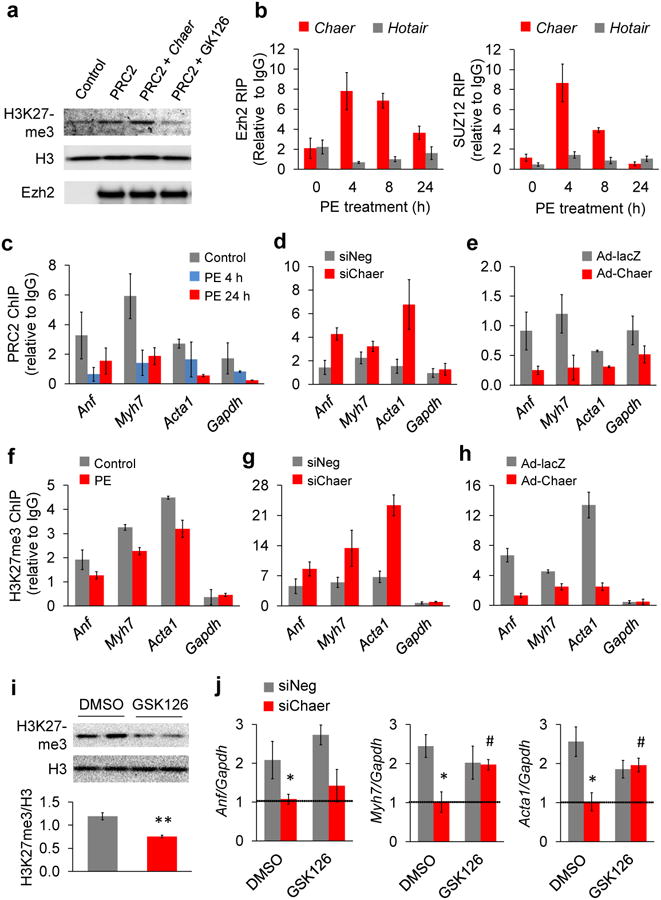
(a) Methyl transferase activity assay for recombinant PRC2 complex with presence or absence of Chaer and Ezh2 inhibitor GSK126 using recombinant histone as substrate. (b) Time-dependent effects of PE on Chaer/Hotair-PRC2 interactions based on RIP assay using antibodies against Ezh2 (left) and SUZ12 (right). Data were mean ± s.d. from triplicates, performed twice. (c-e) Chromatin immunoprecipitation (ChIP) analysis with anti-Ezh2 antibody for the promoter regions of Anf, Myh7 and Acta1 in NRVMs with and without PE treatment (c), PE-treated NRVMs transfected with siNeg or siChaer (d), or NRVMs expressing lacZ (Ad-lacZ) or Chaer (Ad-Chaer) (e). Data were mean ± s.d. from triplicates, and were repeated once. (f-h) ChIP analysis with anti-H3K27me3 antibody for promoter regions of Anf, Myh7 and Acta1 in NRVMs with and without PE treatment (f), PE-treated NRVMs with siNeg or siChaer (g), or NRVMs expressing lacZ (Ad-lacZ) or Chaer (Ad-Chaer) (h). Data were mean ± s.d. from triplicates, performed twice. (i) Immuno-blotting analysis for global H3K27me3 level in NRVMs with or without Ezh2 inhibitor GSK126 (1 μM) treatment. Data were mean ± s.e.m. n = 3. **P < 0.01 (Students' t test). (j). Effects of GSk126 on PE-induced Anf (left), Myh7 (middle) and Acta1 (right) with or without Chaer knockdown. Their expression at basal level was shown as dashed lines. Data were mean ± s.e.m. n = 3. *P < 0.05, versus siNeg; #P < 0.05 versus DMSO.
The transient interaction between Chaer and PRC2 complex upon hypertrophic stimulation indicates a dynamic signaling cascade involved in this process. Concurrent with the enhanced Chaer-PRC2 interaction, the mTOR signaling pathway, indicated by downstream ribosomal protein S6 kinase (S6K) phosphorylation37, was rapidly activated following PE treatment in NRVMs (Fig. 5a and Supplementary Fig. 10a). Indeed, mTOR inhibition by either rapamycin (Fig. 5b) or amino acid starvation38 (Supplementary Fig. 10b) completely blocked the PE-induced enhancement of Chaer-PRC2 interaction (Fig. 5c,d and Supplementary Fig. 10c). Whereas re-feeding the amino-acid-starved NRVMs dramatically enhanced Chaer-PRC2 interaction in a rapamycin-sensitive manner (Supplementary Fig. 10d). Consistently, mTOR inhibition significantly reversed the decrease of H3K27me3 in response to PE treatment (Fig. 5b), and suppressed PE-induced hypertrophic gene expressions (Fig. 5e). These data suggest that the enhancement of Chaer-PRC2 interaction by hypertrophic stimulation is a specific and mTOR-dependent event.
Figure 5. mTORC1 signaling pathway mediates Ezh2-Chaer interaction upon hypertrophic stimulation.
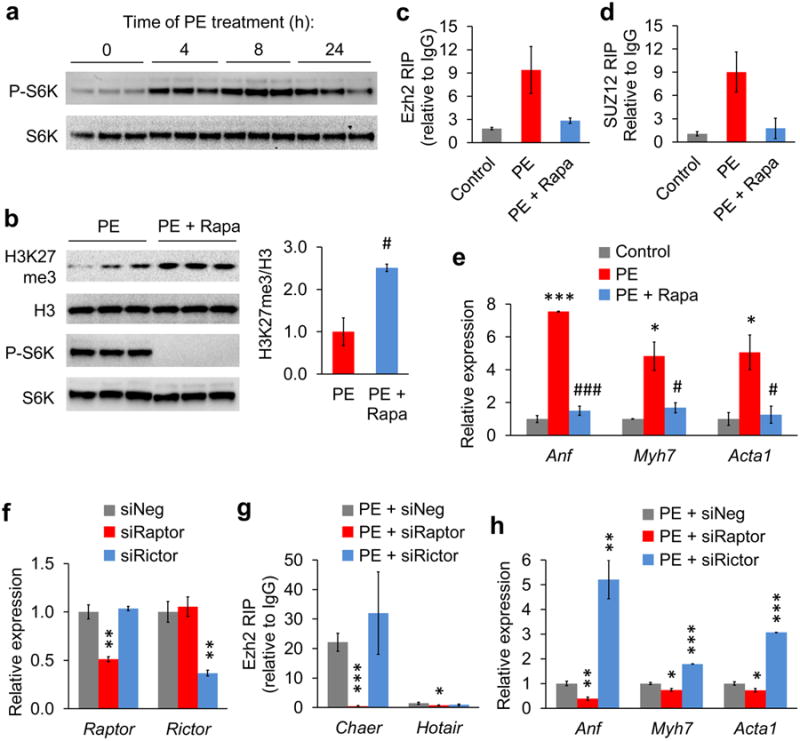
(a) Immunoblotting analysis for ribosomal protein S6 kinase (S6K) phosphorylation in NRVMs treated with PE at different time points. (b) Immunoblotting analysis for H3K27me3, total H3, phosphorylated S6K and total S6K in NRVMs treated with PE only or with PE and mTOR inhibitor rapamycin (Rapa, 20 nM). Quantitative data (right) were mean ± s.e.m. n = 3. #P < 0.01(Students' t test). (c,d) RIP analyses using anti-Ezh2 (c) and anti-SUZ12 (d) antibodies for Chaer-PRC2 interaction in non-treated NRVMs and PE-treated NRVMs (4 h) with or without Rapa. Data were mean ± s.d. from triplicates with one repeat. (e) qRT-PCR for Anf, Myh7 and Acta1 expression in untreated NRVMs and PE-treated NRVMs (4 h) with or without Rapa. Data were mean ± s.e.m. n = 3. *P < 0.05, ***P < 0.001 versus control; #P < 0.05, ###P < 0.001 versus PE (Students' t test). (f) RT-PCR validation of siRNA-mediated knockdown of regulatory-associated protein of mTOR (Raptor) and rapamycin-insensitive companion of mTOR (Rictor). Data were mean ± s.e.m. n = 3. **P < 0.01 versus siNeg (Students' t test). (g) RIP analyses for Chaer/Hotair-PRC2 interaction using anti-Ezh2 antibody in PE-treated NRVMs transfected with siNeg, siRaptor or siRictor. Data were mean ± s.d. from triplicates. (h) qRT-PCR for Anf, Myh7 and Acta1 expression in PE-treated NRVMs transfected with siNeg, siRaptor or siRictor. Data were mean ± s.e.m. n = 3. *P < 0.05, **P < 0.001, ***P < 0.001 versus siNeg (Students' t test).
