

Abstract
Co-regulation of genes has been extensively analyzed, however, rather limited knowledge is available on co-regulations within the miRNome. We investigated differential co-expression of microRNAs (miRNAs) based on miRNome profiles of whole blood from 540 individuals. These include patients suffering from different cancer and non-cancer diseases, and unaffected controls. Using hierarchical clustering, we found 9 significant clusters of co-expressed miRNAs containing 2–36 individual miRNAs. Through analyzing multiple sequencing alignments in the clusters, we found that co-expression of miRNAs is associated with both sequence similarity and genomic co-localization. We calculated correlations for all 371,953 pairs of miRNAs for all 540 individuals and identified 184 pairs of miRNAs with high correlation values. Out of these 184 pairs of miRNAs, 16 pairs (8.7%) were differentially co-expressed in unaffected controls, cancer patients and patients with non-cancer diseases. By computing correlated and anti-correlated miRNA pairs, we constructed a network with 184 putative co-regulations as edges and 100 miRNAs as nodes. Thereby, we detected specific clusters of miRNAs with high and low correlation values. Our approach represents the most comprehensive co-regulation analysis based on whole miRNome-wide expression profiling. Our findings further decrypt the interactions of miRNAs in normal and human pathological processes.
Keywords: Co-expression, Microarray, MicroRNA, Network analysis
Introduction
Microarray experiments have been applied for almost three decades in the detection of disease-relevant changes in gene expression patterns. While in early ages genes have mostly been considered independently from each other, cluster [1], [2] and classification [3], [4], [5] technologies have more recently been applied to find patterns of differentially expressed genes. Finally, gene set analysis approaches [6], [7] and methods integrating pathway topology [8], [9] have been developed to understand the interplay of genes. These approaches have been successfully applied to studies in small non-coding RNAs, e.g., microRNAs (miRNAs).
With the increasing availability of expression profiles for various diseases, differential co-expression of genes moved into the focus of attention. The term “differential co-expression” was firstly coined by Bennets in 1986 [10] studying the co-expression of alpha-actins within the human heart. In 1992, Swiderski reported differential co-expression of long and short form type IX collagen transcripts during avian limb chondrogenesis [11]. Co-expression analysis of genes using microarray technology has also been applied to other human pathologies, including cancer [12]. In 2009, Mo and co-workers presented a stochastic model to identify co-expression patterns of differential gene pairs in prostate cancer progression [13]. Comprehensive methods to detect differential co-expression have been developed by Lai who reported an efficient pattern recognition algorithm [14]. This algorithm used Expected Conditional F-statistic that incorporates statistical information of location and correlation or other scores as proposed by Koska and Spang [15]. Subsequently, several tools and software packages with respective functionality have been developed including CoXpress [16], DiffCoEx [17], dCoxS [18] and differential co-expression framework [19].
Only a few studies have been reported for analysis of differential co-expression for miRNAs. An example is the construction of an miRNA–miRNA synergistic network via co-regulating functional modules and disease miRNA topological features [20]. One reason for the lack of miRNA co-expression studies is certainly the paucity of miRNA expression profiling data. Gene expression profiles have been measured for almost three decades in numerous microarray experiments, of which hundreds of thousands are currently available through the Gene Expression Omnibus [21], [22], however, only a fraction of array data sets are available for miRNAs. The most frequently applied microarray platform is the Agilent miRNA microarray 2.0. Another technology which is frequently applied is the MPEA assay (Febit Biomed, Heidelberg) that has been used to measure several hundred blood-based miRNA profiles which are the source for our meta-analysis.
Previously, Riveros and co-workers reported a comprehensive study for differential co-expression of miRNA that was derived from whole blood of patients with multiple sclerosis [23], providing evidence that differential co-expression from body fluids can be accessed. miRNA expression patterns from human blood cells are increasingly discussed for their potential as a minimal invasive diagnostic tool. Most recently, we reported blood-based miRNA expression patterns for 14 different human pathologies [24] including lung cancer [25], COPD [26], multiple sclerosis [27], ovarian cancer [28], glioblastoma [29], and acute myocardial infarction [30]. Since the various cohorts are relatively small as compared to the large number of potential pair-wise co-expressions, we combined the different data sets into a meta-analysis. Here, we investigate the differential co-expression patterns using the data of a total of 540 blood-based miRNA expression profiles.
