

Abstract
Carfilzomib (CFZ) is a second-generation proteasome inhibitor that is Food and Drug Administration and European Commission approved for the treatment of relapsed or refractory multiple myeloma. CFZ is an epoxomicin derivative with an epoxyketone electrophilic warhead that irreversibly adducts the catalytic threonine residue of the β5 subunit of the proteasome. Although CFZ produces a highly potent, sustained inactivation of the proteasome, the electrophilic nature of the drug could potentially produce off-target protein adduction. To address this possibility, we synthesized an alkynyl analog of CFZ and investigated protein adduction by this analog in HepG2 cells. Using click chemistry coupled with streptavidin based IP and shotgun tandem mass spectrometry (MS/MS), we identified two off-target proteins, cytochrome P450 27A1 (CYP27A1) and glutathione S-transferase omega 1 (GSTO1), as targets of the alkynyl CFZ probe. We confirmed the adduction of CYP27A1 and GSTO1 by streptavidin capture and immunoblotting methodology and then site-specifically mapped the adducts with targeted MS/MS methods. Although CFZ adduction of CYP27A1 and GSTO1 in vitro decreased the activities of these enzymes, the small fraction of these proteins modified by CFZ in intact cells should limit the impact of these off-target modifications. The data support the high selectivity of CFZ for covalent modification of its therapeutic targets, despite the presence of a reactive electrophile. The approach we describe offers a generalizable method to evaluate the safety profile of covalent protein-modifying therapeutics.
Drugs that act by covalently modifying their target molecules through electrophilic reactive groups are being used with an increasing frequency in the clinic (1, 2). These drugs are typically characterized by low treatment doses and long effect durations, as a result of their presumed high specificity for the target molecule (3). Historically, drug designers have been reluctant to create compounds that act via covalent protein modification, because of concerns about potential toxicity arising from off-target binding of the compound to other cellular proteins. However, recent successes with designed covalent inhibitors such as ibrutinib have renewed interest in this class of molecules (1–6).
Carfilzomib (CFZ)1 is a second-generation proteasome inhibitor that has been Food and Drug Administration and European Commission approved for use in relapsed and refractory multiple myeloma (7–9). Structurally, CFZ is an epoxyketone tetrapeptide that irreversibly adducts the proteasome at catalytic threonine residues in the β5 subunits and inhibits chymotrypsin like activity of the proteasome (10). Functionally, CFZ has been demonstrated to overcome resistance to the first-in-class proteasome inhibitor bortezomib and to inhibit the proteasome for a longer duration (11, 12). In the clinic, CFZ has been observed to have a broad tissue distribution with a short half-life of less than 30 min. CFZ is well tolerated with acceptable toxicity profiles and few instances of dose limiting toxicity resulting in cessation of treatment (13, 14). Both single agent and combination therapies with CFZ have demonstrated good response profiles for patients with relapsed/refractory multiple myeloma and CFZ is now undergoing trials for use as a first-line treatment (11, 15–22). However, an unanswered question for CFZ, and for the field of covalent inhibitors in general, is the extent to which the electrophilic warhead in a covalent inhibitor produces off-target damage to proteins. Furthermore, if CFZ does produce protein damage, is that damage a risk for treatment?
Direct mass spectrometry (MS)-based identification of protein adduction by covalent inhibitors provides the most experimentally accessible opportunity to define the potential off-target adductome of a given drug. The integration of alkynyl probes and cleavable click reagents for the purification of adducted proteins into the MS workflow has greatly broadened the utility of this technique. This approach has proven valuable for functional proteomics studies by activity-based proteome profiling (23–25) and to explore cellular protein targets of oxidants and electrophiles (26–31).
Here we describe the application of an alkynyl analog of CFZ to explore off-target binding of CFZ to proteins in a hepatic cell model. We report the identification of 12 off-target proteins following treatment of HepG2 cells with the alkynyl CFZ analog at concentrations close to the maximum plasma concentration of 7.94 μm observed clinically for CFZ at a dose of 36 mg/m2 (32, 33). We further characterized the sites of CFZ adduction on two of the off-target proteins and assessed the effect of adduction on their activities. This study confirms the high specificity of CFZ for its therapeutic targets and provides a useful approach to assess the safety of covalent protein-modifying drugs.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Unless otherwise started, all reagents were purchased from Sigma-Aldrich (St. Louis, MO). CFZ was purchased from ApexBio (Houston, TX). Purified human GSTO1 was purchased from Abcam (Cambridge, MA). Human CYP27A1 was prepared as described (34). Bovine adrenodoxin was expressed in Escherichia coli DH5α cells with the expression plasmid pBA1159 (35, 36). Bovine NADPH-adrenodoxin reductase was expressed in E. coli JM109 cells as described (36, 37), using a pCWori+ expression vector (38).
Primary antibodies used in this study were: PSMB5 (Cell Signaling Technology, Danvers, MA; catalog number 11903S), CYP27A1 (Novus Biologicals, Littleton, CO; catalog number H00001593-D01), GSTO1 (US Biological, Salem, MA; catalog number 127609), and streptavidin (Life Technologies, Carlsbad, CA). Secondary antibodies were from Life Technologies: anti-mouse 680 (catalog number A21058) and anti-rabbit 680 (catalog number A21109).
Click analogs of CFZ were synthesized by Nanosyn (Santa Clara, CA). The syntheses of (S)-6-azido-N-((S)-4-methyl-1-((R)-2-methyloxiran-2-yl)-1-oxopentan-2-yl)-2-((S)-4-methyl-2-((S)-2-(2-morpholinoacetamido)-4-phenylbutanamido)pentanamido)hexanamide (OP-0058830; OP-830), 4-methyl-2-[2-(2-morpholin-4-yl-acetylamino)-4-phenyl-butyrylamino]-pentanoic acid [2-(4-azido-phenyl)-1-[3-methyl-1-(2-methyl-oxiranecarbonyl)-butylcarbamoyl]-ethyl]-amide (OP-0058828; OP-828), and 4-methyl-2-[2-(2-morpholin-4-yl-acetylamino)-4-phenyl-butyrylamino]-pentanoic acid [2-(4-ethynyl-phenyl)-1-[3-methyl-1-(2-methyl-oxiranecarbonyl)-butylcarbamoyl]-ethyl]-amide (OP-0058829; OP-829) are described in Supplementary Methods. The photocleavable azidobiotin click reagent (azido-UV-biotin) was synthesized as described previously (30).
Cell Culture and Treatments
HepG2 cells were obtained from ATCC (Manassas, VA) and grown in Eagle's Minimal Essential Medium (ATCC) supplemented with 10% fetal bovine serum (v/v) (Atlas Biologicals, Fort Collins, CO) at 37 °C in a humidified atmosphere with 5% CO2 (v/v). Cells were plated in 15 cm dishes prior to treatment. Plated cells were treated with OP-829 for 1 h at 37 °C, then washed and harvested in phosphate-buffered saline (PBS). Harvested cells were pelleted at 100 × g for 5 min at 4 °C and then washed twice with PBS. Washed cells were frozen at −80 °C until use.