There are two distinct complexes of mTOR, namely mTORC1 and mTORC2, specified respectively by either regulatory-associated protein of mTOR (Raptor)39 or rapamycin-insensitive companion of mTOR (Rictor)40. To test which complex contributes to the Chaer-mediated hypertrophic transition, we knocked down the expression of Raptor or Rictor in NRVMs with specific siRNAs (Fig. 5f). As shown in Fig 5g, inactivation of Raptor, but not Rictor, completely abolished the enhanced Chaer-PRC2 interaction after PE treatment. In line with this observation, PE-induced expression of Anf, Myh7 and Acta1 were significantly suppressed by Raptor knockdown, but even further enhanced by Rictor knockdown (Fig. 5h). Finally, Chaer over-expression partially restored the expression of hypertrophic genes repressed by rapamycin (Supplementary Fig. 10e) or Raptor knockdown (Supplementary Fig. 10f), suggesting a cross-talk between mTORC1 and Chaer-PRC2 interaction in mediating cardiomyocyte hypertrophic gene expression.
Chaer-mediated epigenetic checkpoint for cardiac hypertrophy
To validate the Chaer-PRC2 pathway in vivo, we further analyzed the impact of Chaer inactivation on mouse heart epigenetic modulation. Indeed, global H3K27me2 and H3K27me3 were significantly increased in the Chaer KO heart after TAC surgery with no significant changes observed on H3K9me2 or H3K9me3 (Fig. 6a). ChIP analysis showed that H3K27me3 levels at promoter regions of Anf, Myh7 and Acta1 were dramatically reduced after 2 weeks of TAC surgery in wild-type heart, but significantly increased in the Chaer KO heart (Fig. 6b). Similar to what we observed in vitro, Chaer-PRC2 interaction in intact mouse heart also showed a dramatic but transient enhancement as early as 1 h after TAC surgery, and began to diminish 4 h later (Fig. 6c). To investigate the significance of the early onset of Chaer-PRC2 interaction, we employed a nanoparticle-mediated transfection method41 to deliver a chemically modified siChaer into heart, which achieved an effective silence of Chaer expression within the experimental timeframe (Supplementary Fig. 11a). Using two delivery strategies with siRNA injection before (Protocol 1) or after (Protocol 2) TAC (Fig. 6d and Supplementary Fig. 11b,c), we observed that pre-emptive Chaer knockdown in mouse heart significantly suppressed cardiac hypertrophy with better-preserved cardiac function and fibrotic remodeling (Fig. 6e and Supplementary Fig. 11d-f). Induction of hypertrophic genes was also significantly blunted in line with attenuated reduction of H3K27me3 levels at their promoter regions (Fig. 6g,h). These results further support the essential role of Chaer-PRC2 interaction in epigenetic regulation during cardiac hypertrophy. In striking contrast, however, delivery of the same Chaer siRNA one day after TAC failed to affect the progression of cardiac hypertrophy, despite of comparable level of Chaer silencing (Fig. 6f and Supplementary Fig. 11b). These data reveal a critical window of epigenetic regulation immediately following the onset of pathological stress in cardiac hypertrophy which functions as an epigenetic checkpoint for the progression of cardiac hypertrophy and pathological remodeling.
Figure 6. Chaer functions as an early check-point of TAC-induced hypertrophy in vivo.
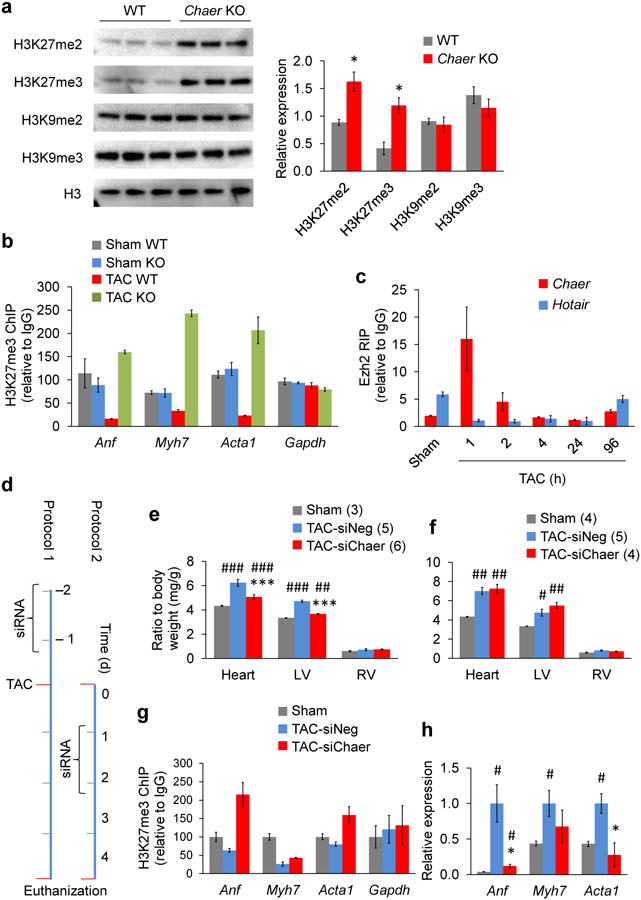
(a) Immunoblotting analyses for H3 modifications as indicated in WT and Chaer-KO hearts 2 weeks after TAC surgery. Data were mean ± s.e.m. n = 3. *P < 0.05 versus WT (Students' t test). (b) ChIP analysis for H3K27me3 level at the promoter regions of Anf, Myh7, Acta1 and Gapdh in WT and Chaer-KO hearts with sham or TAC surgery. Data were mean ± s.d. from triplicates. (c) Time course of Chaer-PRC2 interaction measured by Ezh2-RIP in heart after TAC surgery. Data were mean ± s.d. from triplicates. (d) Protocols of Chaer inactivation with twice siRNA injections performed before (Protocol 1) or after (Protocol 2) TAC surgery. (e,f) Effects of Chaer knockdown in vivo on heart and left ventricle (LV) and right ventricle (RV) weight following protocol 1 (e) or protocol 2 (f). Data were mean ± s.e.m. Sample sizes are labeled as indicated. ***P < 0.001 versus siNeg; #P < 0.05, ##P < 0.01, ###P < 0.001 versus Sham (Students' t test). (g) ChIP analysis for H3K27me3 level at the promoter regions of Anf, Myh7, Acta1 and Gapdh in sham, TAC-siNeg and TAC-siChaer groups of protocol 1 hearts. Data were mean ± s.d. from triplicates. (h) qRT-PCR for Anf, Myh7 and Acta1 expression in sham, TAC-siNeg and TAC-siChaer groups of protocol 1 hearts. Data were mean ± s.e.m. n = 3. *P < 0.05 versus siNeg; #P < 0.05 versus Sham (Students' t test).
