

Abstract
A number of fluorescent dyes based on BODIPY (4,4′-difluoro-4-bora-3a,4a-diaza-s-indacene) have been studied theoretically. This paper presents the results of calculations of these BODIPY dyes in their ground and excited states, performed using DFT and TD-DFT methods, respectively. The influences of N,N-dimethylaminobenzyl, ortho-fluorophenol, and methyl substituents as well as the solvent polarity on the positions of the absorption and emission bands of the dyes were analyzed. The computational data obtained in this work were compared to the corresponding experimental data. The trends in the experimental data were found to agree with those shown by the computational data. Differences between the potential curves obtained when using linear-response (LR) and state-specific (SS) approaches for the ground and excited states are also reported.
Graphical Abstract.
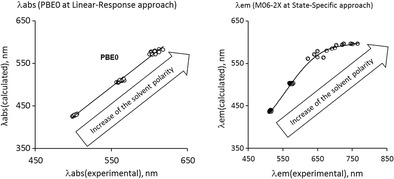
The article shows that the trends of the experimental dependencies λabs = f(Δf) and λem = f(Δf) well described by PBE0 (LR approach) and M06-2X (SS approach) calculations, respectively. The influence of substituents on the spectral characteristics of the BODIPY chromophore are analysed
Electronic supplementary material
The online version of this article (doi:10.1007/s00894-016-3108-8) contains supplementary material, which is available to authorized users.
Keywords: DFT, TD-DFT, BODIPY
Introduction
This paper presents a quantum-mechanical study of fluorescent dyes based on 4,4′-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) [1]. Photophysical studies of these compounds and the design of novel compounds of this type have become very popular over the last decade [2–4]. Such interest is due to the ability to efficiently model the spectral properties of BODIPY derivatives obtained by introducing different types of substituents onto the 4,4′-difluoro-4-bora-3a,4a-diaza-s-indacene core, or by modifying the core in other ways [5]. These compounds are also popular due to their potential application in optoelectronics [6], medicine [7], and biology [8]. Indeed, laborious quantum-mechanical calculations of BODIPY dyes in the excited state using time-dependent density functional theory (TD-DFT) methods are being applied more and more [9–17]. The use of the PBE0 and M06-2X functionals in such studies is motivated by the investigations presented in [14–17].
The task of the work reported in this paper was to computationally study the effects of solvents of various polarities, and the impact of introducing different substituents onto the BODIPY core, on the positions of the absorption (S0→S1) and emission (S1→S0) bands of dyes based on BODIPY using TD-DFT with the PCM approach. The substituents chosen for this study were selected because we obtained relevant experimental data on the corresponding BODIPY derivatives in our previous work [18]. We were able to theoretical derive an accurate description of these dyes, which will make it possible to model such dyes and to determine the nature of the dye–environment interrelations, as well as to identify novel dyes with required spectral characteristics.
Computational details
The calculations were carried out with the Gaussian09 program [19]. The PBE0 [20] and M06-2X [21] functionals and the 6-31 + G(d,p) basis set [22–31] were used for calculations. The structures of the studied compounds were optimized in the ground (S0) and excited (S1) states by DFT [32, 33] and TD-DFT [34, 35] methods, respectively. The absorption (i.e., the S0 → S1 transition) and emission (the S1 → S0 transition) bands were calculated for the compounds in the gas phase and in various solvents with a range of polarities. The absorption spectra were obtained by calculating the first six low-lying excited states within the vertical linear-response, nonequilibrium TD-DFT approximation. The state-specific nonequilibrium solvation approach was applied by saving the solvent reaction field from the ground state. Solvent effects in the ground and excited states were taken into account with the polarizable continuum model [36–38] (PCM), using the integral equation formalism variant (IEFPCM) as a default SCRF method. The states with the largest oscillator force values were optimized in the excited state, and emission spectra were calculated within a linear-response, equilibrium TD-DFT approximation and using state-specific non-equilibrium solvation. The contribution of solvent effects was computed with both the linear-response (LR) [39, 40] and state-specific (SS) [41] quantum mechanical approaches. The selected data and structures are collected in Tables S1–S4 of the “Electronic supplementary material” (ESM).