Human hepatocellular carcinoma SNU-449 cells (ATCC) were cultured in RPMI 1640 Medium (ATCC) supplemented with 10% fetal bovine serum (v/v), 2 mm l-glutamine, and 50 U penicillin and 50 μg streptomycin per ml. The day before treatment, 30,000 cells per well were seeded on a 96-well flat bottom plate. The following day, serial dilutions of test compounds were added to the cells (4 μm maximum final concentration, 0.1% dimethyl sulfoxide (v/v) (DMSO)) for 1 h followed by a PBS wash and then addition of hypotonic lysis buffer (20 mm Tris pH 8.0, 5 mm EDTA).
To assay activity in the SNU-449 cell lysates, succinyl-Leu-Leu-Val-Tyr-7-amino-4-methylcoumarin (Suc-LLVY-AMC; Boston Biochem, Cambridge, MA) was utilized as a specific fluorogenic peptide substrate for detection of the chymotrypsin-like activity of the constitutive (β5) proteasome subunit and immunoproteasome (LMP7/β5i) subunit (39). Ten μl of sample lysate were mixed with 10 μl of Suc-LLVY-AMC substrate (60 μm, 1% DMSO (v/v) final) in a 384-well low volume plate. Signal from the released amino-4-methylcoumarin reaction product was detected using a fluorescent plate reader (Tecan, Männedorf, Switzerland) with excitation and emission wavelengths of 380 and 465 nm, respectively. Activity was quantified at the 60 min reaction time point and normalized to DMSO-treated controls. Data are presented as mean ± S.E.
Metabolic Stability of CFZ Analogs in Hepatocytes
Treatment and Sample Preparation
Cryopreserved human hepatocytes (Xenotech, Kansas City, KS) were quickly thawed in a 37 °C water bath and added to supplemental Dulbecco's Modified Eagle Media (DMEM without penicillin/streptomycin) and isotonic Percoll. Cells were centrifuged and supernatant was aspirated. Cell pellets were re-suspended in supplemental DMEM (without penicillin/streptomycin) and viable cells were counted using trypan blue and a hemocytometer. Hepatocyte incubations were performed with a viable cell concentration of 0.75 × 106 cells/ml and a concentration of 1 μm CFZ, OP-828, OP-829, or OP-830. Positive control incubations with 1 μm 7-ethoxycoumarin and negative control incubations without substrate were also conducted. Total initial incubation volume was 400 μl. Hepatocytes were incubated in a 37 °C incubator containing 5% CO2/95% ambient air (v/v). Aliquots (50 μl) were collected from the plates at 0, 15, 30, 60, 90, and 120 min and were mixed with three volumes of acetonitrile with the internal standard to quench the reaction. After quenching, cells were centrifuged and diluted 1:2 (v/v) with water and 0.1% formic acid before analyzing by liquid chromatography-tandem mass spectrometry (LC-MS/MS) for quantification. For these experiments, incubations were conducted in duplicate.
LC-MS/MS Analysis of CFZ Analogs
LC-MS/MS analyses were performed on an ABSCIEX 5500 QTRAP mass spectrometer (Foster City, CA) equipped with Shimadzu LC-20ADvp pumps, autosampler, and controller system (Columbia, MD) using an XBridge BEH C18, 3.5 μm, 2.1 mm × 100 mm column (Waters, Milford, MA). The instrument was operated in the positive ion electrospray mode using multiple reaction monitoring with parameters individually optimized for each compound and internal standard. The mobile phases (both LC-MS grade) were acetonitrile with 0.1% formic acid (solution A) and water with 0.1% formic acid (solution B), purchased from J.T. Baker (Phillipsburg, NJ). For CFZ and 7-ethoxycoumarin, the following elution method was used at a flow rate of 0.3 ml/min: initial isocratic phase at 25% B (v/v) for the first 0.3 min, a linear gradient from 25 to 95% B over 1.7 min, isocratic phase at 95% B (v/v) for 0.7 min and then re-equilibration to initial conditions. For OP-828, OP-829, and OP-830, the following elution method was employed at a flow rate of 0.4 ml/min: initial isocratic phase at 25% B (v/v) for the first 0.3 min, a linear gradient from 25 to 95% B (v/v) over 1 min, isocratic phase at 95% B (v/v) for 1.4 min and then re-equilibration to initial conditions.
CYP27A1 and GSTO Inhibition Assays
Inhibition assays for CYP27A1 upon CFZ treatment were carried out in 0.5 ml reaction volume containing 1.0 μm CYP27A1, 2.0 μm adrenodoxin, 0.2 μm adrenodoxin reductase, 100 mm potassium phosphate buffer (pH 7.5), 150 μm l-α-1,2-dilauroyl-sn-glycero-3-phosphocholine, and a final cholesterol concentration of 50 μm. The stock solution of cholesterol was prepared in 310 mm (2-Hydroxypropyl)-β-cyclodextrin (45% HPβCD (w/v)). A 5 μl aliquot of stock cholesterol solution was added to the 500 μl final reaction volume. The concentration of CFZ was varied from 2 pm–25 μm. Assays were done with and without preincubation with CFZ. For with pre-incubation, the reaction mixture was kept at 37 °C for 1 h. After that, enzymatic reactions were initiated by the addition of 150 μl of an NADPH-generating system (final concentrations: 20 mm glucose 6-phosphate, 1 mm NADP+, and 2 U/ml yeast glucose 6-phosphate dehydrogenase) (40) and incubated for 15 min at 37 °C. The reaction was quenched with methylene chloride (2 ml), and the products were extracted twice with 2 ml of methylene chloride and centrifuged at 2000 × g for 10 min (23 °C). The organic layers were combined and evaporated to dryness under a nitrogen stream. The residues were dissolved in acetonitrile and subjected to LC-MS (APCI+) analysis. LC-MS was performed on an LTQ mass spectrometer (ThermoFisher Scientific, San Jose, CA) with a Waters Acquity UPLC system and ACQUITY UPLC BEH octadecylsilane (C18) (1.7 μm, 2.1 mm × 100 mm) column at 35 °C at a flow rate of 0.3 ml/min. Mobile phase A consisted of 95:5 (v/v) water and acetonitrile with 0.1% formic acid (v/v); mobile phase B consisted of 5:95 (v/v) water and acetonitrile with 0.1% formic acid (v/v). The LC gradient was programmed in linear steps as follows: 0–8 min, 80% mobile phase B (v/v); 8–9 min, 80% mobile phase B to 100% mobile phase B (v/v); 9–20 min, 100% mobile phase B; 20–21 min, 100% mobile phase B to 80% mobile phase B (v/v); 21–30 min, 80% mobile phase B (v/v). The product 27-hydroxycholesterol was detected at m/z 385 and confirmed by comparing with a commercially available authentic standard (Enzo, Farmingdale, NY). Data were analyzed using Prism software (version 5.04, GraphPad, San Diego, CA).
