

Abstract
Spinal cord injury (SCI) is a debilitating disease, effective prevention measures are in desperate need. Our previous work found that hyperbaric oxygen (HBO) preconditioning significantly protected rats from SCI after stimulated diving, and in vitro study further testified that HBO protected primary cultured rat spinal neurons from oxidative insult and oxygen glucose deprivation injury via heat shock protein (HSP) 32 induction. In this study, underlying molecular mechanisms were further investigated. The results showed that a single exposure to HBO significantly increased intracellular levels of reactive oxygen species (ROS) and nitric oxide (NO) and activated MEK1/2, ERK1/2, p38 MAPK, CREB, Bach1 and Nrf2. The induction of HSP32 by HBO was significantly reversed by pretreatment neurons with ROS scavenger N-Acetyl-L-cysteine, p38 MAPK inhibitor or Nrf2 gene knockdown, enhanced by MEK1/2 inhibitors or gene knockdown but not by ERK1/2 inhibitor. CREB knockdown did not change the expression of HSP32 induced by HBO. N-Acetyl-L-cysteine significantly inhibited the activation of MEK1/2, ERK1/2, p38 MAPK, and Nrf2. Activation of Nrf2 was significantly inhibited by p38 MAPK inhibitor and the nuclear export of Bach1 was significantly enhanced by MEK1/2 inhibitor. The results demonstrated that HBO induces HSP32 expression through a ROS/p38 MAPK/Nrf2 pathway and the MEK1/2/Bach1 pathway contributes to negative regulation in the process. More importantly, as we know, this is the first study to delineate that ERK1/2 is not the only physiological substrates of MEK1/2.
Keywords: Hyperbaric oxygen, Heat shock protein 32, Signal transduction, Negative regulation, Reactive oxygen species
Graphical abstract
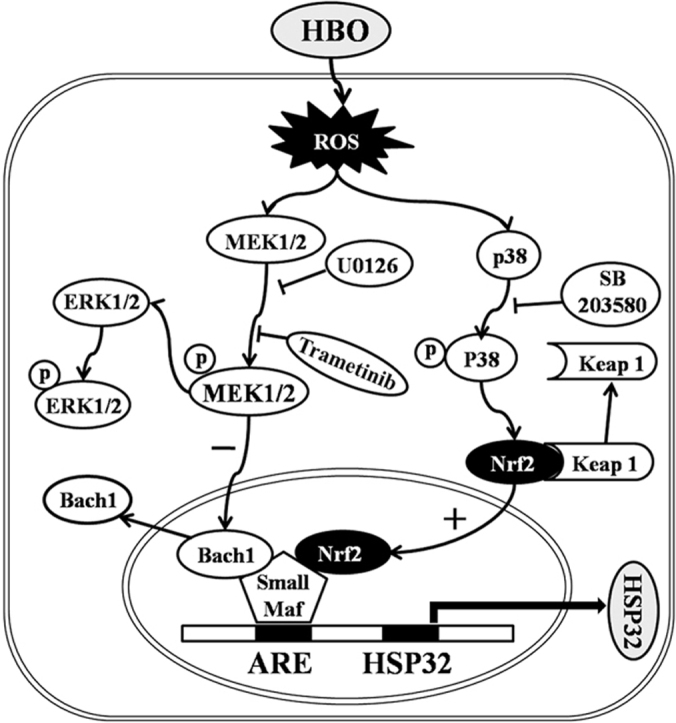
Signaling pathways between HBO exposure and HSP32 expression in rat spinal neurons. Under basal conditions, Bach1 is associated with small Maf proteins and inhibit HSP32 gene expression by binding to the ARE. HBO increases intracellular ROS formation, which activates MEK1/2 and p38 MAPK. The activation of p38 MAPK triggers the dissociation of Nrf2 from keap1 and translocation into the nucleus to form a heterodimer with small Maf proteins and to initiate transcription of HSP32 gene by binding to ARE. At the same time, the activation of MEK1/2 inhibits the Bach1 disassociation from small Maf proteins, which prevents the surge of HSP32 gene transcription. ARE, antioxidant-response element; Keap 1, Kelch-like ECH-associated protein 1.
Highlights
-
•
HBO induces HSP32 through ROS/p38 MAPK/Nrf2 pathway in rat spinal neurons.
-
•
ROS but not RNS participates in HBO induced HSP32 expression.
-
•
MEK1/2/Bach1 contributes to negative regulation in HBO induced HSP32 expression.
-
•
MEK1/2 acts through pathways other than ERK1/2.
1. Introduction
Spinal cord injury (SCI) is an unpredictable and debilitating disease, which may be a complication of surgical operations on the spinal column or thoracoabdominal aorta, or result from decompression sickness associated with sport or commercial diving [1], [2]. The pathological sequence of SCI is mainly mediated by edema, inflammation, excitotoxicity, ischemia-reperfusion injury, and oxidative cell damage [3], [4]. Due to the inadequate blood supply and high lipid content in the spinal cord, which is damaged easily by free radicals, ischemia-reperfusion injury and oxidative stress are two critical mechanisms of SCI [3], [5]. Many therapies have been suggested to protect against SCI, including hypothermia, anti-excitotoxic agents, calcium channel blockers, N-methyl-D-aspartate receptor antagonists, and cerebrospinal fluid drainage, but improvements in outcome are marginal [5]. Novel effective therapies are desperately needed to prevent SCI. Our previous work found that hyperbaric oxygen (HBO) preconditioning significantly protected rat from spinal cord injury after stimulated diving [6], and in vitro study further testified that HBO protected primary cultured rat spinal neurons from oxidative insult and oxygen glucose deprivation injury via heat shock protein (HSP) 32 induction, which peaked at 12 h following HBO exposure [7]. The aim of the present study was to investigate the underlying mechanisms of HBO induced HSP32 expression in primary cultured rat spinal neurons.