Results and discussion
Co-localization and co-expression of miRNAs
As a first approach towards understanding the interplay of miRNAs, we applied hierarchical clustering to the data set containing 540 samples measured for the expression of 863 miRNAs. To reduce the noise, we excluded miRNAs with low expression values (detailed in Material and methods). An average linkage bottom up clustering detected a total of nine significant clusters (P < 0.05). These clusters each contain 2–36 miRNAs (Figure 1). Notably, many clusters contained miRNAs with similar sequences. Good example for co-expression related to similar sequences is Cluster 8 that contains hsa-miR-23a and hsa-miR-23b or Cluster 5 that contains hsa-miR-19a and hsa-miR-19b. The biological mechanism underlying co-expression of miRNAs with similar sequence remains to be elucidated. It is possible that co-expressed miRNAs of similar sequence share similar targets. Other than that, reduced specificity of hybridization-based approaches could partially explain this co-expression. On the other hand, as expected, we also found many miRNAs that clustered together but had different sequences, such as hsa-miR-1260 and hsa-miR-30c in Cluster 3. Respective pair-wise sequence alignments (hsa-miR-19a/hsa-miR-19b, hsa-miR23a/hsa-miR23b, hsa-miR-1260/hsa-miR-30c) are shown in Figure 2.
Figure 1.

Cluster dendrogram of miRNAs Red boxes denote significant clusters as computed by bootstrap re-sampling. The red values were calculated by bootstrap re-sampling and those >95%, corresponding to significance level of 0.05, are considered as significant. Values in green and gray indicate bootstrap probability (BP) and the edge number in the dendrogram, respectively. The significant clusters with approximately unbiased (AU) value greater than 95% (P < 0.05) are labeled with numbers in circle in increasing order from left to right.
Figure 2.

Alignments of co-expression miRNA clusters with similar or different sequences Pairwise sequence alignment indicated that hsa-miR-23a and hsa-miR-23b in Cluster 8 (upper panel) and hsa-miR-19a and hsa-miR-19b in Cluster 5 (middle panel) show high sequence similarity, while there is lower sequence similarity for hsa-miR-1260 and hsa-miR-30c in Cluster 3 (lower panel).
To test the hypothesis that miRNAs belonging to the same polycistronic miRNA cluster or the same miRNA family are co-expressed, we additionally performed enrichment analyses. For each significant set containing more than 5 miRNAs (Clusters 4, 6 and 7 in Figure 1), we performed the enrichment analysis separately to see whether the selected miRNA clusters or families are over-represented. In line with our expectations, the let-7a, miR-106a, miR-106b, miR-15a and miR-17 clusters were significantly enriched (all P ⩽ 0.005) in our Cluster 6, whereas members of the miR-192 polycistronic miRNA cluster were mostly found in Cluster 7 (P = 0.001). Likewise, we also found a strong enrichment of miRNA families in our clusters, such as the let-7 family (P = 0.002), the miR-15 family (P = 0.001), the miR-320 family (P = 0.00002) and the miR-17 family (P = 3E-8) in Cluster 6 and the miR-103 family (P = 0.001) in Cluster 7. Interestingly, no significant enrichment for a known miRNA cluster or family was found in Cluster 4, indicating that our clustering approach groups not only polycistronic (and thus co-transcribed) miRNA clusters or known miRNA families, but also miRNAs that are co-expressed for different reasons. In addition, divergent behavior of individual miRNAs belonging to the same polycistronic cluster or family provides evidence for a significant post-transcriptional component in miRNA expression.
An additional reason for putative co-regulation of miRNAs might be their co-localization in the genome. To this end, we searched for miRNAs that have been clustered together based on the expression data and are located on the same chromosome. Subsequently candidate pairs were mapped to the exact chromosomal position. We found five pairs of miRNAs that showed a high absolute correlation (⩽−0.5 or ⩾0.5) and are located on the same chromosome, as presented in Table 1. Three of those five miRNA pairs showed positive correlation while the remaining two pairs showed negative correlation. The three pairs with positive correlation are located within a distance of 500 base pairs of each other and were each on the same strand. On the other hand, larger genomic distances were found for the two negatively-correlated miRNA pairs. For example, the distance between hsa-miR-423-5p and hsa-miR-144 was about 10 million base pairs (Mb). Moreover, hsa-miR-423-5p was located on the plus strand whereas hsa-miR-144 was located on the minus strand. Figure 3 shows expression values of one pair of positively-correlated miRNAs, namely hsa-miR-20a/hsa-miR-17 (Figure 3A) and one pair of negatively-correlated miRNAs, namely hsa-miR-423-5p/hsa-miR-144 (Figure 3B) for 540 analyzed blood samples. The results showed that the cohorts behaved similarly for each of the pairs.
Table 1.