Results and discussion
Impact of the substituents
As stated in the “Introduction,” the aims of this work were to verify the data afforded by TD-DFT and DFT methods for the dyes of interest in a wide range of solvents (ranging from hexane to DMSO), and to analyze the impact of substitution at position 5 of the BODIPY core with various substituents on the positions of the absorption and emission bands of the dyes. To achieve these aims, three compounds based on BODIPY were studied (Scheme 1).
Scheme 1.

Structures of the studied compounds
These compounds were selected because a considerable amount of experimental data on them is available [18]. Compound 2 plays the role of a reference compound in which a methyl group has been inserted at position 5 (σ R+ = −0.08 [42]). For compounds 1 and 3, N,N-dimethylaminobenzyl and ortho-fluorophenol moieties, respectively, were added at position 5 instead of the methyl group. According to computational and experimental data, the N,N-dimethylaminobenzyl substituent has the strongest effect on the positions of the absorption and emission bands (i.e., the largest bathochromic shift), which is due to the strong electronic effect [42] of this substituent. However, although the ortho-fluorophenol moiety of 3 has a weaker electronic impact than the N,N-dimethylaminobenzyl fragment, 3 does still show bathochromic shifts in the absorption and emission bands with respect to 2. Note that the data calculated using the PBE0 and M06-2X functionals show similar trends to the experimental data (see Fig. 1 and Fig. S1 in the ESM).
Fig. 1.

Comparison of the data for the parameters λ abs and λ em calculated for the compounds of interest using the PBE0 (solid line) and M06-2X (dashed line) functionals in linear-response (LR) and state-specific (SS) approaches with the corresponding data for λ abs and λ em obtained experimentally
Based on the results of the calculations and the corresponding experimental data, we can conclude that the total electronic effect of the –CH=CH–(fluorophenol) substituent is much stronger than the electronic effect of the methyl group (σ R+(CH=CH2) = −0.16, σ R+(C6H5) = −0.22, σ R+(F) = 0.34, and σ R+(OH) = 0.12; all data from [42]); the same is true when the –CH=CH–(N,N-dimethylaminobenzyl) substituent is compared to the methyl substituent (σ R+(CH=CH2) = −0.16, σ R+(C6H5) = −0.22, and σ R+(N(CH3)2) = −0.64; all data from [42]). When the –CH=CH–(fluorophenol) substituent is present, the hydroxyl group of the substituent has only a minor π-electronic effect on the BODIPY core because the hydroxy group is meta with respect to the –CH=CH– bridge. The π-electronic effect of the fluorine atom present on this substituent is also not significant. Indeed, the M+ and M− effects exerted by the fluorine atom and the hydroxyl group cancel each other out to some extent, weakening their influence on the BODIPY core. Therefore, the moiety that has the greatest influence on the bathochromic shifts of the absorption and emission bands of compound 3 with respect to compound 2 is the –CH=CH–C6H5 fragment (σ R+(CH3) = −0.08, σ R+(–CH=CH2) = −0.16, σ R+(−C6H5) = −0.22; all data from [42]).
The substituent that has the strongest effect on the spectral parameters is found to be –CH=CH–(N,N-dimethylaminobenzyl). This substituent causes significant bathochromic shifts of the absorption and emission bands (Δλ ∼ 100–200 nm) for compound 1 with respect to those of compounds 2 and 3. This phenomenon is due to the strong π-electronic effect of the N,N-dialkylamine group on the spectral characteristics of compound 1, which is described in detail in [43, 44].
