

Abstract
The rapid increasing number of completed bacterial genomes provides a good opportunity to compare their proteomes. This study was undertaken to specifically compare and contrast their secretomes—the fraction of the proteome with predicted N-terminal signal sequences, both type I and type II. A total of 176 theoretical bacterial proteomes were examined using the ExProt program. Compared with the Gram-positives, the Gram-negative bacteria were found, on average, to contain a larger number of potential Sec-dependent sequences. In the Gram-negative bacteria but not in the others, there was a positive correlation between proteome size and secretome size, while there was no correlation between secretome size and pathogenicity. Within the Gram-negative bacteria, intracellular pathogens were found to have the smallest secretomes. However, the secretomes of certain bacteria did not fit into the observed pattern. Specifically, the secretome of Borrelia burgdoferi has an unusually large number of putative lipoproteins, and the signal peptides of mycoplasmas show closer sequence similarity to those of the Gram-negative bacteria. Our analysis also suggests that even for a theoretical minimal genome of 300 open reading frames, a fraction of this gene pool (up to a maximum of 20%) may code for proteins with Sec-dependent signal sequences.
Key words: bacteria, secretome, Sec pathway, Tat pathway, ExProt
Introduction
Protein secretion in bacteria plays an important role in the interaction of microbes with each other and with their environments. Bacteria may secrete proteins through the use of a number of specialized secretion systems such as type I (ABC transporters), type III (flagellar-type), and type IV (conjugation-related). Although components of these secretion systems can be found in all microorganisms, they are best described in Gram-negative bacteria 1., 2., 3.. There are other more specialized secretion systems such as autotransporters, prepilin-type, twin-arginine translocation (Tat) pathway 4., 5., and ESAT-6 (6). However, the majority of secreted proteins in bacteria are secreted through the general secretory (Sec) pathway. A protein is tagged for export through the Sec pathway by a signal peptide at its N-terminus. Immediately following this signal peptide, there is a characteristic cleavage site recognized by a signal peptidase that cleaves this peptide following the “threading” of the secreted protein through the Sec complex in the cytoplasmic membrane 1., 2., 3., 7..
Presently there is an increase in bioinformatics tools designed to process entire genomes and their gene products. This increase parallels the increase in the number of sequenced genomes, particularly for bacterial systems. Analysis and comparison of genomes and their products have been extended to predictions of the entire protein complements that make up the secretome of members of the Bacteria. As such, these methods predict proteins targeted for secretion (the secretome) as well as proteins exported to the extracytoplasmic compartment for localization to the periplasm, outer membrane (by means of covalent attachment of lipids), or tethering to the cell wall. One such tool, named ExProt (for Exported Proteins) (8), was designed to predict proteins destined for export through the Sec pathway. This (secretome) analysis was previously tested and validated on a number of putative bacterial proteomes (8). Since then, however, the number of sequenced bacterial genomes has increased significantly. The size of bacterial proteomes varies greatly, and in a significant number of these proteomes, a substantial number of putative proteins do not have assigned functions. Some questions thus arise from these observations: Do bacteria with larger proteomes export a larger repertoire of proteins compared to those with smaller proteomes? Is there a correlation between the size of secretome and the niche of a microorganism? Do pathogens have a characteristically larger secretomes than non-pathogens?
Answering these questions would provide information that enhances our understanding of the evolutionary relationships, interactions with their environment, or the pathogenic life of the various microorganisms. There have been several recent studies utilizing bioinformatics tools to extract information on the life style of microorganisms, including the analysis of the predicted secretome of Lactobacillus plantarum WCFS1 (9) and the analysis of the genome sequence of Natronomonas pharaonis (10). In the present study, we expand the analysis to a larger set of bacterial genomes, with the aim of comparing the results between Gram-negative and Gram-positive bacteria as well as between pathogenic and non-pathogenic bacteria.
Results and Discussion
Gram-negative bacteria
The identity and the number of complete genome sequences selected for this study were determined by taking into considerations of factors such as coverage of the genome database, size of the genome, and duplications of genomes. Some closely related species were not included to minimize data duplication, while others with low representation in the database were included. The range of proteome size selected was wide, ranging in size from 484 open reading frames (ORFs) (Mycoplasma genitalium) to 8,317 ORFs (Bradyrhizobium japonicum).
The results of secretome prediction for Gram-negative bacteria are shown in Table S1 (see Supporting Online Material). Note that the predicted secretome includes both free secreted proteins (periplasmic or extracellular) and lipoproteins. The proportion of secretomes observed in these proteomes ranges from 12.6% to 42.4%. There appears to be a general pattern in which the secretome size increases with the increasing size of proteome for the Gram-negatives (Figure 1; R2=0.24). However, there is no statistically significant difference in secretome size between pathogenic and non-pathogenic Gram-negative bacteria (Figure 2; P>0.05). Of the Gram-negatives tested, the smallest secretomes are those of Buchnera aphidicola (71 proteins; 12.6%) and Wigglesworthia glossinidia (79 proteins; 12.9%). Both organisms have small genomes (564 and 611 ORFs) and are endosymbionts.
Figure 1.