Free heme is produced mainly through the oxidation of hemoproteins, including hemoglobin, myoglobin, neuroglobin, etc [8]. In the center of heme is a Fe atom, which can act to produce highly toxic hydroxyl radicals derived from hydrogen peroxide [8]. Apart from causing oxidative insult, free heme can also promote tumor necrosis factor mediated programmed cell death [8]. HSP32, a stress responsive protein also named heme oxygenase-1, is a rate limiting enzyme in the catabolism of free heme; it degrades heme into three products: carbon monoxide (CO), ferrous iron, and biliverdin [9]. In addition to degrading free heme and neutralizing damage caused by heme, its end products can also exert cytoprotective effects. It is already documented that the HSP32/CO system can exert anti-inflammatory and anti-apoptotic effects [10], ferrous iron released from heme can enhance cell antioxidant capacity via ferritin up-regulation [11], and the beneficial roles of biliverdin and bilirubin are to act as physiological antioxidants [12].
HBO is a treatment modality in which a person breathes 100% oxygen under a pressure greater than one atmosphere absolute in a compression chamber [13]. It is a safe, clinically viable treatment modality, which has been widely applied as the primary treatment for certain injuries, especially for acute CO poisoning, gas gangrene and decompression sickness [14]. In addition to increasing tissue oxygen supply, HBO exposure can cause moderate oxidative stress by slightly increasing intracellular levels of reactive oxygen species (ROS) and/or reactive nitrogen species (RNS), which can induce the expression of cytoprotective proteins and enhance cellular tolerance against harmful stimuli [15]. The protective effects of HSP32 induction by HBO upon organ ischemia-reperfusion injuries are well evidenced, including in the myocardium [16], liver [17], kidney [18], and primary cultured rat spinal neurons [7].
However, the mechanism by which HBO induces the expression of HSP32 in spinal neurons is still unknown. Understanding the mechanism is important for the application of HBO in medical practice and the development of new strategies to prevent SCI.
2. Materials and methods
2.1. Animals
Pregnant Sprague-Dawley rats (14–15 d) were obtained from Shanghai SLAC Laboratory Animal Co., Ltd on the day of use. The study protocols were approved by the Institutional Animal Care and Use Committee of the University and all experimental procedures were performed in accordance with University guidelines.
2.2. Cell culture
Spinal neurons were prepared from embryonic day 14–15 Sprague-Dawley rats as previously described [7]. Briefly, spinal cords were rapidly dissected from embryo and digested with pre-warmed 0.05% trypsin (Invitrogen, USA) at 37 °C for 18 min. After digestion, the resultant cell suspension was adjusted to 1.0×106 cells/ml in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) containing 10% heat-inactivated fetal bovine serum (Invitrogen) and 10% heat-inactivated horse serum (Invitrogen). Cells were plated onto poly-L-lysine (molecular weight 30,000–70,000; Sigma, USA) coated culture plates at a density of 1.2×106 cells/well on 6-well plates or 0.6×106 cells/well on 12-well plates. Four hours later, the medium was completely replaced with neurobasal medium (Invitrogen) supplemented with 2% B27 supplement (Invitrogen), 0.5 mM glutamine (Invitrogen), 100 U/ml penicillin (Invitrogen), and 100 μg/ml streptomycin (Invitrogen). Half the volume of culture media was replaced with fresh media every 3 days.
2.3. Hyperbaric oxygen treatment
HBO exposure was conducted in a temperature and humidity controlled hyperbaric cell incubator (OxyCure 3000, OxyHeal® Health Group, USA). As described in our previous study [7], the neurons were exposed to HBO for 60 min at 280 kPa, which is frequently used in animal and cell studies and 280 kPa is the current upper limit of partial pressure of oxygen applied in clinical HBO therapy [19]. Compression and decompression were both carried out within 5 min. To maintain a physiological pH of the culture, the chamber was flushed and compressed with oxygen containing 1.79% CO2 to achieve a 5 kPa partial pressure of CO2. All pressures described in this text are absolute.
2.4. Bio-Plex phosphoprotein analysis
At specific time points, neurons were collected, and protein lysates were prepared, and content of phosphorylated proteins was detected using the respective Bio-Plex Phosphoprotein Detection kit (Bio-Rad, USA) according to the manufacturer's protocol. Briefly, 50 μl of cell lysate (adjusted to a concentration of 0.6–0.9 μg/ml) was plated in the 96-well filter plate coated with beads coupled to anti-phosphoprotein antibody. The plate was incubated overnight at room temperature on a platform shaker. After a series of washes to remove unbound proteins, biotinylated detection antibodies, each specific for a different epitope, were added to the reaction. This resulted in formation of antibody complexes assembled around the target proteins. Streptavidin-phycoerythrin was then added to bind to the biotinylated detection antibodies on the bead surface. Data were acquired with the Bio-Plex 200 system and analyzed with the Bio-Plex Manager software (Bio-Rad).
2.5. Measurement of intracellular ROS and NO generation
Levels of intracellular ROS and NO were detected using DCFH-DA (a ROS fluorescent probe; Beyotime, China) and DAF-FM DA (a NO fluorescent probe; Beyotime), respectively. Spinal neurons were incubated with DCFH-DA (10 μM) or DAF-FM DA (5 μM) for 20 min at 37 °C. After three washes with PBS to remove unbound fluorescent probes, cultures were exposed to HBO. Fluorescence intensity was observed under a fluorescence microscope (495 nm excitation wave length and 515 nm emission wave length; ZEISS, Germany) immediately after HBO exposure.