Co-localization of correlated miRNAs
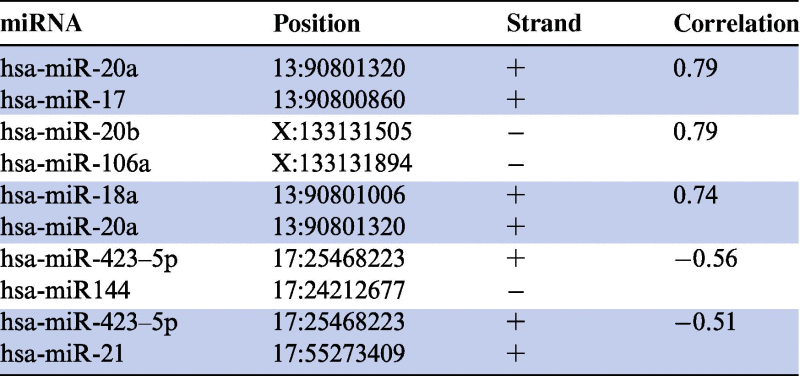 |
Figure 3.

Representative expression profiles of correlated miRNA pairs A. Positive correlation. Expression of two positively-correlated miRNAs, hsa-miR-17 and hsa-miR-20a, was measured for 540 individuals including controls (n = 72, black circle), cancer patients (n = 276, red circle) and non-cancer patients (n = 192, blue circle). B. Negative correlation. Expression of two negatively-correlated miRNAs, hsa-miR-423-5p and hsa-miR-144, was measured for 540 individuals including controls (black circle), cancer patients (red circle) and non-cancer patients (blue circle). A complete list of the disease types and the respective numbers of patients is shown in Table 4.
Differential co-expression of miRNAs
The 540 individuals participating in this study can be grouped in three different cohorts, including unaffected healthy individuals (control), cancer patients (cancer) and non-cancer patients (non cancer). For these three cohorts we asked whether the correlation is equally high in all three groups or whether certain cohorts deviate from the others. To this end, we computed for each pair of miRNAs the correlation values for the three cohorts separately. As a result of the calculation for all pairs, the values of correlation range from −0.67 to 0.89 with average correlation of 0.013. As the slight positive average correlation already indicates, we obtained slightly more positive correlations than negative ones. Thus, we applied different thresholds for positive and negative correlations to acknowledge this non-symmetric distribution. We only considered positively-correlated miRNA pairs with correlation values higher than 0.7 and negatively-correlated miRNA pairs with values lower than −0.5. Using these thresholds we obtained 184 miRNA pairs out of 371,953 pairs in total (0.05%). Of these 184 miRNA pairs, 118 were positively correlated and 66 were negatively correlated. To estimate the extent of differential expression in the 3 cohorts, we computed the variance of the correlation values, ranging from 10−5 to 0.24 with an average of 0.02. The 16 miRNA pairs with the highest variance, corresponding to the most differentially-regulated miRNAs (variance >0.05), are summarized in Table 2 and Figure 4 and the 16 miRNA pairs with the lowest variance (variance ⩽0.00053) are indicated in Table 3 and Figure 5.
Table 2.
Differential co-expression in diseases
| miRNA 1 | miRNA 2 | Overall correlation | Control correlation | Cancer correlation | Non cancer correlation | Variance |
|---|---|---|---|---|---|---|
| hsa-miR-377∗ | hsa-miR-548l | 0.756 | −0.129 | 0.522 | 0.842 | 0.245 |
| hsa-miR-196b | hsa-miR-548l | 0.73 | −0.028 | 0.416 | 0.864 | 0.199 |
| hsa-miR-548l | hsa-miR-135b | 0.72 | −0.023 | 0.373 | 0.863 | 0.197 |
| hsa-miR-423-5p | hsa-miR-144 | −0.556 | 0.052 | −0.651 | −0.462 | 0.132 |
| hsa-miR-595 | hsa-miR-574-5p | 0.743 | 0.121 | 0.78 | 0.62 | 0.118 |
| hsa-miR-363 | hsa-miR-320d | −0.507 | 0.019 | −0.481 | −0.624 | 0.114 |
| hsa-miR-320c | hsa-miR-363 | −0.519 | −0.022 | −0.539 | −0.56 | 0.093 |
| hsa-miR-320b | hsa-miR-144 | −0.575 | −0.078 | −0.619 | −0.582 | 0.091 |
| hsa-miR-106a | hsa-miR-720 | −0.577 | −0.128 | −0.626 | −0.582 | 0.076 |
| hsa-miR-106a | hsa-miR-320c | −0.504 | −0.068 | −0.499 | −0.568 | 0.073 |
| hsa-miR-144 | hsa-miR-320a | −0.571 | −0.143 | −0.642 | −0.542 | 0.07 |
| hsa-miR-126 | hsa-miR-720 | −0.513 | −0.079 | −0.537 | −0.534 | 0.069 |
| hsa-miR-144 | hsa-miR-720 | −0.552 | −0.144 | −0.584 | −0.56 | 0.061 |
| hsa-miR-720 | hsa-miR-20b | −0.58 | −0.187 | −0.599 | −0.602 | 0.057 |
| hsa-miR-23b | hsa-miR-23a | 0.708 | 0.323 | 0.751 | 0.709 | 0.056 |
| hsa-miR-17 | hsa-miR-151-3p | −0.512 | −0.205 | −0.378 | −0.657 | 0.052 |
Figure 4.