All of the calculations performed in this work utilized the two most reliable functionals for boron compounds: PBE0 and M06-2X [45, 46]. A comparison of the data provided by the PBE0 and M06-2X functionals when using the LR approach with the corresponding experimental data shows that the calculated absorption bands are the closest to the experimentally determined absorption bands for compound 1. However, compound 1 also shows the largest deviation between the results obtained using the PBE0 and M06-2X functionals; the deviation for compound 3 is smaller, and there is almost no deviation for compound 2. Both PBE0 and M06-2X give almost the same positions for the absorption bands of compound 2 (Fig. 1). It is also clear that the results obtained using the SS approach (Fig. 1) are more accurate (i.e., they deviate less from the corresponding experimental data) than those yielded by the LR approach. However, there are some problems with the trend in absorption band shifts obtained using the SS approach. The computational results obtained with the LR approach exhibit a satisfactory trend regardless of the functional used (Fig. 1), whereas the results obtained using the SS approach suggest that increasing the solvent polarity causes very small changes in λ abs for compounds 2 and 3 , which is not correct. Increasing the solvent polarity actually increases the bathochromic shifts of the absorption bands, but this phenomenon is barely apparent in the data obtained using the SS approach with both functionals (PBE0 and M06-2X) (Fig. 1).
Before comparing the calculated emission band data with the corresponding experimental data, a conformational analysis was performed. Three rotamers with similar potential energies were calculated for compound 2 in the excited state (Fig. 2). These rotamers differ in the position of the phenyl ring at the meso position. In the first rotamer (A), which is also the least stable, the phenyl ring is almost perpendicular to the core of the BODIPY chromophore, whereas the phenyl ring is significantly twisted in the second (B) and third (C) rotamers (θ A = 89.8°; θ B = 24.9°; θ C = 63.6°). It should be pointed out that the position of this phenyl ring is determined by the relative strengths of two contradictory effects: (1) a steric effect between the phenyl ring and the two methyl groups at positions 1 and 7, which causes the phenyl ring to attempt to adopt a perpendicular position, and (2) π-electronic coupling between the core of the chromophore and the phenyl ring, which leads to a flattening of the molecule.The steric effect predominates in the excited state, resulting in a decrease in the energy of the molecule of 0.99 kcal/mol. Choosing a less stable conformer can lead to an error of about 22 nm in the position of the emission band. The second (B) and third (C) rotamers possess almost the same energies, and the positions of their emission bands are also very similar. However, the structures of these rotamers are different: the torsion angle of the phenyl ring is 24.9° for rotamer B and 63.6° for C. Therefore, the core of the chromophore is flat in C and curved in B. The abovementioned results suggest that it is possible to twist the phenyl ring to transition from one rotamer to another, and that bending the chromophore core leads to a transition from rotamers B and C: ΔE A-B = E S1(A) – E S1(B); ΔE A-C = E S1(A) – E S1(C) = 0.99 kcal/mol. Based on the results of the conformational analysis, conformer C was used in subsequent calculations.
Fig. 2.

Structure of compound 2 in the excited state in DMSO, as calculated using the PBE0 functional
It is important to underline that the calculations performed using the LR approach overestimated the shifts in the emission bands for compounds 2 and 3 (Fig. 1) that occur upon increasing the solvent polarity. In terms of the slope of the curve, the data for compound 1 are the closest to the corresponding experimental emission band data. As also seen for the absorption bands, the positions of the emission bands obtained by the PBE0 method are closer to the corresponding experimental data (630–700 nm for the calculated data vs 640–720 nm for the experimental data; Fig. 1) than the data obtained by the M06-2X method (590–670 nm for the calculated data vs 640–720 nm for the experimental data) when using the LR and SS approaches. However, the correlation between the data and the trend line is better when using M06-2X than when using PBE0. The calculations performed for compounds 2 and 3 using both the M06-2X and PBE0 functionals with the LR approach overestimate the influence of the solvent: note that the data points for each compound in the bottom left plot of Fig. 1 (which represent data obtained for the same compound in various solvents) are distributed in an almost vertical line, indicating that varying the solvent has a far greater effect on the calculated value of λ em for a particular compound than on the corresponding experimental value. On the other hand, when the SS approach is used with either functional, increasing the polarity of the solvent leads to relatively small shifts in the emission bands for compounds 2 and 3, which agrees well with the trend seen in the corresponding experimental data. The shifts seen in the calculated data for compound 1 are only in satisfactory agreement with the corresponding experimentally observed shifts, however.