2.6. Nrf2, CREB and MEK1/2 knockdown by shRNA infection
Lentiviral vectors encoding shRNAs against rat Nrf2 or CREB were designed and manufactured by Obio Technology Co., Ltd (Shanghai, China). The target sequences for rat Nrf2- and CREB-shRNA are 5′-CCATTCCCGAGTTACAGTGTCTTAA-3′ and 5′-GCACTTAAGGACCTTTACT-3′, respectively, whose knockdown effect has been demonstrated previously [20], [21]. For Nrf2 or CREB knockdown, primary cultured rat spinal neurons were infected with CREB-shRNA-lentivirus, Nrf2-shRNA-lentivirus or NC-shRNA-lentivirus at 10–30 Multiplicity of Infection (MOI). For MEK1/2 knockdown, 3 target sequences each for rat MEK1 and MEK2 were designed. The target sequences for MEK1 are Seq1: 5′-GCGAGATCAGCATCTGCAT-3′, Seq2: 5′-GCAGCTCATGGTACATGCT-3′ and Seq3: 5′-GGCAGCTAATTGACTCCAT-3′; for MEK2 are Seq4: 5′-GCTCAAGGACGACGACTTT-3′, Seq5: 5′-GCATCTGCATGGAGCACAT-3′ and Seq6: 5′-GGAACTAGAGGCCAGCTTT-3′. After 8 h infection, the medium was replaced by fresh complete medium for another 4 days prior to further experiments. The knockdown efficiency was validated by western blot.
2.7. Western blot
Cytoplasmic and nuclear proteins were prepared using cell nuclear protein extraction kits (Beyotime) according to the manufacturer’s instructions. The total protein were harvested and lysed in Radio-Immunoprecipitation Assay Lysis Buffer (Beyotime). Protein samples were electrophoresed on 8% SDS-polyacrylamide gels, and transferred onto a polyvinyldifluoridine membrane (Millipore, USA). Membranes were blocked with 5% (w/v) non-fat milk and incubated overnight at 4 °C with rabbit monoclonal primary antibodies directed against rat HSP32 (Abcam, USA), p38 MAPK (Cell Signaling Technology, USA), p-p38 MAPK (Cell Signaling Technology), ERK1/2 (Cell Signaling Technology), p-ERK1/2 (Cell Signaling Technology), CREB (Cell Signaling Technology), p-CREB (Cell Signaling Technology), Bach1 (Cell Signaling Technology), Nrf2 (Abcam), L-amin B (Cell Signaling Technology) or β-actin (Cell Signaling Technology). Proteins were visualized by using HRP-conjugated goat anti-rat IgG (Cell Signaling Technology) and the intensity of each band was measured by Image J software (National Institutes of Health, USA).
2.8. Statistical analysis
Results are presented as mean±standard deviation (SD). Statistical analysis was performed with SPSS software (version 18.0), using one-way ANOVA followed by LSD. p<0.05 was considered statistically significant.
3. Results
3.1. ROS is responsible for HBO induced HSP32 expression in rat spinal neurons
In this study, the effects of HBO on intracellular ROS and nitric oxide (NO) levels were firstly observed immediately after HBO exposure, and their role in HBO induced HSP32 expression were clarified using their respective inhibitor. As showed in Fig. 1, HBO obviously elevated intracellular ROS and NO levels (Fig. 1B and F). Pretreatment of neurons with ROS scavenger N-Acetyl-L-cysteine (NAC 5 mM) [22] or nitric oxide synthase (NOS) inhibitor NG-Nitro-L-arginine methyl ester (L-NAME 100 μM) [23] distinctly inhibited the increase of intracellular ROS and NO, respectively (Fig. 1C and H). To further explore whether there is a crosstalk between ROS and NO or not, we observed the effects of ROS scavenger on NO production and NOS inhibitor on ROS production. We found that ROS scavenger NAC had no effect on NO production, and vice versa (Fig. 1D and G). Next, we investigated the role of ROS and NO in HBO induced HSP32 expression. The results showed that NAC significantly reversed the up-regulation of HSP32 by HBO (Fig. 1I), but L-NAME had no effect on the expression of HSP32 even at higher doses of 500 or 1000 μM (Fig. 1J).
Fig. 1.

Effects of HBO on intracellular levels of ROS and NO and their role in HBO induced HSP32 expression in spinal neurons. ROS scavenger NAC (5 mM) or NOS inhibitor L-NAME (100 μM) was added to the medium 30 min before HBO exposure. The intracellular ROS and NO were detected immediately after HBO exposure using immunofluorescence assay (A-H). The expression of HSP32 was detected by western blot 12 h after HBO exposure (I). The effects of L-NAME at higher concentrations (J). Significance compared with Air group, **p<0.01; significance compared with HBO group ††p<0.01. HBO, hyperbaric oxygen; HSP, heat shock protein; L-NAME, NG-Nitro-L-arginine methyl ester; NAC, N-Acetyl-L-cysteine; NO, nitric oxide; NOS, nitric oxide synthase; ROS, reactive oxygen species.
3.2. Signal molecules possibly involved in HSP32 induction by HBO
To elucidate the molecular mechanisms underlying HBO induced HSP32 expression, we detected the activation of possible involved signal molecules. The phosphorylation of nine important signal molecules was screened using a Bio-Plex Phoshoprotein Detection kit at 0, 6, 12, and 18 h following HBO exposure. As showed in Fig. 2, the phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2), p38 mitogen-activated protein kinase (p38 MAPK), and cAMP-response element binding protein (CREB) were significantly increased immediately after HBO exposure and recovered at 6 h (Fig. 2D, F and G).
Fig. 2.

Activation of signal molecules following HBO exposure in rat spinal neurons. The activation of signal molecules related to HSP32 induction were detected using Bio-Plex Phosphoprotein Detection kit. Significance compared with Air group, *p<0.05, **p<0.01. CREB, cAMP-response element binding protein; ERK1/2, extracellular signal-regulated kinase 1/2; p38 MAPK, p38 mitogen-activated protein kinase.