Correlations of 16 miRNA pairs with variance > 0.05 Differential co-expression of these miRNA pairs is shown separately for cancer patients (red), non-cancer patients (green) and healthy controls (blue). Co-expression of the miRNA pairs was more frequently detected in cancer and non-cancer patients than in healthy controls.
Table 3.
Non-differential co-expression in diseases
| miRNA 1 | miRNA 2 | Overall correlation | Control correlation | Cancer correlation | Non-cancer correlation | Variance |
|---|---|---|---|---|---|---|
| hsa-miR-593∗ | hsa-miR-646 | 0.791 | 0.813 | 0.803 | 0.77 | <0.001 |
| hsa-miR-93 | hsa-miR-20b | 0.737 | 0.758 | 0.751 | 0.721 | <0.001 |
| hsa-miR-593∗ | hsa-miR-214 | 0.834 | 0.815 | 0.824 | 0.852 | <0.001 |
| hsa-miR-330-3p | hsa-miR-621 | 0.858 | 0.887 | 0.859 | 0.851 | <0.001 |
| hsa-miR-593∗ | hsa-miR-331-3p | −0.503 | −0.5 | −0.488 | −0.523 | <0.001 |
| hsa-miR-374b | hsa-miR-374a | 0.722 | 0.729 | 0.734 | 0.702 | <0.001 |
| hsa-miR-621 | hsa-miR-593∗ | 0.838 | 0.852 | 0.849 | 0.822 | <0.001 |
| hsa-miR-330-3p | hsa-miR-214 | 0.801 | 0.825 | 0.793 | 0.815 | <0.001 |
| hsa-miR-452∗ | hsa-miR-593∗ | 0.748 | 0.754 | 0.756 | 0.727 | <0.001 |
| hsa-miR-500 | hsa-miR-195 | −0.532 | −0.509 | −0.54 | −0.52 | <0.001 |
| hsa-miR-1228∗ | hsa-miR-149∗ | 0.719 | 0.755 | 0.731 | 0.73 | <0.001 |
| hsa-miR-107 | hsa-miR-331-3p | −0.67 | −0.685 | −0.658 | −0.675 | <0.001 |
| hsa-miR-330-3p | hsa-miR-452∗ | 0.793 | 0.809 | 0.793 | 0.792 | <0.001 |
| hsa-miR-509-5p | hsa-miR-933 | 0.842 | 0.854 | 0.841 | 0.848 | <0.001 |
| hsa-miR-584 | hsa-miR-362-5p | 0.713 | 0.709 | 0.703 | 0.715 | <0.001 |
| hsa-miR-1184 | hsa-let-7i∗ | 0.792 | 0.786 | 0.788 | 0.792 | <0.001 |
Figure 5.

Correlations of 16 miRNA pairs with variance ⩽ 0.00053 Differential co-expression of these miRNA pairs is shown separately for cancer patients (red), non-cancer patients (green) and healthy controls (blue). Co-expression of the miRNA pairs was comparable in the three groups.
By examining the differential co-expression of the 16 miRNA pairs with variance >0.05, we found that both the cancer patients and the non-cancer patients deviate from the healthy controls. As compared to the healthy controls, co-expression of these 16 miRNA pairs was detected significantly more frequently in both cancer and non-cancer disease groups. Overall the correlation between cancer and non-cancer diseases was 0.95 while decreased correlation was revealed between control and cancer and between control and non-cancer diseases, which is 0.59 and 0.49, respectively. Further analysis identified five miRNA pairs that were positively correlated in patients but not in healthy controls. For example, the pair hsa-miR-23a/hsa-miR-23b showed correlation of 0.71 in non-cancer patients (P < 10−10) and 0.75 in cancer patients (P < 10−10) but only 0.32 in healthy controls (P < 0.01) with the respective 95% confidence intervals as 0.69–0.80, 0.63–0.79 and 0.07–0.54. Moreover, we found 11 miRNAs that were highly anti-correlated, i.e., negatively correlated both in cancer and in non-cancer patients but again not correlated in healthy controls.