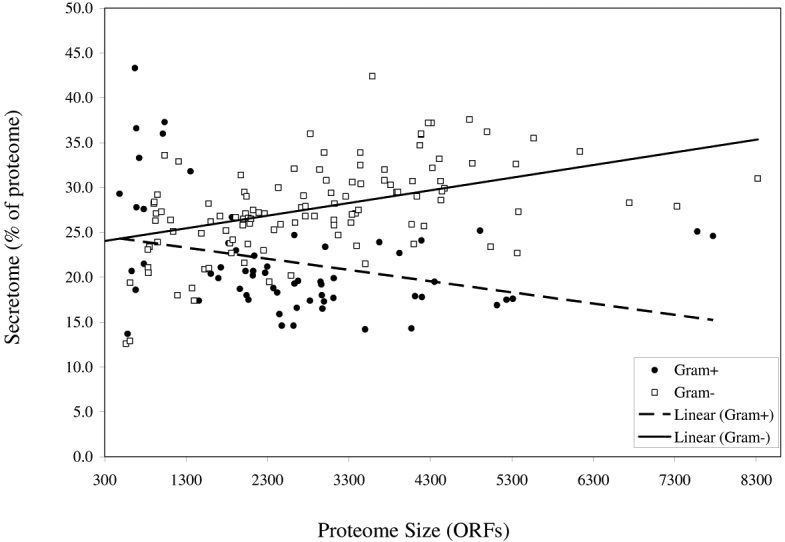
Secretome size in relation to proteome size in Gram-negative and Gram-positive bacteria. Proteomes are represented by the total number of ORFs, and the number of secreted proteins (the secretome) predicted by ExProt is represented as a fraction (percent) of the proteome. The value of R2 for the line fit is 0.24 for the Gram-negative and 0.21 for the Gram-positive data.
Figure 2.
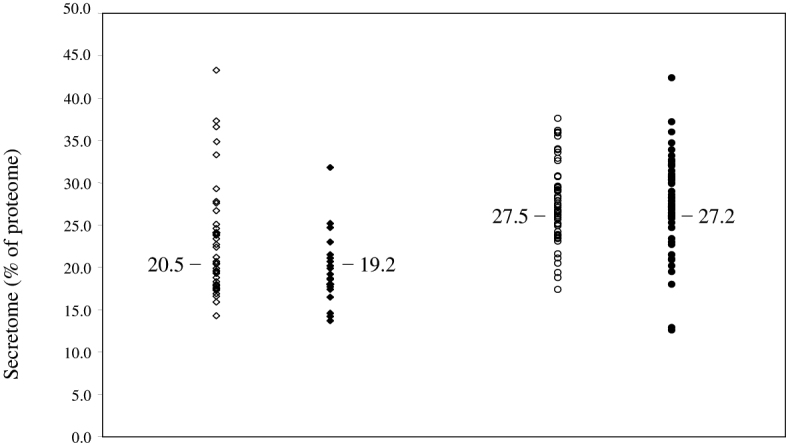
Comparison of secretome size between pathogenic (∘) and non-pathogenic (•) Gram-negative bacteria (right two columns) as well as between pathogenic (⋄) and non-pathogenic (♦) Gram-positive bacteria (left two columns). Numbers beside the columns indicate the median for each of the groups.
It is not surprising to see this low secretome size for organisms with such a small proteome. If one accepts the proposition that these organisms have experienced a significant reduction in genome size through evolution, then it can be argued that the majority of the available genes will code for a “minimal repertoire” of proteins required for core or basic cellular functions. This would mean that such organisms may not be self-sufficient, in that they would require a large number of nutritional sources. Indeed, both organisms are obligate intracellular parasites (11). Thus, their reduced genomes forced them to depend on a host, both for protection and for providing them with essential nutrients that may otherwise be difficult to obtain outside the host. This also seems to be true for Gram-positive bacteria with reduced genomes. For instance, the high G+C actinomycetes Tropheryma wipplei (12) and Mycobacterium leprae (13) have reduced genomes and ExProt predicts small secretomes for both (Table S1).
The line fit for the Gram-negative secretomes in Figure 1 can be described by the equation: Y=0.0017X+23.8. One may interpret this as in a minimal Gram-negative genome (X-intercept at about 300 ORFs), 24.3% of the proteome would represent secreted proteins, including both free secreted proteins (periplasmic or extracellular) and lipoproteins. This proportion seems to be high as a genome with only 300 genes will be expected to utilize most of the minimal gene complement to maintain basic intracellular functions. However, as observed in nature and as discussed above, bacteria with very small genomes are often intracellular parasites or endosymbionts. Our analysis shows that even for very small genomes, a significant fraction will code for secreted proteins. Therefore, we conclude that for a minimal genome, some of the basic intracellular activity, for example certain metabolic pathways, may be sacrificed in favor of maintaining a minimal secretory capability. Indeed, we observed in intracellular parasites several defects in basic intracellular functions such as incomplete metabolic pathways. These organisms will invest some energy to secrete proteins and other polymers to ensure the successful acquisition of essential nutrients from the host, to defend themselves from host defenses, and to maintain their protective surface components.