Inhibition assays for GSTO1 upon CFZ treatment were carried out in 96-well plates using a 0.2 ml reaction volume containing 1.0 nm GSTO1 enzyme, 1.0 mm GSH, 1.0 mm chloro-2,4-dinitrobenzene, 100 mm potassium phosphate buffer (pH 6.5), and variable amounts of CFZ. The concentration of CFZ was varied from 5 nm–25 μm. Assay was done with and without pre-incubation with CFZ. For pre-incubation, reaction mixture containing PBS, enzyme and CFZ, was kept at 37 °C for 1 h. After that, GSH was added to the reaction mixture and enzymatic reactions were initiated by the addition of chloro-2,4-dinitrobenzene, to a final concentration of 1 mm, and a linear increase in absorbance at 340 nm was monitored (41). Data was analyzed using GraphPad Prism software (version 5.04).
Detection of OP-829 Adduction of CYP27A1, GSTO and PSMB5 by Click Capture and Immunoblotting
For each experiment, four plates of cells treated with OP-829 were lysed in modified NETN buffer (1 ml/plate; 50 mm Na+ HEPES, 150 mm NaCl, 0.5% Nonidet Nonidet P-40 (v/v), 0.25% SDS (w/v), pH 7.5) plus protease and phosphatase inhibitors (0.5 μm AEBSF, 10 mm aprotinin, 1.0 mm leupeptin, 5 μm bestatin and 1.0 μm pepstatin, 1.0 mm sodium orthovanadate, 1.0 mm sodium molybdate, 1.0 mm sodium fluoride, and 10 mm β-glycerophosphate). After resuspension in this buffer, the lysates were sonicated for 20 pulses, incubated on ice for 20 min, clarified by centrifugation at 10,000 × g for 10 min, and filtered through a 0.22 μm syringe filter GHP membrane (Pall Life Sciences, Radnor, PA). Protein concentrations were determined using a bicinchoninic acid (BCA) assay (Life Technologies). A total of 16 mg of protein was used for each treatment at a concentration of 2.7 mg/ml; samples were split into two aliquots for ease of sample preparation. Huisgen 1,3-dipolar cycloaddition (click chemistry) was performed as described (29) with 0.2 mm azido-UV-biotin (30), 1 mm sodium ascorbate, 1 mm cupric sulfate, and 0.1 mm tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine to couple the alkynyl analog-adducted proteins to the N3-UV-biotin linker. Proteins were precipitated with methanol to remove excess small molecule reagents, and each sample then was applied to a prewashed (2× modified NETN) 1 ml slurry of streptavidin beads (GE Life Sciences, Pittsburgh, PA) and incubated at room temperature with rotation for 1.5 h. The beads with bound proteins were washed twice with 1% sodium dodecyl sulfate (w/v), twice with 4 m urea, and then once each with 1 m NaCl and PBS. The tagged proteins then were eluted by photorelease with UV light in 1 ml of PBS for 1.5 h. The eluted proteins were passed through a QIAshredder (Qiagen, Valencia, CA) to remove any beads from the elution and then concentrated to a total volume of 60 μl in PBS with a 10 kDa molecular weight cutoff Nanosep centrifugal devices that contain an Omega membrane for low protein binding (VWR, San Dimas, CA). To the 60 μl concentrated elution was added 5 μl of 1 m dithiothreitol (DTT) and 15 μl of lithium dodecyl sulfate (Life Technologies), and the samples were frozen at −80 °C until use.
For immunoblot analysis, sodium dodecyl sulfate-polyacrylamide electrophoresis was performed with either 50 μg of cell lysate or 16 μl of click-labeled, captured protein and run for 50 min at a constant 180 V. Proteins were then electrophoretically transferred to PVDF membranes (Life Technologies) with a BioRad (Hercules, California) wet transfer system operated at a constant 300 mA current for 90 min at 4 °C. Membranes were blocked with a 1:1 mixture (v/v) of Tris-buffered saline plus 0.05% Tween-20 (v/v) (TBST) and blocking buffer (Rockland) for 1 h at room temperature with rocking. Primary antibodies to CYP27A1, GSTO, and PSMB5 were diluted in the same buffer used for blocking, added to the membrane, and incubated at 4 °C overnight with rocking. After incubation, the membranes were washed three times with TBST, incubated with the appropriate secondary antibody diluted 1:10,000 (v/v) in the same buffer used for the primary antibody, and allowed to rock for 30 min at room temperature. The membranes were then washed three times with TBST, visualized using a LiCor Odyssey (Lincoln, Nebraska), and analyzed using Odyssey v3.0 software.
Sample Preparation for LC-MS/MS Analyses
Bead-bound proteins were eluted in LDS buffer and heated at 95 °C for 10 min prior to being loaded on a 10% (w/v) Bis-Tris gel (Life Technologies). The gels were loaded with 30 μl of the eluate (one sample in every other lane) and were then run at a constant 180 V for 3 min and then paused. An additional 30 μl of each sample was loaded on top of the previous sample in the empty loading lanes. The gels were restarted and allowed to run for an additional 7 min to ensure that all of the loaded proteins were inside the gel. Gels were then stained with Simply Blue safe stain (Life Technologies) for 1 min in a microwave oven and then were destained in distilled water for 2–3 h. Each sample lane was cut as a single band and diced into ∼1 mm cubes. The gel pieces were placed into individual Eppendorf tubes and washed twice with 200 μl of 100 mm (Ammonium Bicarbonate (AmBic) buffer). Each sample then was reduced with 5 mm DTT in AmBic buffer for 30 min at 60 °C while shaking at 1000 rpm and then alkylated with 10 mm iodoacetamide in AmBic buffer for 20 min in the dark at room temperature. Excess blue die was then removed with three 200 μl washes of 1:1 (v/v) 50 mm AmBic buffer/acetonitrile and the gel pieces were dehydrated by soaking in 100% acetonitrile for 20 min. The gel pieces were rehydrated in 200 μl of 25 mm AmBic buffer with 500 ng of trypsin per sample and placed in a 37 °C incubator for 16 h. Peptides were extracted with three 20 min incubations of 200 μl of acetonitrile: water (3:2, v/v) containing 1% formic acid and evaporated to dryness in vacuo. The peptides were resuspended in 50 μl of water containing 2% acetonitrile and 0.1% formic acid and injected for LC-MS/MS analysis.