Chaer-PRC2 interaction and regulation in human cardiomyocytes
CHAER is also identified in human genome and its expression was readily detected in human heart (Supplemental Fig 1d). Using two independent pairs of primers, we detected an increase of CHAER expression in dilated cardiomyopathy hearts compared with normal controls, though no significance was reached due to large variations (Supplementary Fig. 12a). To validate the CHAER mediated regulation in human cardiomyocytes, we performed RIP analysis in induced pluripotent stem cell-derived human cardiomyocytes (iPSC-CMs) using anti-Ezh2 antibody. The results showed that human CHAER significantly interacted with Ezh2 in a rapamycin-sensitive manner (Supplementary Fig. 12b). Using nanoparticle-mediated transfection of human CHAER in iPSC-CMs, we observed an increased expression of hypertrophic genes Anf, Myh7 and Acta1 by overexpressing CHAER (Supplementary Fig. 12c,d). To determine the functional conservation of Chaer among mouse, rat and human species, we expressed the human CHAER in rat myocytes (Supplementary Fig. 12e). Similar as the mouse Chaer, over-expression of human CHAER in NRVMs increased the expression of hypertrophic genes (Supplementary Fig. 12f). Over-expression of the mouse Chaer in NRVMs with Chaer (rat) knockdown rescued the suppression on PE-induced expression of hypertrophic genes (Supplementary Fig. 12g-i). Moreover, over-expression of either mouse Chaer or human CHAER in both mouse MEFs and human HEK293 cells led to similar induction of H19 expression (Supplementary Fig. 12j-m), suggesting Chaer has conserved gene regulatory function across rodent and human species.
Discussion
Like protein-coding genes, lncRNAs also contribute significantly to tissue specific responses at least in part via epigenetic regulations22,42. Genome-wide analysis of histone marks has revealed a complex epigenetic landscape that orchestrates gene expression during cardiac hypertrophy9. Here we identified a heart-specific lncRNA Chaer as a critical regulator of cardiac hypertrophy via direct interaction with PRC2. This finding, along with other recent reports21,25-27,30, identify a class of epigenetic regulatory lncRNAs (referred to as Epi-LncRNAs) in cardiac gene regulation24. In addition to Chaer, the PRC2 interacting Fendrr and Braveheart are shown to be important in epigenetic programming during heart development21,26. In contrast, lncRNA Myht regulates cardiac hypertrophy and pathological remodeling through direct interaction with a histone acetylation factor BRG130. Therefore, Epi-LncRNAs may exert their epigenetic regulation via specific protein partners during cardiac development and pathogenesis. To our knowledge, Chaer is the first cardiac specific lncRNA identified with hypertrophic-signaling-dependent interaction with PRC2 and shown by clear genetic evidence to be essential for cardiac pathological hypertrophy and remodeling in an intact animal model.
The molecular basis of lncRNA function is a major challenge in the field. Chaer transcript contains a bi-tetra-loop motif within the 5′ end that is both sufficient and necessary for PRC2 binding. However, the overall structural basis of Chaer function remains speculative which needs to be experimentally validated. Although the predicted structure shares common features with other established PRC2-binding motifs identified in several Ezh2-binding lncRNAs36, Chaer binding appears to repress PRC2 function as demonstrated at both global and promoter-specific H3K27me3 levels. This negative regulation on PRC2 function is also consistent with previous reports that silencing Ezh2 causes cardiac hyeprtrophy43,44. However, Chaer binding does not appear to repress PRC2 enzymatic activity directly based on in vitro assay, but rather likely interferes with PRC2 genomic targeting. Whereas Chaer-PRC2 interaction is only transiently enhanced at the onset of hypertrophy, the impact on down-stream H3K27me3 is long-lasting, given the H3K27me3 levels are progressively decreased even 2 weeks after TAC surgery in heart. Therefore, Chaer contributes to the timing and specificity of cardiac epigenetic reprogramming during hypertrophy by coordinating temporal and spatial specific histone methylation and de-methylation45. It remains to be determined if the effects of Chaer may also involve other histone modifiers, like Hotair, especially the H3K27-specific histone demethylase UTX and jumonji domain containing 3 in cardiac gene regulation46.
Despite an obvious correlation between mTOR pathway and cardiac growth, the role of mTOR in hypertrophic and failing heart remains elusive47-52. One major finding from our study is that Chaer-PRC2 interaction is a stress-induced transient event with exquisite dependence on the mTOR activity. Previously, phosphorylation on Ezh2 (T345) mediated by cyclin-dependent kinase 1 (CDK1) has been shown to enhance its binding affinity with lncRNAs, including Hotair and Xist during cell cycle53. Although we have also observed an enhanced rapamycin-sensitive Ezh2 phosphorylation at T345 in PE-treated NRVMs, this phosphorylation happens at 24 h after PE treatment when Chaer-Ezh2 interaction has already passed its peak (Data not shown). Therefore, other mTORC1-dependent modifications of the PRC2 complex may be involved. Considering the sequence diversity in the binding motifs found in different PRC2 binding lncRNAs, our finding implies that signaling dependent modulation of PRC2 may dictate its lncRNA binding partners under different pathophysiological status, leading to tissue-specific and gene-specific epigenetic reprogramming associated with development and diseases. Clearly, more studies are needed to dissect the structure of of Chaer functional motif and its complex with PRC2, as well as the molecular basis of Chaer mediated PRC2 regulation in response to hypertrophic signaling.
Although our study focuses on cell-autonomous effect of Chaer on cardiomyocyte hypertrophy, the cardiac effect of Chaer expression may also involve other cellular process in non-myocyte cardiac cells. It is intriguing that FISH assay detected significant signal of Chaer within the mouse epicardium where potential progenitor cells for endothelium and fibroblasts have been reported to reside54, and Chaer KO significantly reduced TAC induced cardiac fibrosis. It is also interesting to observe that a significant number of cell cycle genes are regulated by Chaer. It remains to be determined if the broad impact of Chaer expression on cardiac genes is mediated through direct effect of epigenetic regulation or indirectly through other key downstream target genes. Therefore, it would be interesting to investigate whether and how Chaer regulates downstream genes and contribute to other aspects and other types of cardiac remodeling.
In summary, our findings reveal a previously uncharacterized epigenetic switch at the very beginning of cardiac hypertrophy involving mTORC1-dependent interaction between a cardiac enriched lncRNA, Chaer, and the histone modification complex PRC2. This interaction is prerequisite to hypertrophic gene expression, cardiac hypertrophy and pathological remodeling. On the other hand, Chaer-PRC2 interaction is transiently induced immediately following stress stimulation, and this interaction is not necessary for the progression of hypertrophic remodeling beyond the early epigenetic switch point. Therefore, Chaer-PRC2 interaction defines a previously unrecognized early epigenetic checkpoint for stress induced transcriptome reprogramming in heart. Considering the functional conservation of human CHAER in cardiomyocytes, the significant protective effects of Chaer inactivation in stressed heart illustrate that Chaer-PRC2 interaction can serve as a potential therapeutic target to treat cardiac hypertrophy and pathological remodeling in diseased heart.
On-Line Methods
Animal and human studies
All experimental procedures involving animals in this study were reviewed and approved by the Institutional Animal C are and Use Committees (IACUCs) of University of California at Los Angeles (UCLA), and conformed to the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health 55. Experiments with donated human heart tissues were approved by the Ethics Committee Board of Wuhan University, China after obtaining proper informed consent. Male mice in C57BL6 background, age between 8 to 10 weeks, were used in this study with numbers for each experiment as indicated. Animals with each genotype were randomly assigned to Sham or Trans-aortic Constriction (TAC) groups. Echocardiographic analysis was performed blinded to the genotype or procedures operated on the mice.