To consider this further, the data for the Gram-negative secretomes were plotted but only proteomes below 4,000 ORFs were used. The reason for this modification is that obligate intracellular parasites and endosymbionts mostly have proteomes in that range. When the data were analyzed (not shown), the line fit is described by the equation: Y=0.0032X+19.6. In this case, we can see that for a theoretical minimal genome coding for approximately 300 proteins, 20.6% of them are likely to be exported beyond the cytoplasmic compartment. The observed correlation between the size of the secretome and the proteome corroborates the findings of Gomi et al. (14) who analyzed an equivalent number of bacterial proteomes. In that study, the authors employed an in-house program called SOSUIsignal (15) that uses the propensities of occurrence of amino acids for the signal peptides as well as physicochemical parameters of hydrophobicity and amphiphilicity. From their analysis, a minimal genome coding for 300 proteins would have less than 2% (excluding lipoproteins, which are included in our study) potentially secreted proteins.
Bdellovibrio bacteriovorus
Our analysis shows that the proportion of secretome of B. bacteriovorus is the largest (42.4%) amongst the Gram-negative bacteria (Table S1). This bacterium is a parasite of other bacteria, localizing within the periplasm following penetration of the outer membrane and digestion of the peptidoglycan 16., 17.. Its genome shows that a number of metabolic pathways may be incomplete (18), suggesting a dependence on cellular components of the host as a supply for key nutrients. This is further supported by the observations that B. bacteriovorus does not oxidize or ferment organic acids, alcohols or many common carbohydrates 17., 19.. As a result of these two aspects of its life style, namely the breakdown of host macromolecular components and the need to uptake a great variety of macromolecular subunits, this bacterium has the capacity to secrete a large number of hydrolytic enzymes and the capacity to express a large number of membrane-associated nutrient uptake systems. The genome of B. bacteriovorus has recently been analyzed by Krogh et al. (20) for the presence of transport proteins. Using the TMHMM transmembrane helix prediction program 21., 22., they reported that up to 11% (396) of the ORFs are predicted to have one leader sequence/transmembrane segment. They proposed that many of these proteins can potentially have their leader sequence cleaved and the proteins are thus released to the periplasm (secreted, according to our criteria). They have also looked at the subcellular localization of the B. bacteriovorus proteins using the PSORTb program (23). Their findings show that the program was able to predict the localization of about 42% of the proteins, among which 23% were predicted to be cytoplasmic and the remaining 19% were predicted to be membrane, periplasmic, or extracellular proteins.
In our analysis, of the 3,587 ORFs detected by ExProt, 1,520 (42.4%) were identified as potentially having a Sec-dependent signal-like sequence in the N-terminus (Table S1). This is a very large secretome and further examination of the secretome was carried out to uncover a possible reason for this observation. Considering that only 55% of the ORFs of the B. bacteriovorus genome have been assigned a function (18), it would be more informative to look specifically at those with assigned functions within the predicted secretome. Inspection of the proteins in the secretome of B. bacteriovorus shows that 928 proteins are annotated as hypothetical proteins with no assigned function and 154 are annotated as membrane or membrane-associated proteins. If these are removed from the secretome, the number of predicted secreted proteins becomes 438 (22.2% of the proteins with assigned function). From the 438 predicted secreted proteins, ExProt identifies 216 as proteins with a signal peptide type II (lipoproteins). If these were also to be removed from the secretome, that leaves 222 proteins (11.3% of the proteins with assigned function) that are predicted to be free secreted proteins within the periplasm and/or released to the extracellular milieu. In support of the proposition that this microbe depends on hydrolytic enzymes during its parasitic stage, ExProt identifies 104 proteins as potentially secreted hydrolytic/penicillin binding proteins.
Borrelia burgdoferi and Treponema pallidum
The spirochetes form a distinct group of Gram-negative bacteria. Perhaps their most distinct morphologic features are the spiral shape and the periplasmic axial filament, providing this group with a unique form of motility. B. burgdoferi and T. pallidum are two parasitic members of this group. In terms of size and the range of proteome size used in this study, their proteomes can be considered similar, with T. pallidum having 1,036 predicted ORFs (24) and B. burgdoferi having 851 predicted ORFs (25). In this respect, and keeping in mind the correlation between proteome size and predicted secretome size of Gram-negative bacteria, one would expect to see an equivalent or a slightly larger secretome for T. pallidum. However, as can be seen in Table S1, ExProt predicts a significantly larger secretome (348 proteins; 33.6%) for T. pallidum as compared to that for B. burgdoferi (199 proteins; 23.4%).