LC-MS/MS Analyses
LC-MS/MS shotgun proteomics of the single gel fraction samples were carried out on a Q Exactive mass spectrometer (ThermoFisher Scientific) equipped with a Proxeon nLC1000 LC (ThermoFisher Scientific) and a Nanoflex source(ThermoFisher Scientific). Peptides were loaded onto a 15-cm long column with a 75 μm internal diameter (New Objective, Woburn, MA) packed with 3 μm particle size and 120 Å pore size ReproSil-Pur C18-AQ resin (Dr. Maisch GmbH, Ammerbuch-Entringen, Germany). Peptides were eluted over a 150 min gradient with a mobile phase containing aqueous acetonitrile and 0.1% formic acid programmed from 2–5% acetonitrile over 15 min, then 2–35% acetonitrile over 100 min, then 35–90% acetonitrile over 5 min, then 11 min at 90% acetonitrile, then 7 min from 90–2% acetonitrile, followed by holding at 2% acetonitrile for 2 min, all at a flow rate of 300 nl/min. A single MS1 scan from m/z 300–1800 at 70,000 resolution with an automatic gain control (AGC) of 1e6 and max injection time of 5 msec was recorded as profile data. A top 12 method was used whereby the 12 most intense precursors were automatically chosen for MS2 analysis and a dynamic exclusion window of 20 s was employed. For each MS2 scan, a resolution of 17,500, and AGC of 1e5, a max injection time of 250 msec, a 2.5 m/z isolation window, and a normalized collision energy of 27 was used and profile data were recorded.
Parallel reaction monitoring (PRM) assays were performed on a Q Exactive Plus instrument (ThermoFisher Scientific) equipped with a Proxeon nLC1000 LC (ThermoFisher Scientific) and a Nanoflex source (ThermoFisher Scientific), utilizing the same LC column as described above. For PRM analyses, peptides were separated over a 70 min gradient: from 2–5% acetonitrile over 5 min, then 5–35% acetonitrile over 50 min, then 35–90% acetonitrile over 5 min, followed by 10 min at 90% acetonitrile, at a flow rate of 300 nl/min. The PRM method consisted of an MS1 scan at 17,500 resolution with an AGC value of 3e6, max injection time of 64 msec, and scan range from m/z 380–1500 recorded as profile data. This was followed by 14 targeted MS2 scans at a resolution of 17,500 and with an AGC value of 5e5, a max injection time of 80 msec, a 0.5 m/z isolation window, a fixed first mass of 150 m/z, normalized collision energy of 27, and recorded as profile data. The targeted-MS2 methods were controlled via an inclusion list containing the target precursor m/z values and charges (supplemental Tables S1 and S2).
Data Analysis
Thermo .raw datafiles from shotgun LC-MS/MS runs were converted to mzml format using Proteowizard version 3.0.5211 (42). The mzml files were searched using MyriMatch version 2.1.132 (43) and MS-GF+ version 9517 (44) against the human Refseq version 54 database (Sep 25, 2012; 34,590 entries). A semi-tryptic search was employed with a maximum of four missed cleavages allowed. A fixed carbimidomethyl modification on Cys, a variable OP-829 adduction on Thr with a monoisotopic mass of 743.4258, a variable oxidation on Met, and a pyro-Glu on Gln were allowed with a maximum of 3 dynamic mods per peptide with a precursor ion tolerance of 15 ppm and a fragment ion tolerance of 20 ppm. Protein identifications required a minimum of two distinct identified peptides. A target-decoy search was employed using a reverse sequence database to allow calculation of FDR for peptide-spectrum matches (45). Protein-level FDR was calculated by dividing the number of reverse sequence proteins identified by the total number of proteins identified, multiplying by two and converting to a percent. All search result files were parsimoniously assembled in IDPicker 3 version 3.1.643.0 (46).
PRM analyses to detect CFZ-modified peptides were designed and analyzed using Skyline (47). Summed peak areas of the three best transitions of the target adducted and unadducted peptides were normalized to an internal reference peptide (a different unmodified peptide from the same protein) (48) to control for variations in instrument performance and protein loading on the gel. To confirm identification of adducted peptides, manual inspection and annotation of adducted peptide spectra was performed.
Experimental Design and Statistical Rationale
Metabolic stability and activity assays for CFZ and the three analogs were performed in biological duplicate to evaluate reproducibility. Shotgun LC-MS/MS analyses of OP-829 adducted proteins were performed in biological triplicate. Statistical analysis of shotgun data was carried out using Quasitel, which requires at least three replicates in order to effectively identify differences using a generalized linear modeling approach (49). Targeted identification of CFZ adducted peptide spectra were performed once, although each analysis provided several examples of each adducted spectrum. Protein inhibition assays for CYP27A1 and GSTO1 were performed with 3 and 1 replicates respectively. Raw data files can be downloaded at: ftp://massive.ucsd.edu/MSV000079427.
RESULTS
To identify cellular proteins modified by CFZ, we employed a click chemistry-based strategy, which has been used in several recent studies to identify targets of covalent protein modification (23, 24, 28–31, 50). With this technique, a tagged (alkyne or azido group) small molecule is introduced into cells and allowed to react with target proteins. The cells are lysed and the adducted proteins are linked by a copper catalyzed cycloaddition reaction to a linker containing a complementary azido or alkynyl functional group and an affinity tag (usually biotin). This linker-protein adduct conjugate is then purified with streptavidin for identification and analysis.
For this technique, we initially prepared three CFZ analogs (Fig. 1A and supplemental Methods). After verifying the success of the compound synthesis, all three analogs were tested for metabolic stability and proteasome inhibition activity (Fig. 1B and supplemental Fig. S1A). All three CFZ analogs exhibited proteasome inhibitory activity comparable to CFZ and compounds OP-829 and OP-830 exhibited similar stability in hepatocytes. Compound OP-829 was chosen for further analysis based on these data and the ready availability of the photocleavable click biotinylation reagent azido-UV-biotin, which we had used previously for similar analyses (30). MS/MS analyses of both CFZ and OP-829 were performed to further confirm their structures and identify the fragmentation patterns of both compounds (supplemental Fig. S1B–S1C).
Fig. 1.

Carfilzomib off-target identification strategy. A, The structures of the parent carfilzomib compound and three clickable analogs of it. B, Metabolic stability and activity assays comparing the performance of the click analog compounds to the parent compound. C, Workflow for the click capture and LC-MS/MS of the OP-829 analog in HepG2 cells.
HepG2 cells were chosen as our cellular model for carfilzomib adduction as they are derived from human liver cells and thus contain a relevant protein population. Cells were treated (biological triplicates) with 0 μm (DMSO vehicle control 0.05% v/v), 2.5 μm, or 5 μm OP-829 and adducted proteins were biotinylated with azido-UV-biotin, captured with streptavidin, and analyzed by LC-MS/MS (Fig. 1C).
After MS/MS analysis and database search, peptide-spectrum matches were filtered at a 1% FDR and the datasets (three replicates each of the 0, 2.5, and 5 μm OP-829 treatments) were combined to generate a single protein assembly (see supplemental Data set). To control protein-level FDR, a minimum of 10 MS/MS spectra per protein was required for retention in the final dataset. A total of 882 protein groups (indistinguishable proteins) were identified across all conditions at a protein FDR estimated at zero (i.e. no reversed sequence proteins were in the final dataset).