Analysis of the influence of the solvent on the spectral characteristics of the dyes
To describe the influence of the solvent on the positions of both the absorption and emission bands calculated by the TD-DFT method, the dependencies of λ abs and λ em on the Lippert solvent parameter Δf were examined (see Figs. 3 and 4). The Lippert–Mataga parameter can be defined as [48–50]
| 1 |
where the parameters ε and n are the dielectric constant and the refractive index of the solvent, respectively.
Fig. 3.

Experimental (graph in the center) and calculated λ abs values of compounds 1–3 versus the Lippert solvent parameter ∆f (see Eq. 1)
Fig. 4.

Experimental (graph in the center) and calculated λ em values of compounds 1–3 versus the Lippert solvent parameter ∆f (see Eq. 1)
These relationships (derived using the SS and LR approaches) between the positions of the absorption and emission bands and Δf reflect the interactions between ground- and excited-state dye and solvent molecules. According to previous reported experimental data [18], significantly increasing the solvent polarity (i.e., changing the solvent from hexane to DMSO) does not cause a visible shift in the absorption bands of the compounds of interest (see the graph labeled EXP in Fig. 3).
Remarkably, the TD-DFT calculations also indicate that shifts in the absorption bands with increasing solvent polarity are rather small (regardless of whether the PBE0 or M06-2X functional and the SS or LR approach are used; Fig. 3). This effect indicates that significant structural changes (e.g., proton transfer, isomerization, etc.) are absent in the ground state. On the other hand, replacing the substituent (i.e., 2 → 3 → 1) strongly influences the position of the absorption band, causing a bathochromic shift of this band (Fig. 3). As mentioned above, this phenomenon is caused by differences in the electron-donor strengths of the substituents. The order of absorption-band bathochromic shifts (compound 1 > 3 > 2) obtained using TD-DFT calculations is in good agreement with the experimentally observed order [18]; see the graphs in Fig. 3. However, the TD-DFT calculations performed using the SS approach with both functionals [see the (M06-2X, SS) and (PBE0, SS) graphs in Fig. 3] overestimate the influence of the solvent polarity on the position of the absorption band of compound 1.
When it is used to calculate the influence of the solvent on the position of the emission band, the LS approach (with both functionals) indicates that there is a systematic bathochromic shift in the emission band of compound 1 with increasing solvent polarity (i.e., increasing Δf; Fig. 4), but not for compounds 2 and 3. The dependency of λ em on Δf was illustrated more precisely by applying the M06-2X functional and the SS approach. Increasing solvent polarity led to small changes in the positions of the emission bands for compounds 2 and 3 and a significant bathochromic shift for compound 1. Thus, to sum up, calculations performed using the LR approach (with both functionals) gave more accurate results (i.e. data closer to the corresponding experimental data) than the SS approach, although the data obtained using the SS approach (with the M06-2X functional) showed more precise trends. Notably, the LR and SS approaches yield different potential curves in the ground and excited states. The calculations performed using the LR approach led to wider potential curves for the ground and excited states than those obtained using the SS approach (Fig. S2 in the ESM).
It is important to note that the solvent polarity parameter E N T(30) more accurately describes hydrogen bonds than the parameter Δf does; see [47]. When we explored the dependency of the Stokes shift Δν on E N T(30), we obtained rather complicated results, because it was necessary to account for the polarity of the solvent before the positions of the absorption and emission bands could be determined accurately.