These results indicate that the observed overall high variance for the 16 miRNA pairs is mostly due to the healthy controls. While the 16 pairs were only weakly correlated in healthy controls they were correlated or anti-correlated in the patients. The results that may be biased due to slightly different cohort sizes may give first and certainly only preliminary evidence that miRNA expression may be more homogenously coordinated in patients, possibly indicating a change in expression regulation that is common to different types of diseases.
Interestingly, all miRNAs of the miRNA pairs that are negatively correlated in cancer or non-cancer patients but not in healthy controls have been previously related to human diseases according to the Human miRNA and Disease Database (HMDD) [31]. For examples, the known disease-associated miRNAs that were identified as negatively correlated in our study included hsa-miR-17 that was according to the HMDD associated with 33 different diseases, hsa-miR-128 with 18 diseases, hsa-miR-20b with 9 diseases, hsa-miR-423 with 4 diseases and hsa-miR-363 with 3 diseases. This finding is even more profound, since only about one third of all known miRNAs in the HMDD are related to one or more diseases [31]. Obviously, the analysis of co-expression can contribute to the identification of disease-associated miRNAs.
Anti-correlation of expression and co-localization of miRNAs
Besides pairs of miRNAs that were correlated in patients and that were co-localized, we also identified co-localization of miRNA pairs for miRNAs which are anti-correlated in patients. For example, hsa-miR-423-5p and hsa-miR-144 are co-localized on chromosome 17 and are negatively correlated (−0.56). Specifically, the correlation value for this miRNA pair was −0.65 in non-cancer patients and −0.46 in cancer patients, respectively. However, we did not find a negative correlation for this miRNA pair in healthy controls (correlation of 0.05) (Table 1, Table 2).
Putative co-regulation network
Based on the analysis of co-regulated miRNAs, we constructed a network with 184 correlations (correlation value >0.7 or <−0.5). As shown in Figure 6, the derived network encompasses 100 different miRNAs. Roughly, the network can be divided into one large connected component and several small components consisting of 2 to 4 miRNAs each. The large connected component can again be subdivided into two clusters. The upper cluster shown in Figure 6 contains mostly positively correlated miRNA pairs as indicated by blue edges while the cluster shown at the bottom of Figure 6 contains both positive correlations and negative correlations (indicated by red edges). While the miRNAs in the upper cluster do not show obvious sequence similarity as indicated by thin edges, positively correlated miRNAs of the lower cluster show high sequence similarity as indicated by thick edges. The small components with two to four miRNAs are mostly positively correlated. For most of these pairs, the positive correlation is associated with sequence similarities, as for example for the pair of hsa-miR-23a and hsa-miR-23b and the pair of hsa-miR-1247a and hsa-miR-1247b.
Figure 6.

Correlation network Network was constructed with all positive correlations between pairs of miRNAs with a correlation of at least 0.7 (blue edges) and all negative correlations of at least −0.5 (red edges). The thickness of the edges corresponds to the alignment score. The pair-wise sequence alignment was computed using edit distance. Dashed edges indicate correlations that were different in controls and in patients.
Additionally, many of the positive and negative correlations shown in the bottom cluster are different between healthy controls and patients. The differences are visualized as a sub-network in Figure 7. The sub-network separates miRNAs with different correlation between healthy controls and patients (on the left) and miRNAs with similar correlations in healthy controls and patients (on the right). Among the miRNAs with similar correlations are four miRNAs of the let-7 family that have previously been associated with many human malignancies [31]. In addition, each of the remaining miRNAs of the sub-network has previously been associated with at least one human disease according to the HMDD [31].
Figure 7.

Differential co-expression sub-network Sub-network of miRNAs was constructed with correlations that were different between controls and patients (left side) and correlations that were similar between controls and patients (right side). The indication of edges was same as that used in Fig. 6.
Conclusion
Over almost three decades it has been shown that co-expression and specifically differential co-expression of genes play an important role in human pathogenic processes. However, differential co-expression has not been thoroughly analyzed for the miRNome. This is in part due to the lack of respective high-throughput data sets allowing the analysis of miRNA-miRNA interactions. We enlarged a recently published set of 454 whole miRNome profiles [24] to a total of 540 profiles. Analysis of the miRNA co-expression from these profiles provides supporting evidence that genomic localization and sequence similarity are associated with co-expression. In addition, we report a significantly enriched clustering for miRNAs that belong to the same miRNA families or polycistronic miRNA clusters. Moreover, our findings also support that the co-expression may be more pronounced in patients, compared to the healthy controls. Network based analysis allows us to detect specific clusters of miRNAs with high and low correlation. Interestingly, many of the identified differentially co-expressed miRNAs have previously been associated with human pathogenic processes. Notably, the reported data have not been measured from tissues but from blood cells. Since different tissues have specific miRNA profiles, a co-expression analysis would make sense only for one tissue type but not enabling a meta-analysis of different diseases. However, different blood cell compositions in different diseases might influence the overall result of our meta-analysis. Another limitation of our study is certainly the applied microarray technology. Novel approaches such as next-generation small RNA sequencing will likely improve the specificity of respective analyses in the future.