To further determine the activation rule of ERK1/2, p38 MAPK and CREB, we detected the phosphorylation of these signal molecules at 0, 0.5, 1, 2 and 4 h following HBO exposure using western blot. The results clearly showed that the phosphorylation of p38 MAPK, ERK1/2 and CREB significantly increased immediately after HBO exposure and recovered at 0.5 h (Fig. 3A–C). Nuclear factor-E2-related factor-2 (Nrf2) is an important transcription factor in HSP32 induction [24], [25], that cannot be screened by the Bio-Plex Phoshoprotein Detection kit. The nuclear translocation of Nrf2 at 0, 1, 2, 4, and 6 h after HBO were determined by western blot. HBO exposure stimulated nucleus accumulation of Nrf2 in spinal neurons and this peaked at 4 h following HBO exposure (Fig. 3D). Therefore, the 4 h time point following HBO exposure was selected to observe the effects of signaling molecular inhibitors on the activation of Nrf2.
Fig. 3.

Effects of HBO on the activation of ERK1/2, p38 MAPK, CREB and Nrf2. The phosphorylation of ERK1/2 (A), p38 MAPK (B), CREB (C) and the nuclear translocation of Nrf2 (D) were confirmed following HBO exposure. Significance compared with Air group, *p<0.05, **p<0.01. Nrf2, nuclear factor-E2-related factor-2.
3.3. The induction of HSP32 relied on p38 MAPK and Nrf2 activation, and enhanced after “ERK1/2 inhibition”
The roles of p38 MAPK and ERK1/2 in the induction of HSP32 by HBO were studied by pretreatment with their respective inhibitor. The expression of HSP32 was significantly inhibited by p38 MAPK inhibitor SB203580 (Fig. 4A) or Nrf2 gene knockdown (Fig. 4B), but was significantly enhanced by ERK1/2 inhibitor (Fig. 4A). CREB knockdown had no effect on the expression of HSP32 (Fig. 4B). The knockdown efficiency of CREB- and Nrf2-shRNA Lentiviral vectors at different Multiplicity of Infection (MOI) were validated by western blot and the results showed that both CREB and Nrf2 expression were significantly inhibited and there was no significant difference among 10–30 MOI groups (Fig. 4C and D). Therefore, the dose of CREB- and Nrf2-shRNA Lentiviral vectors at 10 MOI were used to knockdown the expression of Nrf2 and CREB. These results indicated that HBO induces HSP32 by p38 MAPK and Nrf2 activation and, meanwhile, ERK1/2 may be negative regulators in this process.
Fig. 4.

Role of ERK1/2, p38 MAPK, CREB, and Nrf2 in HBO induced HSP32 expression in rat spinal neurons. p38 MAPK inhibitor (SB203580, 10 μM) or EKR1/2 inhibitor (U0126, 10 μM) were added to the medium 30 min before HBO exposure and the expression of HSP32 was detected by western blot 12 h after HBO exposure (A). Neurons were transfected with shNrf2, shCREB or vector as described in the Materials and methods section, and the effects on the expression of HSP32 were detected by western blot (B). The knockdown efficiency of shCREB (C) and siNrf2 (D) at different MOI values are presented (C, D). Significance compared with Air group, **p<0.01; significance compared with HBO group, ††p<0.01. MOI, Multiplicity of Infection.
3.4. The roles of MEK1/2-ERK1/2 in the HBO induced HSP32 expression
To confirm the negative regulation of ERK1/2 in HBO induced HSP32 expression, the effects of another inhibitor of ERK1/2 SCH772984 was observed. Unexpectedly, the results showed that inhibition with SCH772984 failed to change HSP32 expression induced by HBO (Fig. 5B), even though it significantly inhibited the activation of ERK1/2 (Fig. 5A).
Fig. 5.

Effects of ERK1/2 direct inhibitor SCH772984 on HBO induced HSP32 expression. Neurons were pretreated with SCH772984 (10 μM) 30 min before HBO exposure. The phosphorylation of ERK1/2 was detected immediately after HBO exposure (A) and the expression of HSP32 was detected 12 h later (B). Significance compared with Air group, **p<0.01; significance compared with HBO group, ††p<0.01.
Although U0126 is frequently used as an ERK1/2 inhibitor, its actual target is mitogen-activated and extracellular signal-regulated kinase 1/2 (MEK1/2), simply because MEK1/2-ERK1/2 is generally considered an integrated signal pathway [26], [27], while SCH772984 is a direct inhibitor to ERK1/2. To confirm the involvement of MEK1/2 in HBO induced HSP32 expression, the activation of MEK1/2 at 0, 0.5, 1, 2 and 4 h following HBO exposure and the effects of another MEK1/2 inhibitor trametinib or MEK1/2 knockdown were observed. As shown in Fig. 6, the phosphorylation of MEK1/2 was significantly increased immediately after HBO exposure and recovered at 0.5 h (Fig. 6A), trametinib significantly enhanced the expression of HSP32 (Fig. 6C) by inhibiting MEK1/2 activation (Fig. 6B) and MEK1/2 gene knockdown (Fig. 7A–C) also significantly enhanced the expression of HSP32 after HBO exposure (Fig. 7D).
Fig. 6.

Effects of MEK1/2 inhibition on the expression of HSP32 induced by HBO. The phosphorylation of MEK1/2 was detected after HBO exposure (A). Neurons were pretreated with U0126 (10 μM) or trametinib (10 μM) 30 min before HBO exposure, and the phosphorylation of MEK1/2 was detected immediately after HBO exposure (B), the expression of HSP32 was detected 12 h after HBO exposure (C). Significance compared with Air group, **p<0.01; significance compared with HBO group ††p<0.01. MEK1/2, mitogen-activated and extracellular signal-regulated kinases; Seq, sequence.