Cell culture, plasmids and Adenoviral vectors
Neonatal rat ventricular myocytes (NRVMs) were prepared as previously described56, and maintained in DMEM supplemented with 1% Insulin-Transferrin-Selenium (ITS-G, BD Biosciences, CA, USA), 100 U/mL penicillin and 100 μg/ml streptomycin for 24h before transfection, infection or drug treatment. Immortalized mouse embryonic fibroblasts (MEFs) and human HEK293 cells, authenticated cell lines from ATCC, were maintained in DMEM supplemented with 10% fetal bovine serum, 2mM L-glutamine, 100 U/mL penicillin and 100 μg/ml streptomycin. The human induced pluripotent stem cell-derived cardiomyocyte (hiPSC-CM) was a gift from Dr. Deepak Srivastava's lab at University of California, San Francisco, and was prepared and maintained as previously reported57. Cardiac fibroblast was a gift from Dr. Arjun Deb's lab at University of California, Los Angeles, and was prepared as previously reported58. Cardiomyocyte from adult mouse heart was isolated based on enzymatic digestion as described previously59. No contamination from mycoplasma was validated by RT-PCR. Wild-type Chaer, truncated Chaer mutants and Hotair were cloned from mouse cDNA with Turbo Pfu DNA polymerase (Thermo Fisher Scientific, MA, USA), and inserted into pcDNA5-CMV vector for transient overxpression purpose. Human CHAER was cloned from donated normal human heart tissues into pcDNA5-CMV vector, and overexpressed in NRVMs with nanoparticle-mediated transfection following the manufacturer's instruction (Altogen). Wild-type Chaer and lacZ were cloned into pShuttle-CMV and recombinated with pAdeasy-1 to generate adenoviruses expressing Chaer (Ad-Chaer) and lacZ (Ad-lacZ). Chaer 66-mer motif and Hotair 89-mer motif were cloned into pBluescript II for in vitro transcription. Mouse Ezh2 was cloned into pT7CFE1 for in vitro coupled transcription and translation. Lipofectamine 2000 (Life Technologies, NY, USA) was used to transfect plasmids into MEFs. Negative control siRNA (siNeg; Bioland Scientific, CA, USA), Chaer siRNA (siChaer; designed and synthesized by Bioland Scientific) and Hotair siRNA (siHotair; Qiagen, Limburg, Netherland) were transfected into NRVMs using Lipofectamine RNAiMAX (Life Technologies). A 1.7 kb long human CHAER homolog was cloned using Zero Blunt TOPO PCR cloning kit (Thermo Fisher Scientific, MA, USA), transferred into pcDNA5-CMV using BamHI and XhoI, and transfected into NRVMs and hiPSC-CMs using Nanoparticle-mediated transfection reagents (Altogen Biosystems, NV, USA) according to the manufacturer's instruction. NRVMs or MEFs were infected with Ad-Chaer (107 PFU/ml) for overexpression with the same amount of Ad-lacZ as a control. Phenylephrine (PE, 50 μM) was used to induce hypertrophy in NRVMs. NRVM starvation was performed by branched chain amino acid depletion medium over night, followed by refeed with normal culture medium.
Generation of Chaer knockout mice
Chaer knockout mice were generated by using CRISPR genome editing system in C57/BL6 background as previously reported60. A pair of single guide RNAs (sgRNAs) flanking the exon 2 of Chaer (majority of the lncRNA) was designed by an online CRISPR Design Tool (http://tools.genome-engineering.org), and constructed into vector pUC57-sgRNA (Addgene). The sgRNA-coding DNA was then amplified together with T7 promoter to generate pure templates for in vitro transcription using MEGA shortscript™ Kit (Life Technologies, NY, USA). Cas9 expression plasmid (Addgene) was linearized with PmeI and used as template for in vitro transcription using the T7 Ultra Kit (Life Technologies). Purified Cas9 mRNA and sgRNAs were mixed and injected into the cytoplasm of fertilized eggs with well-recognized pronuclei in M2 medium (Sigma-Aldrich, MO, USA). Successful knockout was validated by PCR analysis with the following primers:
Chaer WT-F: 5′-CTGAACGCTCTGCAAATCCTA-3′;
Chaer WT-R: 5′-TAAAGCCAGCAAGAACATAAGG-3′;
Chaer KO-F: 5′-GGACAGCATCTTCCCTACCACC-3′;
Chaer KO-R: 5′-CAACCAGTTGGAAGGCCGTAAG-3′.
The wild-type allele yielded an amplicon of 155 bp, while the mutated allele yielded an amplicon of 317 bp. Out of 26 injected embryos, nine pups was generated and six were detected with mutated allele. One was selected to mate with wild-type strain to obtain F1 generation. Heterozygous F1 offspring were interbred to establish Chaer-/- strain.
Transaortic constriction surgery and pressure-volume loop
Transaortic constriction (TAC) surgery was performed as described61. Wild-type and Chaer KO male mice at 2-month age were randomly separated into sham and TAC groups, and group information was double blinded between surgery operator and data analyzer. The mouse was fixed in a supine position with the neck slightly extended. A 20-G catheter was inserted through the larynx into the trachea with care taken not to puncture the trachea or other structures in the pharyngeal region (endotracheal incubation). The ventilation was performed with a tidal volume of 200 ul, respiratory rate of 120/min, 95% oxygen. Body temperature was maintained as close as possible to 37.0°C throughout the experiment by using a self regulating heating pad. After disinfected with 2% iodine, the chest cavity was open by an incision of the left second intercostals space. Aortic arch was dissected from the surrounding tissue. The pericardial sac was opened while a 6-0 suture was passed underneath of the tranverse aortic and ligated against a 27-G needle which was removed latter to provide a lumen. The chest cavity, muscle and skin were closed layer by layer. Sham operated mice underwent similar surgical procedures, including isolation of the aorta and looping of the aorta, but without tying of the suture. Mice were observed until recovery in a 37.0°C heating cage.
For the invasive hemodynamic analysis, mice were anesthetized by 1.5–2% isoflurane, and then a 1.4-FrenchMillar catheter-tip micromanometer catheter (SPR-839; Millar Instruments, Houston, Texas) was inserted through the right carotid artery into the left ventricle of mice. After stabilization for 15 minutes, the heart rate, pressure and volume signals were recorded continuously with an Aria pressure-volume conductance system coupled to a Powerlab/4SP A/D converter and subsequently stored and displayed on computer.
Echocardiography
Transthoracic ultrasonography was performed with a Vevo 2100 system (FUJIFILM VisualSonics, Ontario, Canada). Echocardiography was performed before and 4 days after TAC surgery. The inhalational flow of isoflurane was adjusted to anaesthetize the mice while maintaining their heart rates at 450–550 beats per minute. The peak aortic blood velocity across the aortic constriction was measured at pulsed-wave color Doppler mode. Left ventricular function was assessed by M-mode scanning of the left ventricular chamber, standardized by two-dimensional, short-axis views of the left ventricle at the mid papillary muscle level. Left ventricular chamber size and wall thickness were measured in at least three beats from each projection and averaged. Left ventricular internal dimensions at diastole and systole (LVID;d and LVID;s, respectively) were measured.
In vivo Chaer silence
For in vivo gene knockdown, chemically modified siChaer and siNeg were designed and synthesized by Bioland Scientific (CA, USA), and delivered into heart using nanoparticle transfection reagent (Altogen Biosystems, NV, USA) by two injections through femoral vein each day before (protocol 1) or after (protocol 2) TAC surgery following the manufacturer's instruction62. Chaer knockdown efficiency in heart was evaluated by real-time RT-PCR.