In summary, in human diseases, co-expression and differential co-expression of miRNAs seems to be of similar importance to co-expression of protein-coding genes.
Materials and methods
Patients
The screened cohort contains a total of 540 subjects including healthy controls (n = 72), cancer patients (n = 276) and patients with non-cancer diseases (n = 192). The detailed characteristics of the cohort are listed in Table 4. All blood donors participating in this study signed the informed consent form and the local ethics committee approved the analysis of miRNA expression in blood. Blood samples were collected using PAXgene Blood RNA tubes (BD, Franklin Lakes, New Jersey, USA).
Table 4.
Cohort characteristics
| Class | Disease | No. of samples |
|---|---|---|
| Control | Healthy | 72 |
| Cancer | Lung tumor | 35 |
| Ductal adenocarcinoma | 45 | |
| Melanoma | 35 | |
| Ovarian cancer | 15 | |
| Prostate carcinoma | 35 | |
| Wilms tumor | 50 | |
| Other pancreatic tumors | 48 | |
| Tumor of stomach | 13 | |
| Non cancer | Multiple sclerosis | 23 |
| Sarcoidosis | 45 | |
| Periodontitis | 18 | |
| COPD | 27 | |
| Myocardial infarction | 20 | |
| Pancreatitis | 37 | |
| Benign prostate hyperplasia | 22 | |
| Sum | 540 | |
Note: COPD, chronic obstructive pulmonary disease.
miRNA extraction and microarray screening
Total RNA isolation was performed using miRNeasy Mini Kit (Qiagen) as described previously [25].
Microarray analyses were done on the Geniom RT Analyzer using Geniom miRNA Biochips (Febit Biomed GmbH). Each of the 863 human miRNAs (Sanger miRBase v12.0 to v15.0) was present in at least seven replicates on each of the 540 arrays. The screening was done using micro-fluidics primer extension assay (MPEA) [32]. This assay differs from a standard hybridization assay in that it includes an additional primer extension step. The MPEA assay is verified to be very sequence-specific at the end of miRNAs and thus minimizes the cross-hybridization effect, which is of especially high importance for our cross-hybridization study.
Data processing and bioinformatics analysis
At the first preprocessing step, all arrays were locally background corrected. Then, the replicates of each miRNA on each microarray were merged by computing the median intensity value. To account for between-array effects, standard quantile normalization was performed [33], which showed superior performance as compared to normalization via spike-in or housekeeping miRNAs in previous experiments. All computations then were carried out on the expression matrix containing 863 rows representing 863 miRNAs and 540 columns representing the 540 individuals. The statistical analyses were carried out using R [34] if not mentioned otherwise. To reduce the noise, we excluded miRNAs with low expression values, i.e., the median signal intensity of a miRNA must be great than 500 for a specific miRNA to be considered in our study.
For hierarchical clustering, the pvclust package has been used. The package computes P values for hierarchical clustering based on a multiscale bootstrap resampling, helping to interpret clusters. Specifically, clusters that are highly supported by the data will have low P values while weaker clusters end up with non-significant P values. Significant clusters are enclosed with red boxes in the respective dendrogram. We used 1-abs(cor(x,y)) as a distance measure for the clustering, where cor(x,y) corresponds to the Pearson correlation coefficient of all 540 observations for two miRNAs x and y. By using this distance function, we detect miRNAs that are highly correlated and anti-correlated. In more detail, an average linkage bottom up clustering was carried out.
To compute differential expression and differential co-expression, we again used the pair-wise Pearson correlation of all 863 miRNAs, for all 540 samples together but also for the different groups of controls, cancers and non-cancer diseases, separately. As a result of this analysis, we calculated for miRNA pairs four different correlation values, the overall value and the single values for the three groups. To find the miRNA pairs with different behavior in different groups, we computed the variance of the 371,953 pairs as , where corcontrol,i corresponds to the correlation of control samples for a miRNA pair i, corcancer,i corresponds to the correlation of cancer samples for a miRNA pair i, cornon-cancer,i corresponds to the correlation of control samples for a miRNA pair i, and cori corresponds to the average of the three correlation values for miRNA pair i.