Fig. 7.

Effects of MEK1/2 knockdown on HBO induced HSP32 expression. Neurons were transfected with vector, shMEK1 (Seq1, Seq2 and Seq3) or shMEK2 (Seq4, Seq5 and Seq6) as described in the Materials and Methods section. Transfection efficiency was confirmed by checking MEK1 and MEK2 mRNA transcription levels (A, B). The combined effects of shMEK1 (Seq 2) and shMEK2 (Seq 4) on the expression of MEK1/2 (C) and on the expression of HSP32 (D) was observed by western blot. Significance compared with Air group, **p<0.01; significance compared with HBO group, ††p<0.01.
3.5. HBO and MEK1/2 inhibitor enhanced Bach1 export from the nucleus
Under basal conditions, BTB and CNC homology 1 (Bach1) serves as a repressor of HSP32 gene expression in the nucleus. The effects of HBO and MEK1/2 inhibition on the nucleus and cytosol distribution of Bach1 were detected. HBO exposure significantly increased the translocation of Bach1 outside the nucleus and this peaked at 4–6 h following HBO exposure (Fig. 8A). As there was no significant difference between the 4 and 6 h groups, the 4 h time point following HBO exposure was selected to observe the effects of MEK1/2 inhibition on the translocation of Bach1. The results showed that MEK1/2 inhibition significantly enhanced Bach1 export from the nucleus (Fig. 8B).
Fig. 8.

Effects of HBO and MEK1/2 inhibitor on the nucleus and cytosol distribution of Bach1. The distribution of Bach1 in nucleus and cytosol after HBO exposure was detected by western blot (A). Neurons were pretreated with U0126 (10 μM) 30 min before HBO exposure and the distribution were detected at 4 h after HBO exposure (B). Significance compared with Air group, **p<0.01; significance compared with HBO group, ††p<0.01.
3.6. The relationship between ROS, MEK1/2, p38 MAPK and Nrf2
The ROS scavenger NAC was used to reveal the role of ROS in the activation of MEK1/2, p38 MAPK and Nrf2 by HBO. The phosphorylation of MEK1/2 and p38 MAPK were detected immediately and Nrf2 was detected at 4 h after HBO exposure. NAC significantly inhibited the activation of MEK1/2, p38 MAPK and Nrf2 (Fig. 9A–C). MEK1/2 and p38 MAPK are the usual upstream signal molecules of transcription factor Nrf2. To clarify their relationship, the effects of MEK1/2 inhibitor (U0126) and p38MAPK inhibitor (SB203580) on the activation of Nrf2 were determined. The results showed that only SB203580 but not U0126 inhibited the activation of Nrf2 (Fig. 9F). The relationship between the MEK1/2 and p38 MAPK pathway was also explored. As shown in Fig. 9D and E, neither the MEK1/2 inhibitor nor p38 MAPK inhibitor had any effect on the activation of each other, indicating there was no crosstalk between these two pathways. SB203580 works by inhibiting the activation effects of p38 MAPK to its substrates, but does not change the phosphorylation of p38 MAPK (15). As shown in Fig. 9E, the phosphorylation of p38 MAPK showed no change following SB203580 inhibition.
Fig. 9.

The relationship between ROS, MEK1/2, p38 MAPK and Nrf2. Neurons were pretreated with ROS scavenger (NAC, 5 mM), MEK1/2 inhibitor (U0126, 10 μM) or p38 MAPK inhibitor (SB203580, 10 μM) 30 min before HBO exposure. The phosphorylation of MEK1/2 (A, D), p38 MAPK (B, E), and the nucleus translocation of Nrf2 (C, F) were determined by western blot. Significance compared with Air group, **p<0.01; significance compared with HBO group, ††p<0.01.
4. Discussion
Our previous study indicated that HBO may be an effective prophylactic measure against SCI through induction of HSP32 or HSP70 [6], [7]. The results in this study showed that the induction of HSP32 by HBO is through the ROS/p38 MAPK/Nrf2 signaling cascade and, meanwhile, a MEK1/2/Bach1 mediated negative regulation pathway exists.
HSP32 is a highly inducible protein regulated by a number of transcription factors, and Nrf2 and Bach1 have emerged as the predominant regulators of HSP32 gene expression [25]. Under basal conditions, Nrf2 exists in the cytoplasm through binding to the cytoskeletal-associated protein Kelch-like ECH-associated protein 1 (Keap1) andBach1 is associated with small Maf proteins, inhibiting HSP32 gene expression by binding to the antioxidant-response element (ARE) [25]. After activation, Bach1 disassociates from small Maf proteins and translocates into the cytoplasm. Simultaneously, Nrf2 disassociates from keap1 and translocates into the nucleus to form a heterodimer with small Maf proteins and initiates transcription of the HSP32 gene by binding to ARE [25]. Many chemical and environmental stimuli, including its substrate heme, heavy metals, ultraviolet light, hydrogen peroxide, lipopolysaccharide, and arsenite, are known to induce HSP32 [28]. A common character for those chemical and environmental stimuli, capable of inducing HSP32, is their electrophilic chemistry and the generation of ROS and/or RNS, which can cause oxidative stress and activate Bach1 and Nrf2 [25], [29].