Northern blot analysis
Northern blot was carried out using the DIG Northern Starter Kit (12039672910, Roche). Briefly, the total RNAs were extracted from tissues using TRIzol reagents. RNA was quantified by spectrophotometry (NanoDrop 2000 spectrophotomoter, Thermo Fisher Scientific, MA, USA) and 1 μg RNA was combined with 4 μl formaldehyde, 10 μl deionized formamide and 2 μl 10× MOPS buffer [0.4 M 3 (N-morpholino) propanesulphonic acid, 0.1 M sodiumacetate, 0.01 M EDTA, pH 7.0] in a total volume of 20 μl. Mouse, rat and normal human heart samples were heated at 85 °C for 10 min, cooled on ice and add 2 μl 10× RNA loading buffer [50% glycerol, 0.01 M EDTA, pH 8.0, 0.25% (m/V) bromophenol blue, 0.25% (m/V) Xylene Cyanol FF], then loaded on a 1.5 % agarose, 2.2 M formaldehyde gel. Samples were run at 80 V for 4 h in 1× MOPS buffer. After transferred to a positively charged nylon membrane (Biodyne™ B Nylon Membrane, Thermo Fisher Scientific, MA, USA) and UV cross-linking, 150 ng of a DIG-labelled probe complementary to the target gene was hybridized at 68 °C. Probes were synthesized and quantified using a DIG Northern starter kit (12039672910, Roche) as directed by the manufacturer. Washes and detection were carried out as described by the manufacturer, using the reagents supplied in the DIG Northern starter kit. Blots were visualized using Bio-Rad ChemiDoc™ XRS+ (Bio-Rad). Primers for probe synthesis targeting Chaer RNA are listed below:
-
Mouse: Forward primer: GTCCGATGCCAGTTCCAGTT;
Reverse primer: TAATACGACTCACTATAGGGGCTCCCCTCAGAGTAAAGAG;
-
Rat: Forward primer: GGTGAAAGCCTGTGTAGT;
Reverse primer: TAATACGACTCACTATAGGGGTAGACTGCTTGAGGGAACAG;
-
Human: Forward primer: AGTCACTGCTGTGCTCCATGCCA;
Reverse primer: TAATACGACTCACTATAGGGTGCCAGCTTGGGAGGCCTGTA.
Fluorescent in situ hybridization
The hearts of male C57/B6 mice were dissected, immersed immediately in fresh 10% neutral buffered formalin, fixed for 16 hours at room temperature, rinsed briefly, dehydrated and embedded in paraffin. Heart were cut into 6 μm transverse sections. The TYPE-1 ViewRNA probe against Chaer LncRNA was designed by Affymetrix and was detected by ViewRNA™ ISH Tissue Assay Kit (Affymetrix, QVT0012) according to manufacturer's instructions. Briefly, tissue sections were pretreated in 90-95 °C pretreatment solution for 10 minutes, followed by protease incubation at 40°C for 35 minutes. Hybridization was performed at 40°C for 2-4 hours. After the preamplifier, amplifier and label probe 1-AP hybridization steps, probes were visualized by fast red substrate at 40°C for 40 minutes. Immunofluorescence assays were performed subsequently to detect the cardiac myocytes, endothelial cells and fibroblasts by antibodies against Troponin T Type 2 (Tnnt2; Bioworld Technology, BS6013, 1:100), isolectin b4 (ib4; Enzo Life Sciences, ALX-650-001B-MC05, 1:100) and type I collagen (MD Biosciences, 203002, 1:200), respectively. Images were taken by Olympus FluoView™ FV1000 confocal laser scanning microscope Brochure.
In vitro translation assay
To validate that Chaer is indeed a non-coding RNA, we developed an immunoblotting-based method by combining in vitro translation with puromycin incorporation. Briefly, mouse Chaer and Hotair were cloned into pCFE1-T7 plasmid, and subjected to in vitro translation using a human Hela cell lysate system (1-Step Human High-Yield Mini IVT Kit, Thermo Fisher Scientific). The reaction was mixed following the manufacturer's instruction with the presence of puromycin (Thermo Fisher Scientific, 1 μM), which will be incorporated into any actively translated peptide and facilitates a sensitive detection by immunoblotting using anti-puromycin antibody (Sigma-Aldrich, MO, USA; MABE343, 1:1000). GFP was used as a coding control.
Real-time RT-PCR analysis
Total RNA was extracted from heart or cells using TRIzol reagent (Life Technologies, NY, USA). One μg RNA was reverse transcribed into the first-strand cDNA using Superscript III first-strand synthesis kit (Life Technologies, NY, USA) with random primers. Real-time PCR was performed by CFX96™ Real-Time PCR Detection System (Bio-RAD, CA, USA) using iQ SYBR Green Supermix (Bio-RAD). Values were normalized to Gapdh to calculate the relative expression levels. For fractioning, mouse heart was homogenized in Fraction buffer A containing 10 mM Tris-HCl (pH 7.5), 250 mM Succrose, 0.5 mM EDTA, 0.5 mM EGTA. Cell debris was cleared by centrifuge at 200 g at 4°C for 3 min. Nuclei were pelleted at 2,000 g at 4°C for 5 min. RNAs from nuclear and cytosol fractions were extracted with TRIzol and TRIzol LS reagents (Life Technologies, NY, USA) respectively, and subjected to reverse transcription followed by real-time PCR analysis as described above. Data were shown as percentage and compared to U6 and 18S/Actb as nuclear and cytosol markers respectively. All primers were listed in supplementary Table 5.
RNA deep sequencing and transcriptome analysis
RNA deep-sequencing was performed as described previously61. Total RNAs were extracted from NRVMs with or without PE treatment and with siNeg or siChaer transfections using TRIzol reagents, and then reverse transcribed using TruSeq RNA Library Prep Kit (Illumina, CA, USA). The libraries were subjected to quality validation by Agilent Bioanalyzer 2100, and then paired-end sequenced using HiSeq 2500 (Illumina). The resulting reads were mapped to rn5 database using TopHat263, and visualized on the UCSC browser. Gene ontology analysis was performed with DAVID Bioinformatics Resources 6.7. Genes changed over 1.5 fold were clustered and shown as heat map (log2 scale) using NetWalker64.
Immunoblotting analysis
Immunoblotting analysis was performed as previously described65. Cells were washed twice with ice cold PBS, and harvested in protein lysis buffer (50 mM HEPES [pH7.4], 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA, 1 mM glycerophosphate, 2.5 mM sodium pyrophosphate, 1 mM Na3VO4, 20 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 1 mM DTT, 1× complete protease inhibitor tablet [Roche]). Total cell lysates were separated on 4-12% Bis-Tris gels (Life Technologies), transferred onto PVDF membrane (Merck Millipore). The blots were probed with antibodies for H3, H3K4me2, H3K9me2, H3K27me2 (#9847 from Cell Signaling Technologies for above antibodies; 1:1000), H3K27me3 (Cell Signaling Technologies; #9733, 1:1000), SUZ12 (Cell Signaling Technologies; #3737, 1:1000), Ezh2 (Cell Signaling Technologies; #5246, 1:1000), RpAb46/48 (Santa Cruz Biotechnology; #33170, 1:1000), EED (Santa Cruz Biotechnology; #28701, 1:1000), p-S6K (Thr389, Cell Signaling Technologies; #9234, 1:1000) and S6K (Cell Signaling Technologies, MA, USA; #9202, 1:1000) as indicated in figures. Protein signals were detected using HRP conjugated secondary antibodies and enhanced chemiluminescence (ECL) western blotting detection regents (Thermo Fisher Scientific, MA, USA).
PRC2 histone methyltransferase activity assay
To test the impact of Chaer on PRC2 activity, we performed the histone methyltranferase activity assay as previously described66. Reactions were carried out in a volume of 50 μl and contained 2 pmol nucleosome (New England Biolabs, MA, USA), 2 pmol PRC2 complex, 10 mM HEPES (pH 7.9), 0.25 mM EDTA, 200 mM NaCl, 10% glycerol, 2 mM dithiothreitol, 2.5 mM MgCl2 and 0.8 μM S-adenosyl-methionine (New England Biolabs, MA, USA) with and without 10 nM Chaer RNA or 200 nM Ezh2 inhibitor GSK126 (Merck Millipore, Darmstadt, Germany). Reactions were incubated for 2 h at 30°C, and resolved by SDS–polyacrylamide gel electrophoresis followed by immunoblotting using anti-H3K27me3 antibody (Cell Signaling Technologies, MA, USA; #9756, 1:1000).