The secretome size for T. pallidum is considered high for this sized proteome and to gain an insight into the life style of this microbe, one may consider the differences between the two parasites. B. burgdoferi has a smaller genome with a G+C content of 28.6% (25), while the genome of T. pallidum has a G+C content of 52.8%. At the proteome level, B. burgdoferi has a smaller proteome but with a larger number of predicted lipoproteins, being 132 (24) as compared to 22 putative lipoproteins for T. pallidum (26). Since ExProt identifies lipoproteins as part of the secretome, having 132 putative lipoproteins would be expected to increase the size of the secretome for B. burgdoferi, but that is not the case here. More information may be obtained from examining the secretomes for the two parasites. However, the significance of these differences will not be clear without considering the proteins encoded by plasmid DNA in B. burgdoferi. This organism has extrachromosomal DNA comprising nine circular and twelve linear plasmids (26). The number of ORFs in these plasmids totals 535, giving the organism a total proteome of 1,386 ORFs. Of these, there are 78 putative lipoproteins and 64 other putative exported proteins, for a total of 142 exported proteins from plasmid DNA. Combined with the chromosome, the secretome for B. burgdoferi becomes 341 ORFs (24.6% of the proteome). With the unusually high number of lipoproteins for an organism with a small proteome, its secretome is still smaller than that of T. pallidum.
To explain this difference, a closer look at the secretome of T. pallidum is required. For this purpose, a functional breakdown of proteins in the secretome of T. pallidum was carried out and compared with that of B. burgdoferi (Table 1). It is evident that the two organisms have the capacity to export an equivalent number of flagellar proteins, thus giving credence to the analysis by ExProt. However, it can be seen in Table 1 that significant differences exist in other classes of proteins. Specifically, T. pallidum shows a capacity to export a larger number of transport proteins and at least 50% more hypothetical proteins. The latter class of proteins, at least in part, contributes to increasing the size of the secretome in T. pallidum. Conserved hypothetical proteins are invariably present in the secretomes of all bacteria tested thus far. Although our knowledge of the structure and function of these proteins is very limited, hypothetical proteins in a few cases have been shown to play a role in the physiology and virulence of bacteria. Indeed, analysis of conserved hypothetical proteins in T. pallidum and in several other human pathogens has revealed a set of proteins with unknown function common to all those pathogens tested (27). Those proteins were found to be synthesized by the pathogens, and inactivation of nine of these proteins was found to result in attenuation in a mouse infection model.
Table 1.
Partial functional breakdown of secretome proteins in T. pallidum and B. burgdoferi
| Protein class | T. pallidum | B. burgdoferi |
|---|---|---|
| Lipoproteins | 41 (2 incomplete sequences) | 108 (30 chromosomal, 78 plasmid) |
| Hypothetical proteins | 149 | 92 |
| Transport (including ABC-type) | 23 | 7 |
| Ribosomal proteins | 7 | 3 |
| Flagellar proteins | 8 | 7 |
| Others | 120 | 124 |
| Total | 348 | 341 |
Gram-positive bacteria
The general pattern observed for the secretomes of Gram-positive bacteria is that they are generally smaller than those of the Gram-negative bacteria (Table S1 and Figure 2; P<0.05). In addition, there does not seem to be a positive correlation between secretome size and proteome size as seen in Gram-negative secretome prediction (Figure 1; R2=0.21). However, the Mycoplasma species do not fit into the general pattern. Having some of the smallest of the Gram-positive proteomes, their secretome proportions are the largest (Table S1). This clearly does not fit the pattern seen in this study, where an intracellular parasite with a reduced genome would be expected to export a smaller set of proteins as compared to their free-living counterparts.
To understand why the mycoplasmas have such large secretomes, a detailed analysis of the secretome of M. genitalium was carried out. It was observed that the secretome is dominated by two groups of proteins: conserved hypothetical proteins (41 proteins) and ribosomal proteins (38 proteins). The occurrence of a large number of conserved hypothetical proteins is not unusual in this type of proteome analysis; however, it is highly unusual to see a large number of ribosomal proteins. Clearly, the presence of these proteins in the secretome is the result of false-positive prediction by ExProt. The mycoplasmas are considered Gram-positive based on ribosomal RNA phylogeny. Thus, their proteomes were analyzed by ExProt trained on Gram-positive signal peptide sequences. To see if the N-terminal amino acid sequences of mycoplasma ribosomal proteins differ from the rest of the Gram-positive sequences, their proteomes were analyzed again using ExProt trained on Gram-negative sequences (data not shown). When this was done, only small changes in secretome size were obtained except for two cases: the mycoplasmas and Clostridium perfringens. For the mycoplasmas, their secretomes now fall within the range of Gram-positive bacteria. In the case of M. genitalium, the number of proteins in the secretome falls from 142 when analyzed as a Gram-positive to 113 when analyzed as a Gram-negative. The decrease is mostly due to exclusion of ribosomal proteins, where the number drops from 38 to 4. This observation means that the N-terminal 45 amino acid residues of M. genitalium proteins are more homologous to those from Gram-negative bacteria. To further explore this idea, the N-terminus of the M. genitalium ribosomal protein RL1 was selected to search for homologous sequences in GenBank. Using BLAST analysis (28), of the 17 highest hits, 11 are from Gram-negative bacteria (data not shown). It is therefore concluded that treating the proteomes of mycoplasmas as Gram-negative sequences provides more realistic secretomes.