Initial examination of the spectral count profiles for proteins identified at the two OP-829 treatment levels indicated a steep decline in spectral counts (∼100 fold) from the most highly represented to least highly represented proteins (supplemental Fig. S2A). This relationship contrasts dramatically with that observed in our previous study (29) for protein adduction in RKO cells by the relatively nonselective lipid electrophile probes alkynyl-4-hydroxy-2-nonenal (aHNE) or alkynyl-4-oxo-2-nonenal (aONE), which exhibited a shallow spectral count differential (∼10-fold) between the most and least highly represented proteins (supplemental Fig. S2B). Among the most highly represented OP-829 targets were proteasome subunits that are known therapeutic targets of CFZ.
Inventories from click labeling and streptavidin capture inevitably contain proteins that nonspecifically copurify with true probe targets. To generate the highest confidence inventory of OP-829 targets, we performed spectral count based comparisons by generalized linear modeling with the program Quasitel (49). The generalized linear modeling approach generates quasi-FDR values (multiple comparison adjusted quasi-p values). The two treatment conditions (2.5 and 5 μm) each were compared with the untreated (DMSO) controls to identify proteins that increased significantly in spectral counts relative to the untreated controls. We further assumed that true OP-829 targets should demonstrate concentration-dependent increases in spectral counts with increasing probe dose.
Based on these considerations, we employed a tiered approach to identify the highest confidence OP-829 target proteins, as shown in Table I. In addition to criteria for inclusion into the final protein assembly, we required that protein differential expression between OP-829 treatment and control have a quasi.fdr ≤ 0.1 and that differential proteins display at least fourfold higher spectral counts in OP-829 treated cells than in controls (last column, Table I). This reduced the dataset from over 800 proteins to just 27 in the 2.5 μm to DMSO comparison and 32 in the 5 μm to DMSO comparison.
Table I. Filtering process for candidate OP-829 adducted proteins.
| Comparison | PSM FDR < 1%; ≥ 6 spectra/protein | Proteins |
Proteins |
|---|---|---|---|
| quasi.fdr ≤ 0.10 | ≥ 4-fold change | ||
| DMSO vs | 863 | 31 | 27 |
| OP-829 2.5 μm | |||
| DMSO vs | 845 | 41 | 32 |
| OP-829 5.0 μm |
Overlapping the proteins identified in the filtered dataset above showed that there were a total of fifteen common proteins at the highest stringency criteria for both treatment levels (Table II). Of those fifteen proteins, three (PSMA1, PSMB3, PSMB5) were components of the proteasome, which is the known target of carfilzomib. Of the other twelve, ten were present at a total of 51 spectral counts or less for the data set and thus represent low-level protein adduction candidates. The final two proteins in the list, CYP27A1 and GSTO1, both demonstrated OP-829 concentration-dependent increases in spectral counts with total spectral counts of 184 and 200 respectively and thus constituted potential off-target proteins. We chose to further investigate CYP27A1 and GSTO1 as potential CFZ adduct targets.
Table II. Proteins identified at 2.5 and 5.0 μm OP-829 treatment concentrations or both concentrations.
| OP-829 2.5 μM | Both | OP-829 5.0 μM |
|---|---|---|
| CES2 | AIFM1 | C3 |
| CPNE3 | CYP27A1 | CTSB |
| KIAA0368 | DHRS1 | DDX18 |
| LMAN1 | ERMP1 | GNL3 |
| NOL11 | FBL | H2AFY2 |
| PGAM5 | GSTO1 | ILF3 |
| PSMB8 | PSMA1 | IQGAP1 |
| PTGR1 | PSMB3 | KHSRP |
| RSL1D1 | PSMB5 | MCCC2 |
| SEC24A | RPL19 | METTL7A |
| TXNRD1 | SEC63 | NSUN2 |
| ZMPSTE24 | SGPL1 | PSMB10 |
| TXNDC12 | PSMB7 | |
| UPF1 | PSPC1 | |
| YME1L1 | RRM1 | |
| SAMM50 | ||
| TFB1M |
Comparison of the spectral counts of GSTO1 and CYP27A1 to the known carfilzomib target protein (PSMB5) showed similar concentration-dependent increases in spectral counts with OP-829 treatment concentration (Fig. 2A). To validate our observations of selective, OP-829 concentration-dependent adduction for these three proteins, we performed immunoblot analyses on the UV-photoreleased proteins following click-streptavidin capture (Fig. 2B). We detected both PSMB5 and GSTO1 in the elution lanes of both the 2.5 and 5.0 μm OP-829-treated samples but not in the untreated control samples, thereby confirming the MS results. We were unable to confirm the presence of CYP27A1 in the click UV photoelution samples by immunoblotting; this may have been because of the inability of the antibody to recognize the adducted form of the protein. However, we attempted to probe these samples for CYP27A1 with several different antibodies, but were unable to detect the protein (data not shown).
Fig. 2.

Verification of CFZ adducted proteins. A, Plots of spectral counts for the proteasome and two off-target proteins from the shotgun study demonstrating concentration-dependent increases. B, Immunoblot validation of MS data. See text for discussion of lack of a positive signal for CYP27A1.
To further characterize CYP27A1 and GSTO1 as off-target proteins for CFZ adduction, we performed two additional experiments on purified proteins in vitro. First, we treated the proteins in vitro with OP-829, repeated the adduct click biotinylation, and analyzed them by immunoblot streptavidin detection (Fig. 3). CYP27A1 did indeed exhibit a dose-dependent increase in adduction (Fig. 3A), consistent with the LC-MS/MS results. GSTO1 also showed an OP-829 concentration-dependent increase in adduction (Fig. 3B), which appeared to be more modest than for CYP27A1.
Fig. 3.
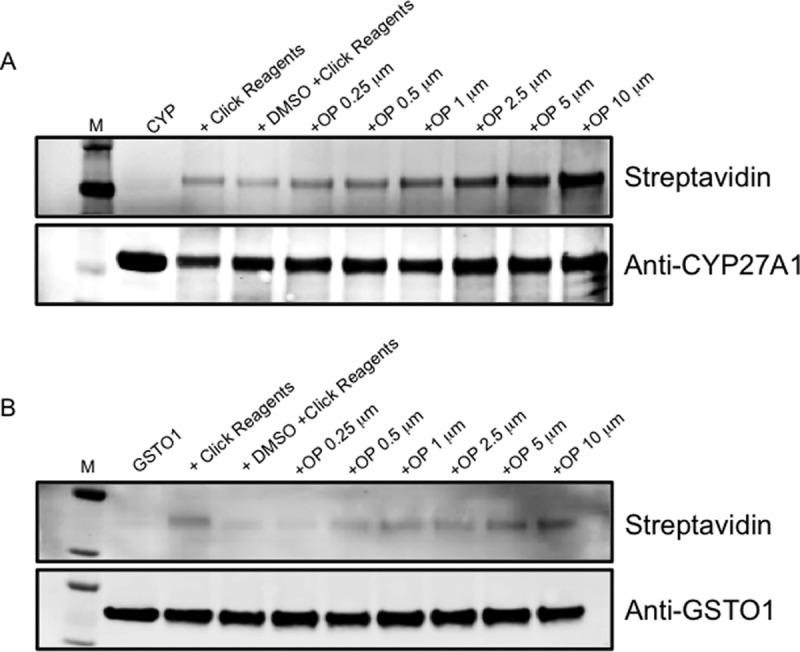
Adduction of CYP27A1 and GTSO1 in vitro. Concentration dependent adduction of (A) CYP27A1 and (B) GSTO1 by OP-829 in vitro as assessed by streptavidin-based immunoblots.