RNA immunoprecipitation
RNA-protein interactions were validated using RNA immunoprecipitation (RIP) essentially as described67. Mouse hearts or cells were homogenized in adequate volumes of polysome lysis buffer (10 mM HEPES-KOH [pH 7.0], 100 mM KCl, 5 mM MgCl2, 25 mM EDTA, 0.5% IGEPAL, 2 mM dithiothreitol [DTT], 0.2 mg/ml Heparin, 50 U/ml RNase OUT [Life Technologies, NY, USA], 50 U/ml Superase IN [Ambion], 1× complete protease inhibitor tablet [Roche]). The suspension was centrifuged at 14,000 g at 4°C for 10 min to remove debris. Lysates containing 1 mg protein were incubated with 500 ng normal IgG (Cell Signaling Technologies, MA, USA; #2729, 1:200), anti-SUZ12 (Cell Signaling Technologies, MA, USA; #3737, 1:200), anti-Ezh2 (Cell Signaling Technologies, MA, USA; #5246, 1:200), anti-WDR5 (Abcam, Cambridge, UK; #56919, 1:200), or anti-LSD1(Cell Signaling Technologies, MA, USA; #2139, 1:200) at 4°C over night on an inverse rotator. Protein A sepharose beads (Life Technologies, 50 μl each) were first blocked in NT2 buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM MgCl2, 0.05% IGEPAL) supplemented with 5% BSA, 0.02% sodium azide and 0.02 mg/ml heparin at 4°C for 1 h, and then added into the lysates followed by 3-h incubation at 4°C on an inverse rotator. The beads were subsequently washed five times in NT2 buffer. RNAs were released by incubating in proteinase K buffer (50 mM Tris [pH 8.0], 100 mM NaCl, 10 mM EDTA, 1% SDS, 1 U/ml proteinase K) for 30 min at 65°C, and pelleted by adding equal volume of isopropyl and centrifuging at 12,000 g at 4°C for 10 min. After washing once with 75% ethanol, RNAs were reverse transcribed into first strand cDNA and used for real-time RT-PCR analysis to detect indicated lncRNAs. Hotair lncRNA was tested as positive controls for SUZ12 and LSD1 bindings, and Hottip lncRNA as a positive control for WDR5 binding. Data were normalized to IgG control groups.
Tagged RNA pull-down
To identify the direct binding partner of Chaer, we employed the tagged RNA pull-down assay as previously described68. Streptavidin-binding S1m DNA was synthesized and cloned into pcDNA5-CMV just ahead of the wild-type Chaer and truncations with roughly 500 bp intervals both from 5′ and 3′. All these constructs together with untagged wild-type Chaer and EGFP were transfected into MEFs for 48 h. Cells were then harvested in SA-RNP lysis buffer (20 mM Tris-HCl [pH7.5], 150 mM NaCl, 1.5 mM MgCl2, 2 mM DTT, 50 U/ml RNase OUT [Life Technologies, NY, USA], 50 U/ml Superase IN [Ambion], 1× complete protease inhibitor tablet [Roche]). Streptavidin sepharose beads were blocked with 500 ng/μl yeast tRNA and 1 mg/ml BSA in SA-RNP lysis buffer before added into cell lysates and incubated at 37°C for 2 h on a rotator. The beads were then pelleted and washed for 5 times with SA-RNP washing buffer (20 mM Tris-HCl [pH7.5], 300 mM NaCl, 5 mM MgCl2, 2 mM DTT, 50 U/ml RNase OUT [Life Technologies, NY, USA], 50 U/ml Superase IN [Ambion], 1× complete protease inhibitor tablet [Roche]). After the last wash, RNA-bound proteins were eluted by addition of 5% RNase A (New England Biolabs, MA, USA) in low salt buffer (20 mM Tris-HCl [pH7.5], 30 mM NaCl, 5 mM MgCl2, 2 mM DTT, 1× complete protease inhibitor tablet [Roche]) for 30 min at 4°C. The eluted proteins were then boiled in 4× LDS sample buffer (Life Technologies) and used for immunoblot analysis.
Rna Emsa
RNA electrophoretic mobility shift assay (EMSA) assay was performed essentially as described69. RNA probes were synthesized from linearized pBluescript-Chaer-66-mer and pBluescript-Hotair-89-mer using RiboMAX Large Scale RNA Production Systems (Promega, WI, USA), and labeled with Biotin using RNA 3′ End Biotinylation Kit (Thermo Fisher Scientific) following the manufacturer's instruction. Recombinant Ezh2 was expressed from linearized pT7CFE1-Ezh2 in a coupled transcription and translation system (1-Step Human In Vitro Protein Expression Kits, Thermo Fisher Scientific) following the manufacturer's instruction. RNA EMSA was performed by using the LightShift Chemiluminescent RNA EMSA Kit (Thermo Fisher Scientific). For each reaction, 10 pmol of labeled probe was incubated with adequate recombinant Ezh2 with the presence of tRNA (1 mg/ml) at room temperature for 30 min. Unlabeled probes at indicated concentrations were used for competition experiments. The reactions were then loaded onto 1% 0.5× TBE agarose gel and transferred to positively charged Nylon membrane (Roche). The membrane was then crosslinked by UV, incubated with HRP-conjugated streptavidin and visualized with ECL reagents. Dissociation constant (Kd) was calculated as the concentration of unlabeled probes when half of the labeled probes were dissociated from the complex with Ezh2.
ChIP-PCR assay
Chromatin immunoprecipitation (ChIP) assay was performed to evaluate PRC2 targeting and H3K27me3 levels at specific promoters as described70,71. Briefly, minced hearts or NRVMs were fixed with 1% formaldehyde for 10 min at room temperature, and then quenched by 125 mM glycine. The samples were homogenized in lysis buffer containing 20 mM Tris-HCl (pH8.0), 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS, and 1× complete protease inhibitor tablet, and sonicated to generate chromatin samples with averaged fragment sizes of 200-1000 bp. After pre-cleared with Protein A sepharose beads, samples were incubated with anti-H3K27me3 antibody (Cell Signaling Technologies, MA, USA), anti-Ezh2 (Cell Signaling Technologies (#5246) for ChIP in mouse hearts ; Santa Cruz Biotechnology (#292275) for ChIP in NRVMs) or normal control IgG at 4°C over night on an inverse rotator. The antibody-chromatin complexes were then pelleted with BSA/Salmon sperm DNA blocked Protein A sepharose beads. After standard washes, the immunoprecipitated DNA was eluted and purified with PCR purification kit (Qiagen). RT-PCR was then performed using primers targeting the promoter regions of hypertrophy-related genes as listed in supplementary Table 5.
Histology, Trichrome staining and Picro Sirius Red staining
Histology (H&E) and trichrome (Masson's) staining were performed as previously described72,73. The mouse hearts from sham, TAC-siNeg or TAC-siChaer groups were perfused and fixed with 10% formalin prior to embedding in paraffin. Embedded hearts were sectioned into 5-μm-thick slices as cross sections at the midpoint of the ventricle. To evaluate the impact of Chaer on fibrosis after TAC surgery, collagen was stained with Picro Sirius Red (PSR) following the manufacturer's instruction (Abcam, Cambridge, UK)74. All the sections were imaged with a SPOT digital camera system (Diagnostic Instruments, Sterling Heights, MI, USA), and measured with a quantitative digital image analysis system (Image-Pro Plus 6.0). Cross section cell size was measured from at least 4 hearts from each group with about 20 cells analyzed per section.
In silicon prediction for RNA structure and protein-RNA interaction
For translational propensity analysis of an RNA sequence, the predicted peptides over 30 amino acids in all three potential reading frames were searched in PFAM 27.075. RNA secondary structure was predicted by RNAfold WebServer (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) based on minimum free energy (MFE) and partition function. The binding propensity between Ezh2 and RNA fragments as indicated was predicted by catRAPID (http://s.tartaglialab.com/page/catrapid_group). Gene ontology (GO) analysis was performed on David Bioinformatics Resources 6.7 (https://david.ncifcrf.gov/).
Statistics
Comparisons in multiple groups were analyzed with one-way ANOVA, followed by Student's t test to calculate the P value between two groups. The sample size was determined holding the probability of a type-I error at α = 0.05. Correlation analysis was done by Pearson's r test. Data were presented as mean ± s.e.m. or s.d. for triplicates.