The experimental dependency reported in [18] shows that for compounds 2 and 3, the Stokes shift remains almost constant with increasing solvent polarity (see the EXP graph in Fig. 4). This phenomenon indicates that the molecule is weakly polarized during the transition from the ground state to the excited one. However, the Stokes shift is observed to increase significantly for compound 1 with increasing solvent polarity. That said, these changes in the Stokes shift are not stable, because the Stokes shift value for compound 1 decreases notably when this compound is placed in a protic solvent. This decrease in Stokes shift is the result of hydrogen bonding between the N,N-diethylamine moiety and the protic solvent. These interactions reduce the effect of the π-conjugated coupling between the N,N-diethylamine fragment and the BODIPY core, which, in turn, leads to an increase in the polarization of the dye and thus an increase in the Stokes shift. Unfortunately, not all of the calculations give results that agree well with the corresponding experimental results. The calculations performed using the M06-2X functional with the LR approach reveal sudden decreases in the Stokes shift for all three compounds (Fig. 5). However, satisfactory results were obtained using the M06-2X functional with the SS approach.
Fig. 5.

Dependency of the Stokes shift on the solvent polarity parameter E N T(30) for each of the studied compounds
We also report here on the difference between the potential curves obtained using the SS and LR approaches for the ground and excited states (Fig. S2 in the ESM). We found that that potential curves obtained using the SS approach were wider for the ground and excited states than those obtained using the LR approach.
Conclusions
This paper presents a comparison of the results of calculations of BODIPY dyes performed using the M06-2X and PBE0 functionals and the LR and SS approaches with the corresponding experimental data.
An analysis of the influence of the substituent (either N,N-dimethylaminobenzyl, ortho-fluorophenol, or methyl) at position 3 of the BODIPY core was accomplished. It was found that the presence of the N,N-dimethylaminobenzyl substituent induced the largest shifts of the absorption and emission bands of the dyes to the bathochromic region, in good agreement with the experimental data [18, 51, 52].
A conformational analysis of the studied compounds in the excited state was performed, based on TD-DFT calculations. It was found that the three identified conformers do not differ significantly in energy. A similar conclusion was drawn by Mennucci and coworkers [53] following calculations of BODIPY dyes performed using the LR and cLR approaches.
The positions of the absorption and emission bands calculated using the PBE0 and M06-2X functionals with both approaches tend to show bathochromic shifts, where the shifts increase according to the sequence: compound 2 < compound 3 < compound 1, which is in accordance with the experimental results. As for the dependencies of the spectral parameters (λ abs and λ em) on the Lippert solvent parameter Δf, we found that (1) the trend for λ abs vs Δf observed experimentally is accurately reproduced by calculations utilizing the PBE0 and M06-2X functionals and the LR approach (the SS approach provides only satisfactory results), and (2) the trend in λ em vs Δf observed experimentally is well described by calculations performed using the M06-2X functional and the SS approach. It is a remarkable fact that only calculations carried out using the M06-2X functional and the SS approach were able to describe the dependency of Δν on the solvent polarity parameter E N T(30) even fairly well. Therefore, the influence of the solvent on the absorption and emission bands of the dyes of interest should be determined using two functionals: the PBE0 functional (with the LR approach) and the M06-2X functional (with the SS approach).
Electronic supplementary material
Below is the link to the electronic supplementary material.
(DOCX 2261 kb)
Acknowledgments
The authors gratefully acknowledge the Wroclaw Centre for Networking and Supercomputing (WCSS) for their use of its computational facilities. This work was supported by RFBR (no: 16-03-00405) and KNOW-9 grants.