It is well known that moderate oxidative stress is fundamental to HBO induced cytoprotective proteins, which can enhance the cellular tolerance against harmful stimuli [15]. This study began with observation of the effects of HBO on intracellular ROS and NO levels after HBO exposure using DCFH-DA [30] and DAF-FM DA [31], respectively. The results showed that HBO exposure obviously elevated intracellular ROS and NO levels immediately after HBO exposure, which could be inhibited by NAC or L-NAME. DCFH-DA is the most widely used probe for detecting intracellular ROS. However it reacts with many types of ROS, so the specific ROS increased after HBO exposure was not clear in this study [32], [33]. In addition to cause oxidative stress, intracellular ROS and NO can also act as signal molecules, and in some cases, they can regulate each other’s production [34], [35]. However, in the present experimental scenario, no interaction between them was found.
Since ROS and NO are two well established inducers of HSP32 [25], [36], their involvements in HBO induced HSP32 expression were further observed. The results indicate that ROS but not NO participated in HBO induced HSP32 expression. A similar finding has been reported in human umbilical vein endothelial cells [37].
Recent studies suggested that the induction of HSP32 requires activation of several signaling pathways, including Akt, ATF-2, BCR-Abl-2, MEK1/2-ERK1/2, JNK, p38 MAPK, STAT3, NF-κB, CREB, and Nrf2 [25], [38]. Apart from these signal molecules, under hypoxic conditions, HIF-1α is another important regulator of HSP32 gene expression, which, however, will rapidly degrade at normal oxygen tension or in a hyperoxic environment [25]. To elucidate the signal pathway(s) mediated between HBO and HSP32 expression in spinal neurons, the activation of the above signal molecules and the upstream and downstream relationship were observed, and revealed that HBO induced HSP32 expression is mediated via the ROS/p38 MAPK/Nrf2 pathway.
One finding of significance and interesting in this study was the discovery that a negative regulation mechanism simultaneously worked with the positive induction pathway for HSP32, which was further highlighted by the finding that MEK1/2 acted through a non-ERK1/2 pathway. The existing data showed that activation of ERK1/2 played a mostly positive role in the regulation of HSP32 [25], [39], and ERK1/2 is the only known physiological substrates of MEK1/2 [26]. U0126 is a specific inhibitor of MEK1/2 and is commonly used to inhibit the activation of the MEK1/2-ERK1/2 cascade [27], [40]. As far as we know, this is the first finding that MEK1/2 may work via pathways other than ERK1/2.
How did MEK1/2 exert inhibition of the expression of HSP32 induced by HBO? Bach1 is the main suppressive transcription factor of ARE regulated genes, including HSP32 [25]. The translocation from nucleus to cytoplasm was increased following HBO exposure, which was further enhanced by MEK1/2 inhibition. This suggests that Bach1 was the nuclear factor mediating the negative regulation downstream to MEK1/2. However, whether other molecules were involved between MEK1/2 and Bach1 needs further study.
A growing body of data indicates that, although HSP32 serves a cytoprotective function, the surge of heme metabolites can also result in neuronal cell death [41], [42]. The existence of a MEK1/2 negative regulation pathway may be a self-protection mechanism in spinal neurons. Meanwhile, the present results show that the activation of all cytosol signal molecules occurred and recovered within 0.5 h, activation of the Nrf2 and Bach1 lasted for more than 6 h, and in our previous study, the activated expression of HSP32 lasted for more than 30 h with the peak appearing at 12 h [7]. The negative regulation may play pivotal role on the suspension of activation. The physiological significance of this in vivo deserves further study.
In conclusion, this study revealed that HBO induced HSP32 expression in primary cultured rat spinal neurons through ROS/p38 MAPK/Nrf2 pathway is synchronized by MEK1/2/Bach1 mediated negative regulation, and the effects of MEK1/2 was not exerted via ERK1/2, which are generally regarded as its only physiological substrates.
Conflict of interest
The authors declare that they have no competing interests.
Author contributions
WGX conceived the study, designed the experiments, and edited the manuscript. GYH, JLD and LX performed the experiments and wrote the manuscript. HJY and JJX analyzed and interpreted the data. All the authors approved the final version of the manuscript.
Acknowledgments
This work was supported by the National Natural Science Foundation of China, China, No. 81171873. Great thanks to Dr. Peter Buzzacott for the help in preparation the manuscript.
References
- 1.O’Callaghan A., Mastracci T.M., Eagleton M.J. Staged endovascular repair of thoracoabdominal aortic aneurysms limits incidence and severity of spinal cord ischemia. J. Vasc. Surg. 2015;61:347–354. doi: 10.1016/j.jvs.2014.09.011. (e341) [DOI] [PubMed] [Google Scholar]
- 2.Vann R.D., Butler F.K., Mitchell S.J., Moon R.E. Decompression illness. Lancet. 2011;377:153–164. doi: 10.1016/S0140-6736(10)61085-9. [DOI] [PubMed] [Google Scholar]