When cultured in vitro, mycoplasmas require complex culture media, often supplemented with horse serum. In vivo, mycoplasmas depend on their host to provide them with certain essential nutrients. Therefore, one may ask why do these microorganisms need such large secretomes? Although this question may not be readily answered, one possibility is that a large set of exported proteins is required for the assembly, maintenance, and operation of the unusual polar structure. This polar structure has been shown to be elaborate and involved in the gliding motility of mycoplasmas (29). Other possibilities include the extensive cell envelope, typical of those comprised of polysaccharides, visualized with negative staining of certain mycoplasma (30). So far in our analysis, there appears to be a correlation between secretome size and cell complexity. While difficult to define, this complexity can be related to such cellular characteristics as structures for attachment and motility, extracytoplasmic structures, as in outer membranes and appendages, as well as cellular communication with the environment. For example, bacteria with very small genomes are often either intracellular pathogens or endosymbionts. This means that they depend on nutrient supplies from the host and do not require the secretion of a variety of enzymes to process and uptake extracellular nutrients. In saprophytic bacteria, the secretion of a large number of proteases and glycosidases aids them in acquiring needed nutrients. Although this is a simplification of the extremely complex nature of the interactions between microbes and their environments, it provides a reasonable explanation for our observations.
Our finding that the Gram-positive bacteria, on average, have smaller secretomes than the Gram-negatives (Figure 2) supports the findings of Gomi et al. (14) who used a program very different than ours. They suggested in their study that the Gram-negatives will likely depend more on secreted proteins to support the biochemical reactions in the periplasm and the outer membrane, two compartments not found in Gram-positive bacteria. This may be interpreted as a correlation between secretome size and cell complexity. Similar with the Gram-negative secretomes, Gomi et al. (14) found a strong correlation between the secretome size and the proteome size in the Gram-positive bacteria. However, our analysis using ExProt shows only a very weak correlation for the Gram-positive secretomes excluding the mycoplasmas (Figure 3; R2=0.08). The line fit for the Gram-positive data in Figure 3 can be described by the equation: Y=0.0004X+18.1. As suggested for the Gram-negative data mentioned above, this equation suggests that in a minimal Gram-positive genome with 300 ORFs (X-intercept in the plot), up to 18.2% of the ORFs may code for secreted proteins (including lipoproteins). This value is comparable with the value (20.6%) obtained for the Gram-negative secretomes (for genomes <4,000 ORFs). This again suggests that a bacterium with a very small genome is expected to dedicate a fraction of its genome, even at the expense of intracellular activity, for secreted proteins.
Figure 3.

Secretome size in relation to proteome size in Gram-positive bacteria excluding the mycoplasmas (R2=0.08).
Ribosomal proteins in the secretomes
Close inspection of the putative secretomes obtained in this work revealed that each secretome contains at least one putative ribosomal protein. The special case is that in the secretomes of the mycoplasmas, significant numbers of ribosomal proteins were found within the predicted secretomes (as discussed above). Ribosomal proteins are considered cytoplasmic proteins and were thus treated as false positives in the prediction of the secretomes. Why would ribosomal proteins have N-terminal sequences so similar to cleavable signal peptides? Are there examples of proteins normally considered cytoplasmic that are found to be exported? There is no obvious answer to the first question but it is related to the second question. The answer to the second question is yes. In fact, there have been several reports showing association of proteins, traditionally recognized as cytoplasmic, with membranes/cell walls in various microorganisms. The earliest is perhaps the work of Jacobson and Rosenbusch (31), who reported on the association of elongation factor Tu (EF-Tu) with the cytoplasmic membrane in Escherichia coli. Since then, EF-Tu has been found to be associated with the periplasm of Neisseria gonorrhoeae (32) and in the cell wall of Mycobacterium leprae (33). More recently, similar findings were reported for Mycoplasma pneumoniae (34), Lactobacillus johnsonii (35), Listeria monocytogenes (36), and most recently for Anaplasma marginale (37).
Other examples of cytoplasmic proteins found to be exported across the cytoplasmic membrane include the ribosomal proteins themselves. In Helicobacter pylori, ribosomal protein L11 was found by Kim et al. (38) to be secreted to the culture media. They confirmed that the presence of this protein in the culture supernatant was not due to non-specific cell lysis but rather by active secretion. Similar findings were reported by Korem and co-workers (39) for the RNAIII activating protein (RAP), a quorum sensing activator of Staphylococcus aureus, which is an ortholog of the ribosomal protein L2. Moreover, three ribosomal proteins (L4, L12, and S6) were identified in the culture supernatant of Listeria monocytogenes (40). At first inspection, one would offer the logical explanation that the source of these proteins is cell lysis, a normal process during the culturing of bacteria. On the other hand, however, one may offer the explanation that certain cytoplasmic proteins may in fact have dual functions and can be targeted by the cell to different subcellular sites. Although speculative, this proposal is the result of observations from both experimental and bioinformatic methods. As a result, the inclusion of certain ribosomal proteins in the putative secretomes reported here may reflect predictions of true positives by ExProt rather than false positives.