Footnotes
This paper belongs to Topical Collection 7th Conference on Modeling & Design of Molecular Materials in Trzebnica (MDMM 2016)
References
- 1.Treibs A, Kreuzer F-H. Difluorboryl-Komplexe von Di- und Tripyrrylmethenen. Liebigs Ann Chem. 1968;718:208–223. doi: 10.1002/jlac.19687180119. [DOI] [Google Scholar]
- 2.Loudet A, Burgess K (2007) Dyes and their derivatives: syntheses and spectroscopic properties. Chem Rev 107:4891–4932 [DOI] [PubMed]
- 3.Boens N, Leen V, Dehaen W. Fluorescent indicators based on BODIPY. Chem Soc Rev. 2012;41:1130–1172. doi: 10.1039/C1CS15132K. [DOI] [PubMed] [Google Scholar]
- 4.Ulrich G, Ziessel R, Harriman A. The chemistry of fluorescent BODIPY dyes: versatility unsurpassed. Ang Chem Int Ed. 2008;47:1184–1201. doi: 10.1002/anie.200702070. [DOI] [PubMed] [Google Scholar]
- 5.Benstead M, Mehl GH, Boyle RW (2011) 4,4′-Difluoro-4-bora-3a,4a-diaza-s-indacenes (BODIPYs) as components of novel light active materials. Tetrahedron 67:3573–3601
- 6.Knut R, Resch-Genger U. Rigidization, preorientation and electronic decoupling—the ‘magic triangle’ for the design of highly efficient fluorescent sensors and switches. Chem Soc Rev. 2002;31:116–127. doi: 10.1039/b100604p. [DOI] [PubMed] [Google Scholar]
- 7.Yuming Y, Zhao Q, Feng W, Li F. Luminescent chemodosimeters for bioimaging. Chem Rev. 2013;113:192–270. doi: 10.1021/cr2004103. [DOI] [PubMed] [Google Scholar]
- 8.Kobayashi H, Ogawa M, Alford R, Choyke PL, Urano Y. New strategies for fluorescent probe design in medical diagnostic imaging. Chem Rev. 2010;110:2620–2640. doi: 10.1021/cr900263j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le Guennic B, Chibani S, Charaf-Eddin A, Ziessel R, Ulrich G, Jacquemin D. The NBO pattern in luminescent chromophores: unravelling excited-state features using TD-DFT. Phys Chem Chem Phys. 2013;15:7534–7540. doi: 10.1039/c3cp50669j. [DOI] [PubMed] [Google Scholar]
- 10.Chibani S, Le Guennic B, Charaf-Eddin A, Maury O, Andraud C, Jacquemin D. On the computation of adiabatic energies in azaboron-dipyrromethene dyes. J Chem Theory Comput. 2012;8:3303–3313. doi: 10.1021/ct300618j. [DOI] [PubMed] [Google Scholar]
- 11.Jiao L, Yu C, Wang J, Briggs EA, Besley NA, Robinson D, Ruedas-Rama MJ, Orte A, Crovetto L, Talavera EM, Alvarez-Pez JM, Van der Auweraer M, Boens N (2015) Unusual spectroscopic and photophysical properties of meso-tert-butylBODIPY in comparison to related alkylated BODIPY dyes. RSC Adv 5:89375–89388
- 12.Chibani S, Le Guennic B, Charaf-Eddin A, Laurent AD, Jacquemin D. Revisiting the optical signatures of BODIPY dyes with theoretical tools. Chem Sci. 2013;4:1950–1963. doi: 10.1039/c3sc22265a. [DOI] [Google Scholar]
- 13.Adamo C, Jacquemin D (2013) The calculations of excited-state properties with time-dependent density functional theory. Chem Soc Rev 42:845–856 [DOI] [PubMed]
- 14.Alberto ME, De Simone BC, Mazzone G, Quartarolo AD, Russo N (2014) Theoretical determination of electronic spectra and intersystem spin–orbit coupling: the case of isoindole-BODIPY dyes. J Chem Theory Comput 10:4006–4013 [DOI] [PubMed]
- 15.Ji S, Ge J, Escudero D, Wang Z, Zhao J, Jacquemin D (2015) Molecular structure–intersystem crossing relationship of heavy-atom-free BODIPY triplet photosensitizers. J Org Chem 80:5958–5963 [DOI] [PubMed]