We also considered the possibility that, even though the alkynyl CFZ analog OP-829 adducted these proteins, CFZ may not. We thus treated the pure proteins with carfilzomib and then performed parallel reaction monitoring (PRM) targeted MS analyses for cysteinyl peptides in both unadducted and CFZ-adducted forms. We identified CFZ adducts on three peptides for CYP27A1 (Cys228, Cys427, and Cys476; supplemental Fig. S3A) and two peptides for GSTO1 (Cys32 and Cys192; supplemental Fig. S3B), thus confirming that CFZ itself can adduct these proteins. In addition to the CFZ mass-shifted precursor and peptide fragment ion signals, the MS/MS spectra also contained characteristic fragments of CFZ itself (see supplemental Fig. S1B), which increased confidence in the spectral annotation. Interestingly, Cys476 on CYP27A1 has been reported to be important for binding of the heme group to this enzyme (51) and Cys32 on GSTO1 is the known active site cysteine residue (52). These adduct site localizations suggested that adduction of these proteins may impact enzymatic activity.
Having established sites of CFZ adduction on CYP27A1 and GSTO, we quantified the unadducted and CFZ-adducted cysteinyl peptides following treatment of the purified proteins in vitro with 0, 1, and 10 μm CFZ (supplemental Fig. S4, supplemental Table S3). Although the adducted peptides increased with CFZ treatment concentration, the levels of the unadducted peptides decreased only modestly. This result, in incubations of CFZ with purified protein, suggests that, although CYP27A1 and GSTO1 are significantly adducted in treated cells, the fractional consumption of these enzymes is expected to be low. In contrast, PRM analysis of the OP-829-adducted and unadducted PSMB5 N-terminal Thr-containing peptide (the therapeutic target of CFZ) from OP-829-treated HepG2 lysates indicated near-complete consumption of the unadducted peptide (supplemental Fig. S4).
Having confirmed that CYP27A1 and GSTO1 were both alkylated by CFZ on potentially important residues, we determined whether these modifications inhibit catalytic activities of these proteins. We tested for functional inhibition of each protein by analysis of cholesterol 27-hydroxylation activity (CYP27A1) and of chloro-2,4-dinitrobenzene-glutathione conjugation (GSTO1) in vitro. CFZ produced concentration-dependent inhibition of both proteins (Fig. 4).
Fig. 4.
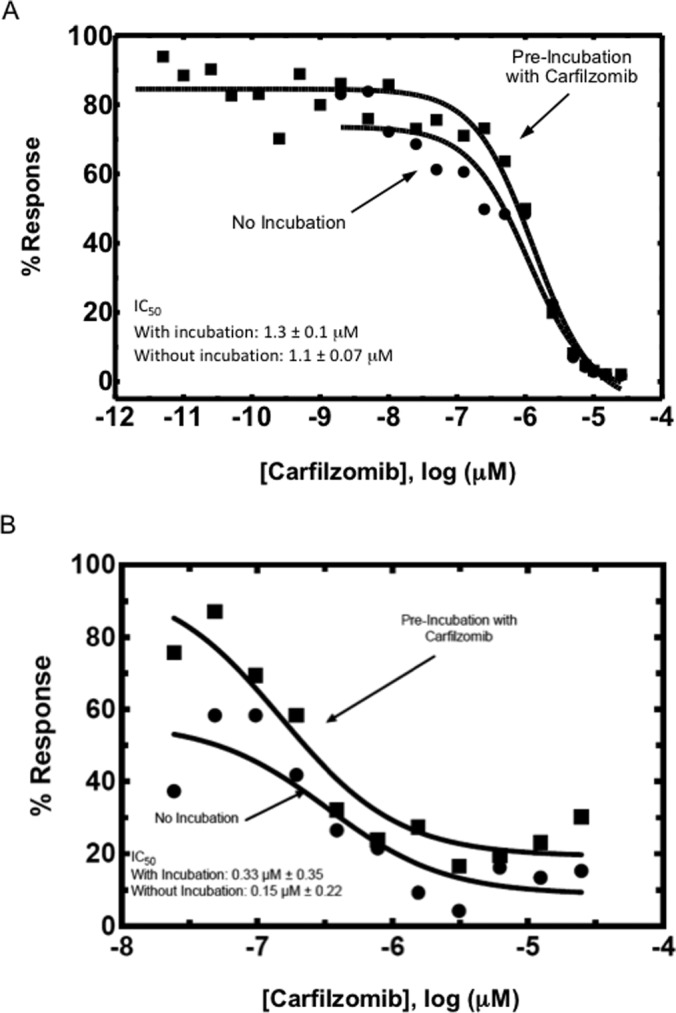
Functional effect of protein adduction by CFZ. (A) CYP27A1 and (B) GSTO1 were adducted by CFZ in vitro and activity assays were performed to assess the consequences of adduction. Both proteins show concentration-dependent decreases in activity in response to adduction.
DISCUSSION
An important problem presented by the emergence of electrophilic covalent protein-modifying drugs is the scope and impact of off-target protein binding. We chose to address this question for the drug CFZ by utilizing the alkynyl analog OP-829, which allowed us to specifically purify and identify adducted proteins through LC-MS/MS shotgun assays. In addition to the known proteasome covalent targets of OP-829, we identified CYP27A1 and GSTO1 as off-target proteins with which the probe formed covalent adducts. We validated the adduction of CYP27A1 and GTSO1 by OP-829 through immunoblot analysis and directly identified peptide sequences adducted in vitro by CFZ itself. We demonstrated a concentration-dependent decrease in enzymatic function of both CYP27A1 and GSTO1 in vitro upon CFZ treatment. Nevertheless, our data suggest that the impact of inhibition of these enzymes by CFZ at therapeutically relevant concentrations would be minimal and that, despite our identification of off-target binding, CFZ displays impressive selectivity for its therapeutic targets.