Twin-arginine signal peptides
In addition to the Sec-dependent signal peptides, microorganisms utilize another type of N-terminal signal peptide, which contains a conserved twin-arginine motif. Proteins tagged with this type of signals are targeted to the Tat complex for export through the plasma membrane. These leader peptides appear to be more common in, but are not restricted to, proteins containing complex redox cofactors. The distinguishing feature of Tat signal peptides is the presence of the (S/T)RRxFLK consensus sequence within the n-region of the leader peptide (41). The Tat signal peptides, however, resemble those of the Sec-dependent peptides in many aspects, such as distinct n-, h-, and c-regions, a particular signal peptidase cleavage site, a net positive charge in the n-region, and similar lengths. Because of these extensive similarities, it was relevant to our analysis to determine the level of false positives in the predicted secretomes due to the presence of Tat-type secreted proteins.
It had been pointed out previously that the E. coli genome contains at least 29 putative secreted proteins with Tat-like signal peptides having the twin-arginine motif (42). Analysis of the E. coli secretome predicted in our study shows that out of 1,468 proteins with putative Sec-dependent signal peptides (Table S1), no proteins in the secretome were found to have the Tat consensus in the leader peptide. However, there are two proteins with similar motifs: FdnG and NapA proteins. FdnG is the major subunit of the nitrate-inducible formate dehydrogenase and contains the sequence SRRQFFK starting at residue 4 in the N-terminus of the protein. NapA is a periplasmic nitrate reductase precursor and contains the sequence SRRSFMK, also starting at residue 4 in the N-terminus of the protein. Similar analysis was carried out on a number of other secretomes reported in this work. Take Bacillus subtilis for example, out of 737 proteins with putative Sec-dependent signal peptides (Table S1), only one protein, YkuE, has the Tat consensus sequence. This protein is a putative metallophosphoesterase and the Tat consensus starts at residue 5 in the N-terminus of the protein. On average, the putative secretomes reported in this work contain only 1–2 proteins with Tat-like consensus sequences. Considering the similarities between the two types of signal peptides pointed out above, the ExProt program used in our analysis is clearly proficient at discriminating between them and that the contribution of the Tat signal peptides to the putative secretomes reported here is negligible.
Membrane proteins
Membrane proteins can be considered as exported proteins being processed through the Sec complex, but are not secreted proteins since they remain within the membranes of the bacterium. Membrane proteins may contain a number of topogenic sequences to target them to and incorporate them within the cytoplasmic or the outer membrane, in the case of Gram-negative bacteria. These sequences include a leader or cleavable signal sequence, a non-cleavable signal sequence, a stop transfer sequence, and finally a reverse signal sequence. Those that have a cleavable signal sequence are processed through the signal peptidase to cleave the signal peptide. This class of membrane proteins will also have a stop transfer (signal anchor) sequence that is released laterally from the Sec complex to integrate the protein within the hydrophobic acyl phase of the membrane (3).
Because of the presence of the cleavable signal sequence at the N-terminus of this class of membrane proteins, the ExProt program would identify them as secreted proteins and constitute false positives in our analysis. That was indeed the case when the secretomes were screened for the presence of membrane proteins. In the secretome of Bacillus anthracis, out of 936 predicted secreted proteins, 17 (1.8%) were annotated as being putative membrane proteins. Similarly, the numbers of membrane proteins for the secretomes of B. subtilus and Staphylococcus aureus were 8 (out of 737) and 3 (out of 509), respectively. Larger numbers for these false positives were obtained in the secretomes of the Gram-negatives, potentially due to additional membrane proteins destined for the outer membrane in these bacteria. In E. coli, for example, 107 membrane proteins (out of 1,468) were detected within its predicted secretome. However, even with a margin of error of up to 10% in the size of the secretome, the relationship between the secretome size and the proteome size would still hold. As mentioned earlier, the purpose of using the ExProt program was not to identify potential secreted proteins in bacteria with high accuracy and precision, which should be achieved using a combination of different programs and stringent identification criteria. Rather, it was the ability of the ExProt program to rapidly (in seconds) process entire proteomes and identify, with relatively accurate prediction (8), the Sec-dependent secretome potential of bacteria. This feature of the program allowed us to analyze 176 bacterial genomes.
In addition, it should be pointed out that since analysis through the ExProt program utilizes the translated gene sequences within each of the genomes, it will be affected by the quality of the annotation for each of the genomes. Errors in identifying the correct gene start codon, presence of pseudogenes, and other gene anomalies will directly affect our predictions.
Conclusion
The utilization of a large and diverse number of genomes in our study allowed us to examine the relative Sec-dependent secretome potential between different bacteria. It also allowed us to draw certain conclusions regarding the secretome size and the interactions between microbes and their environments. On average, Gram-negative bacteria were found to contain a larger number of potential Sec-dependent sequences than the Gram-positives do. Within the former, there is stronger correlation between genome size and secretome size. In both groups, no correlation was found between secretome size and pathogenicity. However, it was observed that within the Gram-negatives, intracellular pathogens have the smallest secretomes.
Materials and Methods
Sequenced genomes were necessary to determine proteomes and secretomes for all microorganisms in this study. Published genomes were obtained from the National Center for Biotechnology Information (NCBI) online database (http://www.ncbi.nlm.nih.gov). A total of 176 genomes were used for our analysis. The type strain was selected where possible and incomplete sequences were ignored. Complete listing of the secretomes, including annotations and predicted cleavage sites for each putative secreted protein, is available at http://oldwebsite.laurentian.ca/biology/msaleh/exprot.htm. The files are in text format and can be downloaded directly. Table S1 showing secretome size for the 176 analyzed proteomes and related references is included as Supporting Online Material.