- 16.Mazzone G, Quartarolo AD, Russo N. PDT-correlated photophysical properties of thienopyrrole BODIPY derivatives. Theoretical insights. Dyes Pigments. 2016;130:9–15. doi: 10.1016/j.dyepig.2016.02.021. [DOI] [Google Scholar]
- 17.Santoro F, Jacquemin D (2016) Going beyond the vertical approximation with timedependent density functional theory. WIREs Comput Mol Sci. doi:10.1002/wcms.1260
- 18.Filarowski A, Kluba M, Cieślik-Boczula K, Koll A, Kochel A, Pandey L, De Borggraeve W, Van der Auweraer M, Catalán J, Boens N. Generalized solvent scales as a tool for investigating solvent dependence of spectroscopic and kinetic parameters. Application to fluorescent BODIPY dyes. Photochem Photobiol Sci. 2010;9:996–1008. doi: 10.1039/c0pp00035c. [DOI] [PubMed] [Google Scholar]
- 19.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA et al (2009) Gaussian 09, revision D.01. Gaussian, Inc., Wallingford
- 20.Adamo C, Barone V (1999) Toward reliable density functional methods without adjustable parameters: the PBE0 model. J Chem Phys 110:6158–6170
- 21.Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Acc. 2008;120:215–241. doi: 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- 22.Ditchfield R, Hehre WJ, Pople JA (1971) Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J Chem Phys 54:724–728
- 23.Hehre WJ, Ditchfield R, Pople JA (1972) Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J Chem Phys 56:2257–2261
- 24.Hariharan PC, Pople JA. Influence of polarization functions on molecular-orbital hydrogenation energies. Theor Chem Acc. 1973;28:213–222. doi: 10.1007/BF00533485. [DOI] [Google Scholar]
- 25.Hariharan PC, Pople JA (1974) Accuracy of AHn equilibrium geometries by single determinant molecular orbital theory. Mol Phys 27:209–214
- 26.Gordon MS. The isomers of silacyclopropane. Chem Phys Lett. 1980;76:163–168. doi: 10.1016/0009-2614(80)80628-2. [DOI] [Google Scholar]
- 27.Francl MM, Pietro WJ, Hehre WJ, Binkley JS, DeFrees DJ, Pople JA, Gordon MS. Self‐consistent molecular orbital methods. XXIII. A polarization‐type basis set for second‐row elements. J Chem Phys. 1982;77:3654–3665. doi: 10.1063/1.444267. [DOI] [Google Scholar]
- 28.Binning RC, Jr, Curtiss LA. Compact contracted basis sets for third-row atoms: Ga–Kr. J Comp Chem. 1990;11:1206–1216. doi: 10.1002/jcc.540111013. [DOI] [Google Scholar]
- 29.Blaudeau J-P, McGrath MP, Curtiss LA, Radom L (1997) Extension of Gaussian-2 (G2) theory to molecules containing third-row atoms K and Ca. J Chem Phys 107:5016–5021
- 30.Rassolov VA, Pople JA, Ratner MA, Windus TL. 6-31G* basis set for atoms K through Zn. J Chem Phys. 1998;109:1223–1229. doi: 10.1063/1.476673. [DOI] [Google Scholar]
- 31.Rassolov VA, Ratner MA, Pople JA, Redfern PC, Curtiss LA. 6-31G* basis set for third-row atoms. J Comp Chem. 2001;22:976–984. doi: 10.1002/jcc.1058. [DOI] [Google Scholar]
- 32.Hohenberg P, Kohn W (1964) Inhomogeneous electron gas. Phys Rev 136:B864–B871
- 33.Kohn W, Sham LJ (1965) Self-consistent equations including exchange and correlation effects. Phys Rev 140:A1133–A1138
- 34.Petersilka M, Gossmann UJ, Gross EKU (1996) Excitation energies from time-dependent density-functional theory. Phys Rev Lett 76:1212–1215 [DOI] [PubMed]
- 35.Casida ME (1995) In: Chong DP (ed) Recent advances in density functional methods. World Scientific, Singapore, 1:155–192
- 36.Miertuš S, Scrocco E, Tomasi J (1981) Electrostatic interaction of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects. Chem Phys 55:117–129