Empirically, we considered miRNA pairs with correlations above 0.7 as highly co-expressed and with correlations below -0.5 as anti-correlated. Using Cytoscape [35] we visualized the network of these miRNA pairs and visualized the results using Cytoscapes mapping functionality with an organic layout.
Authors’ contributions
CS contributed to the study design, data evaluation and wrote the manuscript; AK designed the study, wrote the manuscript and carried out computations; PL carried out the experiments; CB contributed to data analysis and revision of the manuscript; AC contributed to the analysis of miRNA clusters and families; JW contributed to study design and revision of the manuscript; BM assisted the data analysis and reviewed the manuscript; EM designed the study, wrote the manuscript. All authors read and approved the final manuscript.
Conflict of interest
CS and AK are affiliates of Siemens Healthcare, Erlangen, Germany.
Acknowledgements
We acknowledge all patients participating in this study. Financial support was granted by HOMFOR, Deutsche Forschungsgemeinschaft (DFG, LE 2783/1-1), and Hedwig-Stalter Foundation.
References
- 1.Eisen M.B., Spellman P.T., Brown P.O., Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Bin R., Risso D. A novel approach to the clustering of microarray data via nonparametric density estimation. BMC Bioinformatics. 2011;12:49. doi: 10.1186/1471-2105-12-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang J., Liu B., Jiang X., Zhao H., Fan M., Fan Z. A systems biology-based gene expression classifier of glioblastoma predicts survival with solid tumors. PLoS ONE. 2009;4:e6274. doi: 10.1371/journal.pone.0006274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Golub T.R., Slonim D.K., Tamayo P., Huard C., Gaasenbeek M., Mesirov J.P. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 5.Gill R., Datta S. A statistical framework for differential network analysis from microarray data. BMC Bioinformatics. 2010;11:95. doi: 10.1186/1471-2105-11-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Backes C., Keller A., Kuentzer J., Kneissl B., Comtesse N., Elnakady Y.A. GeneTrail – advanced gene set enrichment analysis. Nucleic Acids Res. 2007;35:W186–W192. doi: 10.1093/nar/gkm323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Al-Shahrour F., Minguez P., Vaquerizas J.M., Conde L., Dopazo J. BABELOMICS: a suite of web tools for functional annotation and analysis of groups of genes in high-throughput experiments. Nucleic Acids Res. 2005;33:W460–W464. doi: 10.1093/nar/gki456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rahnenfuhrer J., Domingues F.S., Maydt J., Lengauer T. Calculating the statistical significance of changes in pathway activity from gene expression data. Stat Appl Genet Mol Biol. 2004 doi: 10.2202/1544-6115.1055. 3:Article 16. [DOI] [PubMed] [Google Scholar]
- 9.Keller A., Backes C., Gerasch A., Kaufmann M., Kohlbacher O., Meese E. A novel algorithm for detecting differentially regulated paths based on gene set enrichment analysis. Bioinformatics. 2009;25:2787–2794. doi: 10.1093/bioinformatics/btp510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bennetts B.H., Burnett L., dos Remedios C.G. Differential co-expression of alpha-actin genes within the human heart. J Mol Cell Cardiol. 1986;18:993–996. doi: 10.1016/s0022-2828(86)80013-x. [DOI] [PubMed] [Google Scholar]
- 11.Swiderski R.E., Solursh M. Differential co-expression of long and short form type IX collagen transcripts during avian limb chondrogenesis in ovo. Development. 1992;115:169–179. doi: 10.1242/dev.115.1.169. [DOI] [PubMed] [Google Scholar]
- 12.Choi J.K., Yu U., Yoo O.J., Kim S. Differential coexpression analysis using microarray data and its application to human cancer. Bioinformatics. 2005;21:4348–4355. doi: 10.1093/bioinformatics/bti722. [DOI] [PubMed] [Google Scholar]
- 13.Mo W.J., Fu X.P., Han X.T., Yang G.Y., Zhang J.G., Guo F.H. A stochastic model for identifying differential gene pair co-expression patterns in prostate cancer progression. BMC Genomics. 2009;10:340. doi: 10.1186/1471-2164-10-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai Y., Wu B., Chen L., Zhao H. A statistical method for identifying differential gene-gene co-expression patterns. Bioinformatics. 2004;20:3146–3155. doi: 10.1093/bioinformatics/bth379. [DOI] [PubMed] [Google Scholar]
- 15.Kostka D., Spang R. Finding disease specific alterations in the co-expression of genes. Bioinformatics. 2004;20:i194–i199. doi: 10.1093/bioinformatics/bth909. [DOI] [PubMed] [Google Scholar]