Supplementary Material
Acknowledgments
This work was supported in part by grants from National Institute of Health, USA (HL070079, HL103205, HL108186 and HL110667) for YW; HL115238 for TV, R01HG006264 and U01HG007013 for XX; and University of California, at Los Angeles, CTSI-Cardiovascular Pilot Team Research Grant UL1TR000124 (YW, XX, TV); The National Science Fund for Distinguished Young Scholars (No. 81425005), the Key Project of the National Natural Science Foundation (No. 81330005), the National Science and Technology Support Project (No. 2013YQ030923-05, 2014BAI02B01, and 2015BAI08B01) for HL; ZW was supported by American Heart Association Western States Affiliate Post-doctoral Fellowship (15POST24970034). CG was a recipient of UCLA Eli & Edythe Broad Center Pre-Doctoral Fellowship in Stem Cell Science. HW was supported by China Scholarship Council (201406280042) and a recipient of Jennifer S. Buchwald Graduate Fellowship in Physiology. A.A.C. was supported by National Institute of Health predoctoral training grant T90DE022734. The authors wish to acknowledge outstanding technical support from H. Pu and Core support from UCLA Cardiovascular Research Laboratories and Division of Molecular Medicine at UCLA.
Footnotes
Author Contributions: ZW designed all the experiments with input from HTY, HL and YW, performed majority of the experiments and drafted the manuscript; YW designed and supervised the study; XJZ performed the Northern blot, ChIP, FISH, Immunoblotting and PV loop assays for Chaer KO mice with input from YXJ, PZ and HL; KQD, JG and HL developed the Chaer knockout mice and performed some of the in vivo experiments; XW and GL participated in the tagged RNA pulldown assay and RIP assays; IC, CG and HW participated in the lncRNA screening and cloning experiments; TY participated in the histology assay; AC, XGX and TMV participated in the transcriptome analysis; SR performed the TAC surgery; YSG and DS provided iPSC derived human cardiomyocytes; SL and AD provided primary cardiac fibroblasts and myocytes from mouse heart.
Competing Financial Interests: Authors state no competing financial interest related to this study.
ACCESSION CODE for RNA-Seq Dataset:: ZGSE82339
References
- 1.Sala V, et al. Signaling to cardiac hypertrophy: insights from human and mouse RASopathies. Mol Med. 2012;18:938–947. doi: 10.2119/molmed.2011.00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuwahara K, Nishikimi T, Nakao K. Transcriptional regulation of the fetal cardiac gene program. J Pharmacol Sci. 2012;119:198–203. doi: 10.1254/jphs.12r04cp. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y. Mitogen-activated protein kinases in heart development and diseases. Circulation. 2007;116:1413–1423. doi: 10.1161/CIRCULATIONAHA.106.679589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013;381:242–255. doi: 10.1016/S0140-6736(12)60397-3. [DOI] [PubMed] [Google Scholar]
- 5.Joh RI, Palmieri CM, Hill IT, Motamedi M. Regulation of histone methylation by noncoding RNAs. Biochim Biophys Acta. 2014;1839:1385–94. doi: 10.1016/j.bbagrm.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gould A. Functions of mammalian Polycomb group and trithorax group related genes. Curr Opin Genet Dev. 1997;7:488–494. doi: 10.1016/s0959-437x(97)80075-5. [DOI] [PubMed] [Google Scholar]
- 7.Kaneda R, et al. Genome-wide histone methylation profile for heart failure. Genes to cells : devoted to molecular & cellular mechanisms. 2009;14:69–77. doi: 10.1111/j.1365-2443.2008.01252.x. [DOI] [PubMed] [Google Scholar]
- 8.Angrisano T, et al. Epigenetic switch at atp2a2 and myh7 gene promoters in pressure overload-induced heart failure. PLoS One. 2014;9:e106024. doi: 10.1371/journal.pone.0106024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Papait R, et al. Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc Natl Acad Sci U S A. 2013;110:20164–20169. doi: 10.1073/pnas.1315155110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishii N, et al. Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J Hum Genet. 2006;51:1087–1099. doi: 10.1007/s10038-006-0070-9. [DOI] [PubMed] [Google Scholar]
- 11.Friedrichs F, et al. HBEGF, SRA1, and IK: Three cosegregating genes as determinants of cardiomyopathy. Genome Res. 2009;19:395–403. doi: 10.1101/gr.076653.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burd CE, et al. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010;6:e1001233. doi: 10.1371/journal.pgen.1001233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumarswamy R, et al. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ Res. 2014;114:1569–1575. doi: 10.1161/CIRCRESAHA.114.303915. [DOI] [PubMed] [Google Scholar]
- 14.Wang K, et al. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ Res. 2014;114:1377–1388. doi: 10.1161/CIRCRESAHA.114.302476. [DOI] [PubMed] [Google Scholar]
- 15.Jiang F, Zhou X, Huang J. Long Non-Coding RNA-ROR Mediates the Reprogramming in Cardiac Hypertrophy. PLoS One. 2016;11:e0152767. doi: 10.1371/journal.pone.0152767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Viereck J, et al. Long noncoding RNA Chast promotes cardiac remodeling. Sci Transl Med. 2016;8:326ra322. doi: 10.1126/scitranslmed.aaf1475. [DOI] [PubMed] [Google Scholar]
- 17.Liu L, et al. The H19 long noncoding RNA is a novel negative regulator of cardiomyocyte hypertrophy. Cardiovasc Res. 2016 doi: 10.1093/cvr/cvw078. [DOI] [PubMed] [Google Scholar]
- 18.Peters T, et al. Long Non-Coding RNA Malat-1 Is Dispensable during Pressure Overload-Induced Cardiac Remodeling and Failure in Mice. PLoS One. 2016;11:e0150236. doi: 10.1371/journal.pone.0150236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matkovich SJ, Edwards JR, Grossenheider TC, de Guzman Strong C, Dorn GW., 2nd Epigenetic coordination of embryonic heart transcription by dynamically regulated long noncoding RNAs. Proc Natl Acad Sci U S A. 2014;111:12264–9. doi: 10.1073/pnas.1410622111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ounzain S, et al. Genome-wide profiling of the cardiac transcriptome after myocardial infarction identifies novel heart-specific long non-coding RNAs. Eur Heart J. 2014;36:353–68a. doi: 10.1093/eurheartj/ehu180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grote P, et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell. 2013;24:206–214. doi: 10.1016/j.devcel.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JH, et al. Analysis of transcriptome complexity through RNA sequencing in normal and failing murine hearts. Circ Res. 2011;109:1332–1341. doi: 10.1161/CIRCRESAHA.111.249433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kurian L, et al. Identification of novel long noncoding RNAs underlying vertebrate cardiovascular development. Circulation. 2015;131:1278–1290. doi: 10.1161/CIRCULATIONAHA.114.013303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, Wang Y. Dawn of the Epi-LncRNAs: new path from Myheart. Circ Res. 2015;116:235–236. doi: 10.1161/CIRCRESAHA.114.305490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li L, et al. Targeted disruption of Hotair leads to homeotic transformation and gene derepression. Cell Rep. 2013;5:3–12. doi: 10.1016/j.celrep.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klattenhoff CA, et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. 2013;152:570–583. doi: 10.1016/j.cell.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang KC, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–124. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Z, et al. The Long Noncoding RNA HOTAIR Contributes to Cisplatin Resistance of Human Lung Adenocarcinoma Cells via downregualtion of p21(WAF1/CIP1) Expression. PLoS One. 2013;8:e77293. doi: 10.1371/journal.pone.0077293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li F, et al. Characterization of Sucrose transporter alleles and their association with seed yield-related traits in Brassica napus L. BMC Plant Biol. 2011;11:168. doi: 10.1186/1471-2229-11-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han P, et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514:102–106. doi: 10.1038/nature13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kallen AN, et al. The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol Cell. 2013;52:101–112. doi: 10.1016/j.molcel.2013.