- 37.Scalmani G, Frisch MJ, Mennucci B, Tomasi J, Cammi R, Barone V (2006) Geometries and properties of excited states in the gas phase and in solution: theory and application of a time-dependent density functional theory polarizable continuum model. J Chem Phys 124:094107–094121 [DOI] [PubMed]
- 38.Tomasi J, Mennucci B, Cammi R. Quantum mechanical continuum solvation models. Chem Rev. 2005;105:2999–3093. doi: 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- 39.Cossi M, Barone V. Time-dependent density functional theory for molecules in liquid solutions. J Chem Phys. 2001;115:4708–4717. doi: 10.1063/1.1394921. [DOI] [Google Scholar]
- 40.Cammi R, Mennucci B, Tomasi J (2000) Fast evaluation of geometries and properties of excited molecules in solution: a Tamm–Dancoff model with application to 4-dimethylaminobenzonitrile. J Phys Chem A 104:5631–5637
- 41.Cossi M, Barone V. Solvent effect on vertical electronic transitions by the polarizable continuum model. J Chem Phys. 2000;112:2427–2435. doi: 10.1063/1.480808. [DOI] [Google Scholar]
- 42.Hansch C, Leo A, Taft RW. A survey of Hammett substituent constants and resonance and field parameters. Chem Rev. 1991;91:165–195. doi: 10.1021/cr00002a004. [DOI] [Google Scholar]
- 43.Grabowski ZR, Rotkiewicz K, Rettig W. Structural changes accompanying intramolecular electron transfer: focus on twisted intramolecular charge-transfer states and structures. Chem Rev. 2003;103:3899–4032. doi: 10.1021/cr940745l. [DOI] [PubMed] [Google Scholar]
- 44.Chulvi K, Costero AM, Ochando LE, Gil S, Vivancos J-L, Gavina P (2015) Solvatochromic and single crystal studies of two neutral triarylmethane dyes with a quinone methide structure. Molecules 20:20688–20698 [DOI] [PMC free article] [PubMed]
- 45.Chibani S, Charaf-Eddin A, Mennucci B, Le Guennic B, Jacquemin D. Optical signatures of OBO fluorophores: a theoretical analysis. J Chem Theory Comput. 2014;10:805–815. doi: 10.1021/ct4009848. [DOI] [PubMed] [Google Scholar]
- 46.Chibani S, Budzak S, Medved M, Mennucci B, Jacquemin D. Full cLR-PCM calculations of the solvatochromic effects on emission energies. Phys Chem Chem Phys. 2014;47:26024–26029. doi: 10.1039/C4CP03919J. [DOI] [PubMed] [Google Scholar]
- 47.Lippert EZ (1955) Dipolmoment und Elektronenstruktur von angeregten Molekülen. Z Naturforsch A 10:541–545
- 48.Mataga N, Kaifu Y, Koizumi M (1955) The solvent effect on fluorescence spectrum. Change of solute–solvent interaction during the lifetime of excited solute molecule. Bull Chem Soc Jpn 28:690–691
- 49.Mataga N, Kaifu Y, Koizumi M (1956) Solvent effects upon fluorescence spectra and the dipole-moments of excited molecules. Bull Chem Soc Jpn 29:465–470
- 50.Reichardt C (1994) Solvatochromic dyes as solvent polarity indicators. Chem Rev 94:2319–2358
- 51.Deniz E, Isbasar GC, Bozdemir OA, Yildirim LT, Siemiarczuk A, Akkaya EU (2008) Bidirectional switching of near IR emitting boradiazaindacene fluorophores. Org Lett 16:3401–3403 [DOI] [PubMed]
- 52.Rurack K, Kollmannsberger M, Daub J (2001) Molecular switching in the near infrared (NIR) with a functionalized boron–dipyrromethene dye. Angew Chem Int Ed 40:385–387 [DOI] [PubMed]
- 53.Jacquemin D, Chibani S, Le Guennic B, Mennucci B (2014) Solvent effects on cyanine derivatives: a PCM investigation. J Phys Chem A 118:5343–5348 [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 2261 kb)