Our studies with CFZ and OP-829 provide a generalizable strategy to identify potential off-target protein adducts of a drug and to assess the potential for off-target protein modifications to contribute to toxicity. The creation of a clickable analog compound with a similar activity profile offers a straightforward way to test for off-target adduction in relevant model systems. Combining the click purification of protein adducts with high throughput quantitative LC-MS/MS assays makes identifying off-target proteins fast and efficient. We synthesized the alkynyl CFZ analog OP-829, which was suitable for click chemistry-based immunopurification and had a similar activity profile to the parent CFZ molecule. Two other CFZ analogs were not further evaluated but could have produced reinforcing datasets. Subsequent LC-MS/MS assays of adducted proteins identified a small number of potential off-target proteins. We then used orthogonal methods to validate the adduction of two of these proteins (CYP27A1 and GSTO1) by both the alkynyl analog and the parent CFZ. This additional validation of the two most abundantly adducted proteins in the shotgun analysis by the parent chemical structure serves as a proof of concept for the utility of this approach for the investigation of off-target binding by covalent inhibitors.
We note that this approach is distinct from activity-based protein profiling with chemoselective probes (23–25), where the objective is functional protein classification based on probe reactivity. The use of click analog molecular probes to assess target selectivity of candidate proteasome inhibitors has been described (53, 54), but the assessment of global target specificity is a novel, but important extension of this approach. Indeed, recent studies of antitumor drugs that covalently modify the epidermal growth factor receptor have employed clickable analogs to profile off-target reactivities (55).
A potential concern with probe capture strategies is nonspecific binding of proteins to the capture beads, which produces false-positive target identifications. This can be controlled in several ways, including the use of a biologically inactive clickable analog of the probe compound. However, this type of control may not be appropriate for studies of covalent modifiers, where the issues of structure specificity and covalent modification are difficult to separate. Thus, we employed a vehicle control, together with two concentrations of the probe. Proteins that are true targets of the probe should be modified in higher amounts with increasing probe concentration, as we have shown in previous studies of electrophile targets (29). This requirement combined with a tiered data analysis strategy provided a stringent filtering of proteins identified in the capture experiments.
The limited number of off-target CFZ-adducted proteins identified by our shotgun study highlights the specificity of CFZ for its target. Evidence of specificity is indicated by ranking OP-829 protein targets by spectral counts, where a few proteins had high spectral counts but most were represented by few spectral counts, which is consistent with the profile for modification by a nonspecific electrophile (supplemental Fig. S2). Of the proteins with higher spectral counts, proteasome components represented known therapeutic targets for CFZ modification. However, this group also included CYP27A1 and GSTO1, which we validated as targets of OP-829 and CFZ. However, adduction by a covalent inhibitor does not necessarily imply any functional consequences. To evaluate the impact of the CFZ off-target adduction, we treated CYP27A1 and GSTO1 in vitro with increasing amount of CFZ and evaluated the impact of adduction on protein function (Fig. 4). We measured inhibition of enzymatic activities for both proteins by CFZ treatment with IC50 values in the micromolar range. However, our quantitative estimates of adduction of CYP27A1 and GSTO in vitro suggests that these proteins are much less efficiently modified than proteasomal targets and that fractional inhibition of these off-targets would be minimal in intact cells.
Given that CFZ has been well tolerated in the clinic with few reports of dose-limiting toxicity, it is perhaps not surprising that the number of off-target proteins identified in our study was low. The data presented here demonstrate that this electrophilic drug displays high specificity for its therapeutic target and adds further mechanistic support to the clinical safety profile of CFZ.
Supplementary Material
Footnotes
Author contributions: J.D.F., S.G.C., H.L.W., F.P.G., and D.C.L. designed research; J.D.F., S.G.C., S.G., M.E.A., E.L., and J.T. performed research; J.D.F., S.G.C., M.E.A., E.L., J.T., H.L.W., F.P.G., and D.C.L. analyzed data; J.D.F., E.L., J.T., H.L.W., F.P.G., and D.C.L. wrote the paper.
* J. D. F. and M. E. A. were supported by the National Institutes of Health Training Program in Environmental Toxicology (T32 ES007028). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental material.
This article contains supplemental material.
Disclosure of Potential Conflicts of Interest: D.C.L. received research funding from Onyx Pharmaceuticals; E.L., J.T, and H.W. are employed by Onyx Pharmaceuticals.
1 The abbreviations used are:
- CFZ
- carfilzomib
- AmBic
- ammonium bicarbonate
- AP-MS
- affinity purification mass spectrometry
- BSA
- bovine serum albumin
- co-IP
- co-immunoprecipitation
- CYP
- cytochrome P450
- DMEM
- Dulbecco's modified eagle's medium
- DMSO
- dimethylsulfoxide
- DTT
- dithiothreitol
- GST
- glutathione transferase (O1: omega 1)
- IP
- immunoprecipitation
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- LDS
- lithium dodecyl sulfate
- PBS
- phosphate buffered saline
- PRM
- parallel reaction monitoring
- TBST
- tris-buffered saline plus 0.05% Tween-20 (v/v).
REFERENCES
- 1. Noe M. C., and Gilbert A. M. (2012) Targeted covalent enzyme inhibitors. Ann. Reports Med. Chem. 47, 413–439 [Google Scholar]
- 2. Singh J., Petter R. C., Baillie T. A., and Whitty A. (2011) The resurgence of covalent drugs. Nat. Rev. Drug Discov. 10, 307–317 [DOI] [PubMed] [Google Scholar]
- 3. Mah R., Thomas J. R., and Shafer C. M. (2014) Drug discovery considerations in the development of covalent inhibitors. Bioorg. Med. Chem. Lett. 24, 33–39 [DOI] [PubMed] [Google Scholar]
- 4. Gayko U., Fung M., Clow F., Sun S., Faust E., Price S., James D., Doyle M., Bari S., and Zhuang S. H. (2015) Development of the Bruton's tyrosine kinase inhibitor ibrutinib for B cell malignancies. Ann. N.Y. Acad. Sci. [DOI] [PubMed] [Google Scholar]
- 5. Liu Q., Sabnis Y., Zhao Z., Zhang T., Buhrlage S. J., Jones L. H., and Gray N. S. (2013) Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol. 20, 146–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Johnson D. S., Weerapana E., and Cravatt B. F. (2010) Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med. Chem. 2, 949–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Z., Yang J., Kirk C., Fang Y., Alsina M., Badros A., Papadopoulos K., Wong A., Woo T., Bomba D., Li J., and Infante J. R. (2013) Clinical pharmacokinetics, metabolism, and drug-drug interaction of carfilzomib. Drug Metab. Dispos. 41, 230–237 [DOI] [PubMed] [Google Scholar]