- 3.Juurlink B.H., Paterson P.G. Review of oxidative stress in brain and spinal cord injury: suggestions for pharmacological and nutritional management strategies. J. Spinal Cord Med. 1998;21:309–334. doi: 10.1080/10790268.1998.11719540. [DOI] [PubMed] [Google Scholar]
- 4.Norenberg M.D., Smith J., Marcillo A. The pathology of human spinal cord injury: defining the problems. J. Neurotrauma. 2004;21:429–440. doi: 10.1089/089771504323004575. [DOI] [PubMed] [Google Scholar]
- 5.Wan I.Y., Angelini G.D., Bryan A.J., Ryder I., Underwood M.J. Prevention of spinal cord ischaemia during descending thoracic and thoracoabdominal aortic surgery. Eur. J. Cardiothorac. Surg. 2001;19:203–213. doi: 10.1016/s1010-7940(00)00646-1. [DOI] [PubMed] [Google Scholar]
- 6.Ni X.X., Ni M., Fan D.F., Sun Q., Kang Z.M., Cai Z.Y., Liu Y., Liu K., Li R.P., Xu W.G. Heat-shock protein 70 is involved in hyperbaric oxygen preconditioning on decompression sickness in rats. Exp. Biol. Med. 2013;238:12–22. doi: 10.1258/ebm.2012.012101. [DOI] [PubMed] [Google Scholar]
- 7.Huang G.Y., Xu J.J., Xu L., Wang S., Li R.P., Liu K., Zheng J., Cai Z.Y., Zhang K., Luo Y.D., Xu W.G. Hyperbaric oxygen preconditioning induces tolerance against oxidative injury and oxygen-glucose deprivation by up-regulating heat shock protein 32 in rat spinal neurons. PLoS One. 2014;9:e85967. doi: 10.1371/journal.pone.0085967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gozzelino R., Jeney V., Soares M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010;50:323–354. doi: 10.1146/annurev.pharmtox.010909.105600. [DOI] [PubMed] [Google Scholar]
- 9.Maines M.D. The heme oxygenase system: a regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 10.Tsuchihashi S., Fondevila C., Kupiec-Weglinski J.W. Heme oxygenase system in ischemia and reperfusion injury. Ann. Transplant. 2004;9:84–87. [PubMed] [Google Scholar]
- 11.Baranano D.E., Wolosker H., Bae B.I., Barrow R.K., Snyder S.H., Ferris C.D. A mammalian iron ATPase induced by iron. J. Biol. Chem. 2000;275:15166–15173. doi: 10.1074/jbc.275.20.15166. [DOI] [PubMed] [Google Scholar]
- 12.Stocker R., Yamamoto Y., McDonagh A.F., Glazer A.N., Ames B.N. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 13.Thom S.R. Hyperbaric oxygen: its mechanisms and efficacy. Plast. Reconstr. Surg. 2011;127(Suppl 1):131S–141S. doi: 10.1097/PRS.0b013e3181fbe2bf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ding Z., Tong W.C., Lu X.X., Peng H.P. Hyperbaric oxygen therapy in acute ischemic stroke: a review. Int. Neurol. 2014;2:201–211. doi: 10.1159/000362677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thom S.R. Oxidative stress is fundamental to hyperbaric oxygen therapy. J. Appl. Physiol. 2009;106:988–995. doi: 10.1152/japplphysiol.91004.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin X., Wang X., Fan Z., Peng C., Ren Z., Huang L., Liu Z., Zhao K. Hyperbaric oxygen preconditioning attenuates myocardium ischemia-reperfusion injury through upregulation of heme oxygenase 1 expression: pi3k/akt/nrf2 pathway involved. J. Cardiovasc. Pharmacol. Ther. 2015;20:428–438. doi: 10.1177/1074248414568196. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y., Sun X.J., Liu J., Kang Z.M., Deng X.M. Heme oxygenase-1 could mediate the protective effects of hyperbaric oxygen preconditioning against hepatic ischemia-reperfusion injury in rats. Clin. Exp. Pharmacol. Physiol. 2011;38:675–682. doi: 10.1111/j.1440-1681.2011.05560.x. [DOI] [PubMed] [Google Scholar]
- 18.He X., Xu X., Fan M., Chen X., Sun X., Luo G., Chen L., Mu Q., Feng Y., Mao Q., Chao Z. Preconditioning with hyperbaric oxygen induces tolerance against renal ischemia-reperfusion injury via increased expression of heme oxygenase-1. J. Surg. Res. 2011;170:e271–e277. doi: 10.1016/j.jss.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 19.Xu W., Liu W., Huang G., Zou Z., Cai Z., Xu W. Decompression illness: clinical aspects of 5278 consecutive cases treated in a single hyperbaric unit. PLoS One. 2012;7:e50079. doi: 10.1371/journal.pone.0050079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogawa Y., Saito Y., Nishio K., Yoshida Y., Ashida H., Niki E. Gamma-tocopheryl quinone, not alpha-tocopheryl quinone, induces adaptive response through up-regulation of cellular glutathione and cysteine availability via activation of ATF4. Free Radic. Res. 2008;42:674–687. doi: 10.1080/10715760802277396. [DOI] [PubMed] [Google Scholar]
- 21.Peng G., Han M., Du Y., Lin A., Yu L., Zhang Y., Jing N. SIP30 is regulated by ERK in peripheral nerve injury-induced neuropathic pain. J. Biol. Chem. 2009;284:30138–30147. doi: 10.1074/jbc.M109.036756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aruoma O.I., Halliwell B., Hoey B.M., Butler J. The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic. Biol. Med. 1989;6:593–597. doi: 10.1016/0891-5849(89)90066-x. [DOI] [PubMed] [Google Scholar]
- 23.Jendzjowsky N.G., DeLorey D.S. Role of neuronal nitric oxide in the inhibition of sympathetic vasoconstriction in resting and contracting skeletal muscle of healthy rats. J. Appl. Physiol. 2013;115:97–106. doi: 10.1152/japplphysiol.00250.2013. [DOI] [PubMed] [Google Scholar]