The ExProt program (8) was used for analysis of translated genome sequences and for assigning putative secretomes. This program searches for signal peptide sequences in the N-terminal 45 amino acid residues of each protein; where a signal sequence is identified, it assigns a most probable signal peptidase cleavage site. The architecture of this program contains a combined algorithm and a neural network that are trained separately on Gram-negative and Gram-positive signal sequences. Validation of ExProt against specific signal peptide datasets and its application to secretome prediction are detailed in the original study describing the program (8). Statistical analysis was carried out using the Statistica program (StatSoft, Inc., Tulsa, USA).
Authors’ contributions
CS collected the datasets, conducted data analyses, and drafted the manuscript. AK assisted in the data analyses and co-wrote the manuscript. MS supervised the project. All authors read and approved the final manuscript.
Competing interests
The authors have declared that no competing interests exist.
Supporting Online Material
Table S1
References
- 1.Papanikou E. Bacterial protein secretion through the translocase nanomachine. Nat. Rev. Microbiol. 2007;5:839–851. doi: 10.1038/nrmicro1771. [DOI] [PubMed] [Google Scholar]
- 2.Saier M.H., Jr. Protein secretion and membrane insertion systems in Gram-negative bacteria. J. Membr. Biol. 2006;214:75–90. doi: 10.1007/s00232-006-0049-7. [DOI] [PubMed] [Google Scholar]
- 3.Dalbey R.E., Kuhn A. Evolutionarily related insertion pathways of bacterial, mitochondrial, and thylakoid membrane proteins. Annu. Rev. Cell Dev. Biol. 2000;16:51–87. doi: 10.1146/annurev.cellbio.16.1.51. [DOI] [PubMed] [Google Scholar]
- 4.Lee P.A. The bacterial twin-arginine translocation pathway. Annu. Rev. Microbiol. 2006;60:373–395. doi: 10.1146/annurev.micro.60.080805.142212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Müller M., Klösgen R.B. The Tat pathway in bacteria and chloroplasts. Mol. Membr. Biol. 2005;22:113–121. doi: 10.1080/09687860500041809. [DOI] [PubMed] [Google Scholar]
- 6.Berthet F.X. A Mycobacterium tuberculosis operon encoding ESAT-6 and a novel low-molecular-mass culture filtrate protein (CFP-10) Microbiology. 1998;144:3195–3203. doi: 10.1099/00221287-144-11-3195. [DOI] [PubMed] [Google Scholar]
- 7.de Keyzer J. The bacterial translocase: a dynamic protein channel complex. Cell. Mol. Life Sci. 2003;60:2034–2052. doi: 10.1007/s00018-003-3006-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saleh M.T. Identification of putative exported/secreted proteins in prokaryotic proteomes. Gene. 2001;269:195–204. doi: 10.1016/s0378-1119(01)00436-x. [DOI] [PubMed] [Google Scholar]
- 9.Boekhorst J. The predicted secretome of Lactobacillus plantarum WCFS1 sheds light on the interaction with its environment. Microbiology. 2006;152:3175–3183. doi: 10.1099/mic.0.29217-0. [DOI] [PubMed] [Google Scholar]
- 10.Falb M. Living with two extremes: conclusions from the genome sequence of Natronomonas pharaonis. Genome Res. 2005;15:1336–1343. doi: 10.1101/gr.3952905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gil R. The genome sequence of Blochmannia floridanus: comparative analysis of reduced genomes. Proc. Natl. Acad. Sci. USA. 2003;100:9388–9393. doi: 10.1073/pnas.1533499100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bentley S.D. Sequencing and analysis of the genome of the Whipple’s disease bacterium Tropheryma whipplei. Lancet. 2003;361:637–644. doi: 10.1016/S0140-6736(03)12597-4. [DOI] [PubMed] [Google Scholar]
- 13.Honoré N. Nucleotide sequence of the first cosmid from the Mycobacterium leprae genome project: structure and function of the Rif-Str regions. Mol. Microbiol. 1993;72:207–214. doi: 10.1111/j.1365-2958.1993.tb01112.x. [DOI] [PubMed] [Google Scholar]
- 14.Gomi M. Comparative proteomics of the prokaryota using secretory proteins. Chem-Bio. Inform. J. 2005;5:56–64. [Google Scholar]
- 15.Gomi M. High performance system for signal peptide prediction: SOSUIsignal. Chem-Bio Inform. J. 2004;4:142–147. [Google Scholar]
- 16.Tudor J.J. A new model for the penetration of prey cells by bdellovibrios. J. Bacteriol. 1990;172:2421–2426. doi: 10.1128/jb.172.5.2421-2426.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stolp H. The bdellovibrios: bacterial parasites of bacteria. Annu. Rev. Phytopathol. 1973;11:53–76. [Google Scholar]
- 18.Rendulic S. A predator unmasked: life cycle of Bdellovibrio bacteriovorus from a genomic perspective. Science. 2004;303:689–692. doi: 10.1126/science.1093027. [DOI] [PubMed] [Google Scholar]