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monnier P, et al. H19 lncRNA controls gene expression of the Imprinted Gene Network by recruiting MBD1. Proc Natl Acad Sci U S A. 2013;110:20693–20698. doi: 10.1073/pnas.1310201110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finn RD, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell. 2011;44:667–678. doi: 10.1016/j.molcel.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leppek K, Stoecklin G. An optimized streptavidin-binding RNA aptamer for purification of ribonucleoprotein complexes identifies novel ARE-binding proteins. Nucleic Acids Res. 2013;42:e13. doi: 10.1093/nar/gkt956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu L, Murat P, Matak-Vinkovic D, Murrell A, Balasubramanian S. Binding Interactions between Long Noncoding RNA HOTAIR and PRC2 Proteins. Biochemistry. 2013;52:9519–9527. doi: 10.1021/bi401085h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang S, Chemaly ER, Hajjar RJ, Lebeche D. Resistin promotes cardiac hypertrophy via the AMP-activated protein kinase/mammalian target of rapamycin (AMPK/mTOR) and c-Jun N-terminal kinase/insulin receptor substrate 1 (JNK/IRS1) pathways. J Biol Chem. 2011;286:18465–18473. doi: 10.1074/jbc.M110.200022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sancak Y, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim DH, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 40.Sarbassov DD, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 41.Li Q, Wang P, Ye K, Cai H. Central role of SIAH inhibition in DCC-dependent cardioprotection provoked by netrin-1/NO. Proc Natl Acad Sci U S A. 2015;112:899–904. doi: 10.1073/pnas.1420695112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cabili MN, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delgado-Olguin P, et al. Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat Genet. 2012;44:343–347. doi: 10.1038/ng.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang T, et al. MicroRNA-214 provokes cardiac hypertrophy via repression of EZH2. Biochem Biophys Res Commun. 2013;436:578–584. doi: 10.1016/j.bbrc.2013.05.079. [DOI] [PubMed] [Google Scholar]
- 45.Shi L, et al. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc Natl Acad Sci U S A. 2011;108:7541–7546. doi: 10.1073/pnas.1017374108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Agger K, et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449:731–734. doi: 10.1038/nature06145. [DOI] [PubMed] [Google Scholar]
- 47.Song X, et al. mTOR attenuates the inflammatory response in cardiomyocytes and prevents cardiac dysfunction in pathological hypertrophy. Am J Physiol Cell Physiol. 2010;299:C1256–1266. doi: 10.1152/ajpcell.00338.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu X, Wang X, Bi Y, Bu P, Zhang M. The histone demethylase PHF8 represses cardiac hypertrophy upon pressure overload. Exp Cell Res. 2015;335:123–134. doi: 10.1016/j.yexcr.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 49.Xu N, et al. The alteration of protein prenylation induces cardiomyocyte hypertrophy through Rheb-mTORC1 signalling and leads to chronic heart failure. J Pathol. 2015;235:672–685. doi: 10.1002/path.4480. [DOI] [PubMed] [Google Scholar]
- 50.Li Y, et al. AMPK inhibits cardiac hypertrophy by promoting autophagy via mTORC1. Arch Biochem Biophys. 2014;558:79–86. doi: 10.1016/j.abb.2014.06.023. [DOI] [PubMed] [Google Scholar]
- 51.Shende P, et al. Cardiac mTOR complex 2 preserves ventricular function in pressure-overload hypertrophy. Cardiovasc Res. 2016;109:103–114. doi: 10.1093/cvr/cvv252. [DOI] [PubMed] [Google Scholar]
- 52.Shende P, et al. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation. 2011;123:1073–1082. doi: 10.1161/CIRCULATIONAHA.110.977066. [DOI] [PubMed] [Google Scholar]
- 53.Kaneko S, et al. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev. 2010;24:2615–2620. doi: 10.1101/gad.1983810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marketou ME, Parthenakis F, Vardas PE. Pathological Left Ventricular Hypertrophy and Stem Cells: Current Evidence and New Perspectives. Stem cells international. 2016;2016:5720758. doi: 10.1155/2016/5720758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.National Research Council Committee for the Update of the Guide for the, C & Use of Laboratory, A. Guide for the Care and Use of Laboratory Animals. National Academies Press (US) National Academy of Sciences; Washington (DC): 2011. The National Academies Collection: Reports funded by National Institutes of Health. [Google Scholar]
- 56.MacLellan WR, Xiao G, Abdellatif M, Schneider MD. A novel Rb- and p300-binding protein inhibits transactivation by MyoD. Mol Cell Biol. 2000;20:8903–8915. doi: 10.1128/mcb.20.23.8903-8915.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Laake LW, et al. Reporter-based isolation of induced pluripotent stem cell- and embryonic stem cell-derived cardiac progenitors reveals limited gene expression variance. Circ Res. 2010;107:340–347. doi: 10.1161/CIRCRESAHA.109.215434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ubil E, et al. Mesenchymal-endothelial transition contributes to cardiac neovascularization. Nature. 2014;514:585–590. doi: 10.1038/nature13839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou YY, et al. Culture and adenoviral infection of adult mouse cardiac myocytes: methods for cellular genetic physiology. Am J Physiol Heart Circ Physiol. 2000;279:H429–436. doi: 10.1152/ajpheart.2000.279.1.H429. [DOI] [PubMed] [Google Scholar]
- 60.Wang H, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee JH, et al. Analysis of transcriptome complexity through RNA sequencing in normal and failing murine hearts. Circulation research. 2011;109:1332–1341. doi: 10.1161/CIRCRESAHA.111.249433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li Q, Wang P, Ye K, Cai H. Central role of SIAH inhibition in DCC-dependent cardioprotection provoked by netrin-1/NO. Proc Natl Acad Sci U S A. 2015;112:899–904. doi: 10.1073/pnas.1420695112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Komurov K, Dursun S, Erdin S, Ram PT. NetWalker: a contextual network analysis tool for functional genomics. BMC Genomics. 2012;13:282. doi: 10.1186/1471-2164-13-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu G, et al. Protein phosphatase 2Cm is a critical regulator of branched-chain amino acid catabolism in mice and cultured cells. J Clin Invest. 2009;119:1678–1687. doi: 10.1172/JCI38151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nekrasov M, Wild B, Muller J. Nucleosome binding and histone methyltransferase activity of Drosophila PRC2. EMBO Rep. 2005;6:348–353. doi: 10.1038/sj.embor.7400376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Baroni TE, Chittur SV, George AD, Tenenbaum SA. Advances in RIP-chip analysis : RNA-binding protein immunoprecipitation-microarray profiling. Methods Mol Biol. 2008;419:93–108. doi: 10.1007/978-1-59745-033-1_6. [DOI] [PubMed] [Google Scholar]
- 68.Leppek K, Stoecklin G. An optimized streptavidin-binding RNA aptamer for purification of ribonucleoprotein complexes identifies novel ARE-binding proteins. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Han P, et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514:102–106. doi: 10.1038/nature13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sdek P, et al. Rb and p130 control cell cycle gene silencing to maintain the postmitotic phenotype in cardiac myocytes. J Cell Biol. 2011;194:407–423. doi: 10.1083/jcb.201012049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Young MD, et al. ChIP-seq analysis reveals distinct H3K27me3 profiles that correlate with transcriptional activity. Nucleic Acids Res. 2011;39:7415–7427. doi: 10.1093/nar/gkr416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Streicher JM, Kamei K, Ishikawa TO, Herschman H, Wang Y. Compensatory hypertrophy induced by ventricular cardiomyocyte-specific COX-2 expression in mice. J Mol Cell Cardiol. 2010;49:88–94. doi: 10.1016/j.yjmcc.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mitchell-Jordan SA, et al. Loss of Bmx nonreceptor tyrosine kinase prevents pressure overload-induced cardiac hypertrophy. Circ Res. 2008;103:1359–1362. doi: 10.1161/CIRCRESAHA.108.186577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jiang DS, et al. Interferon regulatory factor 7 functions as a novel negative regulator of pathological cardiac hypertrophy. Hypertension. 2014;63:713–722. doi: 10.1161/HYPERTENSIONAHA.113.02653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Finn RD, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.