- 24.Kim W., Kim H.U., Lee H.N., Kim S.H., Kim C., Cha Y.N., Joe Y., Chung H.T., Jang J., Kim K., Suh Y.G., Jin H.O., Lee J.K., Surh Y.J. Taurine chloramine stimulates efferocytosis through upregulation of Nrf2-mediated heme oxygenase-1 expression in murine macrophages: possible involvement of carbon monoxide. Antioxid. Redox Signal. 2015;23:163–177. doi: 10.1089/ars.2013.5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jazwa A., Cuadrado A. Targeting heme oxygenase-1 for neuroprotection and neuroinflammation in neurodegenerative diseases. Curr. Drug Targets. 2010;11:1517–1531. doi: 10.2174/1389450111009011517. [DOI] [PubMed] [Google Scholar]
- 26.Roskoski R.J. MEK1/2 dual-specificity protein kinases: structure and regulation. Biochem. Biophys. Res. Commun. 2012;417:5–10. doi: 10.1016/j.bbrc.2011.11.145. [DOI] [PubMed] [Google Scholar]
- 27.Seow S.L., Eik L.F., Naidu M., David P., Wong K.H., Sabaratnam V. Lignosus rhinocerotis (cooke) ryvarden mimics the neuritogenic activity of nerve growth factor via MEK/ERK1/2 signaling pathway in PC-12 cells. Sci. Rep. 2015;5:16349. doi: 10.1038/srep16349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elbirt K.K., Whitmarsh A.J., Davis R.J., Bonkovsky H.L. Mechanism of sodium arsenite-mediated induction of heme oxygenase-1 in hepatoma cells. Role of mitogen-activated protein kinases. J. Biol. Chem. 1998;273:8922–8931. doi: 10.1074/jbc.273.15.8922. [DOI] [PubMed] [Google Scholar]
- 29.Cuadrado A., Moreno-Murciano P., Pedraza-Chaverri J. The transcription factor Nrf2 as a new therapeutic target in Parkinson’s disease. Expert Opin. Ther. Targets. 2009;13:319–329. doi: 10.1517/13543780802716501. [DOI] [PubMed] [Google Scholar]
- 30.Malhotra P., Adhikari M., Singh S.K., Kumar R. N-acetyl tryptophan glucopyranoside (NATG) provides radioprotection to murine macrophage J774A.1 cells. Free Radic. Res. 2015;49:1488–1498. doi: 10.3109/10715762.2015.1095295. [DOI] [PubMed] [Google Scholar]
- 31.Liu M., Zollbrecht C., Peleli M., Lundberg J.O., Weitzberg E., Carlstrom M. Nitrite-mediated renal vasodilatation is increased during ischemic conditions via cGMP-independent signaling. Free Radic. Biol. Med. 2015;84:154–160. doi: 10.1016/j.freeradbiomed.2015.03.025. [DOI] [PubMed] [Google Scholar]
- 32.Forman H.J., Augusto O., Brigelius-Flohe R., Dennery P.A., Kalyanaraman B., Ischiropoulos H., Mann G.E., Radi R., Roberts L.J. 2nd, Vina J., Davies K.J. Even free radicals should follow some rules: a guide to free radical research terminology and methodology. Free Radic. Biol. Med. 2015;78:233–235. doi: 10.1016/j.freeradbiomed.2014.10.504. [DOI] [PubMed] [Google Scholar]
- 33.Banskota S., Regmi S.C., Kim J.A. NOX1 to NOX2 switch deactivates AMPK and induces invasive phenotype in colon cancer cells through overexpression of MMP-7. Mol. Cancer. 2015;14:123. doi: 10.1186/s12943-015-0379-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maines M.D. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988;2:2557–2568. [PubMed] [Google Scholar]
- 35.Minetti M., Mallozzi C., Di Stasi A.M., Pietraforte D. Bilirubin is an effective antioxidant of peroxynitrite-mediated protein oxidation in human blood plasma. Arch. Biochem. Biophys. 1998;352:165–174. doi: 10.1006/abbi.1998.0584. [DOI] [PubMed] [Google Scholar]
- 36.Motterlini R., Foresti R., Intaglietta M., Winslow R.M. NO-mediated activation of heme oxygenase: endogenous cytoprotection against oxidative stress to endothelium. Am. J. Physiol. 1996;270:H107–H114. doi: 10.1152/ajpheart.1996.270.1.H107. [DOI] [PubMed] [Google Scholar]
- 37.Liu X.M., Peyton K.J., Shebib A.R., Wang H., Durante W. Compound C stimulates heme oxygenase-1 gene expression via the Nrf2-ARE pathway to preserve human endothelial cell survival. Biochem. Pharmacol. 2011;82:371–379. doi: 10.1016/j.bcp.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsieh H.L., Wang H.H., Wu C.Y., Yang C.M. Reactive oxygen species-dependent c-Fos/activator protein 1 induction upregulates heme oxygenase-1 expression by bradykinin in brain astrocytes. Antioxid. Redox Signal. 2010;13:1829–1844. doi: 10.1089/ars.2009.2957. [DOI] [PubMed] [Google Scholar]
- 39.Chuang J.I., Huang J.Y., Tsai S.J., Sun H.S., Yang S.H., Chuang P.C., Huang B.M., Ching C.H. FGF9-induced changes in cellular redox status and HO-1 upregulation are FGFR-dependent and proceed through both ERK and AKT to induce CREB and Nrf2 activation. Free Radic. Biol. Med. 2015;89:274–286. doi: 10.1016/j.freeradbiomed.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 40.Kim B., Sullivan K.A., Backus C., Feldman E.L. Cortical neurons develop insulin resistance and blunted Akt signaling: a potential mechanism contributing to enhanced ischemic injury in diabetes. Antioxid. Redox Signal. 2011;14:1829–1839. doi: 10.1089/ars.2010.3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keep R.F., Hua Y., Xi G. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. 2012;11:720–731. doi: 10.1016/S1474-4422(12)70104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kwon K.J., Kim J.N., Kim M.K., Kim S.Y., Cho K.S., Jeon S.J., Kim H.Y., Ryu J.H., Han S.Y., Cheong J.H., Ignarro L.J., Han S.H., Shin C.Y. Neuroprotective effects of valproic acid against hemin toxicity: possible involvement of the down-regulation of heme oxygenase-1 by regulating ubiquitin-proteasomal pathway. Neurochem. Int. 2013;62:240–250. doi: 10.1016/j.neuint.2012.12.019. [DOI] [PubMed] [Google Scholar]