- 16.Watson M. CoXpress: differential co-expression in gene expression data. BMC Bioinformatics. 2006;7:509. doi: 10.1186/1471-2105-7-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tesson B.M., Breitling R., Jansen R.C. DiffCoEx: a simple and sensitive method to find differentially coexpressed gene modules. BMC Bioinformatics. 2010;11:497. doi: 10.1186/1471-2105-11-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cho S.B., Kim J., Kim J.H. Identifying set-wise differential co-expression in gene expression microarray data. BMC Bioinformatics. 2009;10:109. doi: 10.1186/1471-2105-10-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chia B.K., Karuturi R.K. Differential co-expression framework to quantify goodness of biclusters and compare biclustering algorithms. Algorithms Mol Biol. 2010;5:23. doi: 10.1186/1748-7188-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu J., Li C.X., Li Y.S., Lv J.Y., Ma Y., Shao T.T. MiRNA-miRNA synergistic network: construction via co-regulating functional modules and disease miRNA topological features. Nucleic Acids Res. 2011;39:825–836. doi: 10.1093/nar/gkq832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barrett T., Troup D.B., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F. NCBI GEO: archive for functional genomics data sets – 10 years on. Nucleic Acids Res. 2011;39:D1005–D10010. doi: 10.1093/nar/gkq1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riveros C., Mellor D., Gandhi K.S., McKay F.C., Cox M.B., Berretta R. A transcription factor map as revealed by a genome-wide gene expression analysis of whole-blood mRNA transcriptome in multiple sclerosis. PLoS ONE. 2010;5:e14176. doi: 10.1371/journal.pone.0014176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keller A., Leidinger P., Bauer A., ElSharawy A., Haas J., Backes C. Toward the blood-borne miRNome of human diseases. Nat Methods. 2011;8:841–843. doi: 10.1038/nmeth.1682. [DOI] [PubMed] [Google Scholar]
- 25.Keller A., Leidinger P., Borries A., Wendschlag A., Wucherpfennig F., Scheffler M. MiRNAs in lung cancer – studying complex fingerprints in patient’s blood cells by microarray experiments. BMC Cancer. 2009;9:353. doi: 10.1186/1471-2407-9-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leidinger P., Keller A., Borries A., Huwer H., Rohling M., Huebers J. Specific peripheral miRNA profiles for distinguishing lung cancer from COPD. Lung Cancer. 2011;74:41–47. doi: 10.1016/j.lungcan.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 27.Keller A., Leidinger P., Lange J., Borries A., Schroers H., Scheffler M. Multiple sclerosis: microRNA expression profiles accurately differentiate patients with relapsing-remitting disease from healthy controls. PLoS ONE. 2009;4:e7440. doi: 10.1371/journal.pone.0007440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hausler S.F., Keller A., Chandran P.A., Ziegler K., Zipp K., Heuer S. Whole blood-derived miRNA profiles as potential new tools for ovarian cancer screening. Br J Cancer. 2010;103:693–700. doi: 10.1038/sj.bjc.6605833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roth P., Wischhusen J., Happold C., Chandran P.A., Hofer S., Eisele G. A specific miRNA signature in the peripheral blood of glioblastoma patients. J Neurochem. 2011;118:449–457. doi: 10.1111/j.1471-4159.2011.07307.x. [DOI] [PubMed] [Google Scholar]
- 30.Meder B., Keller A., Vogel B., Haas J., Sedaghat-Hamedani F., Kayvanpour E. MicroRNA signatures in total peripheral blood as novel biomarkers for acute myocardial infarction. Basic Res Cardiol. 2011;106:13–23. doi: 10.1007/s00395-010-0123-2. [DOI] [PubMed] [Google Scholar]
- 31.Lu M., Zhang Q., Deng M., Miao J., Guo Y., Gao W. An analysis of human microRNA and disease associations. PLoS ONE. 2008;3:e3420. doi: 10.1371/journal.pone.0003420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vorwerk S., Ganter K., Cheng Y., Hoheisel J., Stahler P.F., Beier M. Microfluidic-based enzymatic on-chip labeling of miRNAs. Nat Biotechnol. 2008;25:142–149. doi: 10.1016/j.nbt.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 33.Bolstad B.M., Irizarry R.A., Astrand M., Speed T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 34.R Development Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2008.
- 35.Lopes C.T., Franz M., Kazi F., Donaldson S.L., Morris Q., Bader G.D. Cytoscape Web: an interactive web-based network browser. Bioinformatics. 2010;26:2347–2348. doi: 10.1093/bioinformatics/btq430. [DOI] [PMC free article] [PubMed] [Google Scholar]