- 19.Seidler R.J., Starr M.P. Isolation and characterization of host-independent bdellovibrios. J. Bacteriol. 1969;100:769–785. doi: 10.1128/jb.100.2.769-785.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barabote R.D. Comprehensive analysis of transport proteins encoded within the genome of Bdellovibrio bacteriovorus. Genomics. 2007;90:424–446. doi: 10.1016/j.ygeno.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krogh A. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001;305:576–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 22.Sonnhammer E.L. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1998;6:175–182. [PubMed] [Google Scholar]
- 23.Gardy J.L. PSORTb v.2.0: expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics. 2005;21:617–623. doi: 10.1093/bioinformatics/bti057. [DOI] [PubMed] [Google Scholar]
- 24.Fraser C.M. Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science. 1998;281:375–378. doi: 10.1126/science.281.5375.375. [DOI] [PubMed] [Google Scholar]
- 25.Fraser C.M. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature. 1997;390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- 26.Casjens S. A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the Lyme disease spirochete Borrelia burdgoferi. Mol. Microbiol. 2000;35:490–516. doi: 10.1046/j.1365-2958.2000.01698.x. [DOI] [PubMed] [Google Scholar]
- 27.Garbom S. Identification of novel virulence-associated genes via genome analysis of hypthetical genes. Infect. Immun. 2004;72:1333–1340. doi: 10.1128/IAI.72.3.1333-1340.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altschul S.F. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 29.Krause D.C., Balish M.F. Structure, function, and assembly of the terminal organelle of Mycoplasma pneumoniae. FEMS Microbiol. Lett. 2001;198:1–7. doi: 10.1111/j.1574-6968.2001.tb10610.x. [DOI] [PubMed] [Google Scholar]
- 30.Miyata M., Uenoyama A. Movement on the cell surface of the gliding bacterium, Mycoplasma mobile, is limited to its head-like structure. FEMS Microbiol. Lett. 2002;215:285–289. doi: 10.1111/j.1574-6968.2002.tb11404.x. [DOI] [PubMed] [Google Scholar]
- 31.Jacobson G.R., Rosenbusch J.P. Abundance and membrane association of elongation factor Tu in E. coli. Nature. 1976;261:23–26. doi: 10.1038/261023a0. [DOI] [PubMed] [Google Scholar]
- 32.Porcella S.F. Identification of an EF-Tu protein that is periplasm-associated and processed in Neisseria gonorrhoeae. Microbiology. 1996;142:2481–2489. doi: 10.1099/00221287-142-9-2481. [DOI] [PubMed] [Google Scholar]
- 33.Marques M.A. Mapping and identification of the major cell wall-associated components of Mycobacterium leprae. Infect. Immun. 1998;66:2625–2631. doi: 10.1128/iai.66.6.2625-2631.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dallo S.F. Elongation factor Tu and E1β subunit of pyruvate dehydrogenase complex act as fibronectin binding proteins in Mycoplasma pneumoniae. Mol. Microbiol. 2002;46:1041–1051. doi: 10.1046/j.1365-2958.2002.03207.x. [DOI] [PubMed] [Google Scholar]
- 35.Granato D. Cell surface-associated elongation factor Tu mediates the attachment of Lactobacillus johnsonii NCC533 (La1) to human intestinal cells and mucins. Infect. Immun. 2004;72:2160–2169. doi: 10.1128/IAI.72.4.2160-2169.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaumburg J. The cell wall subproteome of Listeria monocytogenes. Proteomics. 2004;4:2991–3006. doi: 10.1002/pmic.200400928. [DOI] [PubMed] [Google Scholar]
- 37.Lopez J.E. Identification of novel antigenic proteins in a complex Anaplasma marginale outer membrane immunogen by mass spectrometry and genomic mapping. Infect. Immun. 2005;73:8109–8118. doi: 10.1128/IAI.73.12.8109-8118.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim N. Proteins released by Helicobacter pylori in vitro. J. Bacteriol. 2002;184:6155–6162. doi: 10.1128/JB.184.22.6155-6162.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Korem M. Characterization of RAP, a quorum sensing activator of Staphylococcus aureus. FEMS Microbiol. Lett. 2003;223:167–175. doi: 10.1016/S0378-1097(03)00326-4. [DOI] [PubMed] [Google Scholar]
- 40.Trost M. Comparative proteome analysis of secretory proteins from pathogenic and non-pathogenic Listeria species. Proteomics. 2005;5:1544–1557. doi: 10.1002/pmic.200401024. [DOI] [PubMed] [Google Scholar]
- 41.Berks B.C. A common export pathway for proteins binding complex redox cofactors? Mol. Microbiol. 1996;22:393–404. doi: 10.1046/j.1365-2958.1996.00114.x. [DOI] [PubMed] [Google Scholar]
- 42.Tullman-Ercek D. Export pathway selectivity of Escherichia coli twin arginine translocation signal peptides. J. Biol. Chem. 2007;282:8309–8316. doi: 10.1074/jbc.M610507200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1