- 8. McBride A., Klaus J. O., and Stockerl-Goldstein K. (2015) Carfilzomib: a second-generation proteasome inhibitor for the treatment of multiple myeloma. Am. J. Health Syst. Pharm. 72, 353–360 [DOI] [PubMed] [Google Scholar]
- 9. Kumar S. K., Rajkumar S. V., Dispenzieri A., Lacy M. Q., Hayman S. R., Buadi F. K., Zeldenrust S. R., Dingli D., Russell S. J., Lust J. A., Greipp P. R., Kyle R. A., and Gertz M. A. (2008) Improved survival in multiple myeloma and the impact of novel therapies. Blood 111, 2516–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Demo S. D., Kirk C. J., Aujay M. A., Buchholz T. J., Dajee M., Ho M. N., Jiang J., Laidig G. J., Lewis E. R., Parlati F., Shenk K. D., Smyth M. S., Sun C. M., Vallone M. K., Woo T. M., Molineaux C. J., and Bennett M. K. (2007) Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 67, 6383–6391 [DOI] [PubMed] [Google Scholar]
- 11. Redic K. (2013) Carfilzomib: a novel agent for multiple myeloma. J. Pharm. Pharmacol. 65, 1095–1106 [DOI] [PubMed] [Google Scholar]
- 12. Kuhn D. J., Chen Q., Voorhees P. M., Strader J. S., Shenk K. D., Sun C. M., Demo S. D., Bennett M. K., van Leeuwen F. W., Chanan-Khan A. A., and Orlowski R. Z. (2007) Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 110, 3281–3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. O'Connor O. A., Stewart A. K., Vallone M., Molineaux C. J., Kunkel L. A., Gerecitano J. F., and Orlowski R. Z. (2009) A phase 1 dose escalation study of the safety and pharmacokinetics of the novel proteasome inhibitor carfilzomib (PR-171) in patients with hematologic malignancies. Clin. Cancer Res. 15, 7085–7091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Badros A. Z., Vij R., Martin T., Zonder J. A., Kunkel L., Wang Z., Lee S., Wong A. F., and Niesvizky R. (2013) Carfilzomib in multiple myeloma patients with renal impairment: pharmacokinetics and safety. Leukemia 27, 1707–1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jagannath S., Vij R., Stewart A. K., Trudel S., Jakubowiak A. J., Reiman T., Somlo G., Bahlis N., Lonial S., Kunkel L. A., Wong A., Orlowski R. Z., and Siegel D. S. (2012) An open-label single-arm pilot phase II study (PX-171–003-A0) of low-dose, single-agent carfilzomib in patients with relapsed and refractory multiple myeloma. Clin. Lymphoma Myeloma Leuk. 12, 310–318 [DOI] [PubMed] [Google Scholar]
- 16. Siegel D. S., Martin T., Wang M., Vij R., Jakubowiak A. J., Lonial S., Trudel S., Kukreti V., Bahlis N., Alsina M., Chanan-Khan A., Buadi F., Reu F. J., Somlo G., Zonder J., Song K., Stewart A. K., Stadtmauer E., Kunkel L., Wear S., Wong A. F., Orlowski R. Z., and Jagannath S. (2012) A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood 120, 2817–2825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vij R., Wang M., Kaufman J. L., Lonial S., Jakubowiak A. J., Stewart A. K., Kukreti V., Jagannath S., McDonagh K. T., Alsina M., Bahlis N. J., Reu F. J., Gabrail N. Y., Belch A., Matous J. V., Lee P., Rosen P., Sebag M., Vesole D. H., Kunkel L. A., Wear S. M., Wong A. F., Orlowski R. Z., and Siegel D. S. (2012) An open-label, single-arm, phase 2 (PX-171-004) study of single-agent carfilzomib in bortezomib-naive patients with relapsed and/or refractory multiple myeloma. Blood 119, 5661–5670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hajek R., Bryce R., Ro S., Klencke B., and Ludwig H. (2012) Design and rationale of FOCUS (PX-171-011): a randomized, open-label, phase 3 study of carfilzomib versus best supportive care regimen in patients with relapsed and refractory multiple myeloma (R/R MM). BMC Cancer 12, 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang M., Martin T., Bensinger W., Alsina M., Siegel D. S., Kavalerchik E., Huang M., Orlowski R. Z., and Niesvizky R. (2013) Phase 2 dose-expansion study (PX-171-006) of carfilzomib, lenalidomide, and low-dose dexamethasone in relapsed or progressive multiple myeloma. Blood 122, 3122–3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Niesvizky R., Martin TG 3rd, Bensinger W. I., Alsina M., Siegel D. S., Kunkel L. A., Wong A. F., Lee S., Orlowski R. Z., and Wang M. (2013) Phase Ib dose-escalation study (PX-171–006) of carfilzomib, lenalidomide, and low-dose dexamethasone in relapsed or progressive multiple myeloma. Clin. Cancer Res. 19, 2248–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jakubowiak A. J., Dytfeld D., Griffith K. A., Lebovic D., Vesole D. H., Jagannath S., Al-Zoubi A., Anderson T., Nordgren B., Detweiler-Short K., Stockerl-Goldstein K., Ahmed A., Jobkar T., Durecki D. E., McDonnell K., Mietzel M., Couriel D., Kaminski M., and Vij R. (2012) A phase 1/2 study of carfilzomib in combination with lenalidomide and low-dose dexamethasone as a frontline treatment for multiple myeloma. Blood 120, 1801–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khan M. L., and Stewart A. K. (2011) Carfilzomib: a novel second-generation proteasome inhibitor. Future Oncol. 7, 607–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barglow K. T., and Cravatt B. F. (2007) Activity-based protein profiling for the functional annotation of enzymes. Nat. Methods 4, 822–827 [DOI] [PubMed] [Google Scholar]
- 24. Niphakis M. J., and Cravatt B. F. (2014) Enzyme inhibitor discovery by activity-based protein profiling. Annu. Rev. Biochem. 83, 341–377 [DOI] [PubMed] [Google Scholar]
- 25. Sanman L. E., and Bogyo M. (2014) Activity-based profiling of proteases. Annu. Rev. Biochem. 83, 249–273 [DOI] [PubMed] [Google Scholar]
- 26. Yang J., Gupta V., Carroll K. S., and Liebler D. C. (2014) Site-specific mapping and quantification of protein S-sulphenylation in cells. Nat. Commun. 5, 4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang J., Gupta V., Tallman K. A., Porter N. A., Carroll K. S., and Liebler D. C. (2015) Global, in situ, site-specific analysis of protein S-sulfenylation. Nat. Protoc. 10, 1022–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang J., Tallman K. A., Porter N. A., and Liebler D. C. (2015) Quantitative chemoproteomics for site-specific analysis of protein alkylation by 4-hydroxy-2-nonenal in cells. Anal. Chem. 87, 2535–2541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Codreanu S. G., Ullery J. C., Zhu J., Tallman K. A., Beavers W. N., Porter N. A., Marnett L. J., Zhang B., and Liebler D. C. (2014) Alkylation damage by lipid electrophiles targets functional protein systems. Mol. Cell. Proteomics 13, 849–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim H. Y., Tallman K. A., Liebler D. C., and Porter N. A. (2009) An azido-biotin reagent for use in the isolation of protein adducts of lipid-derived electrophiles by streptavidin catch and photorelease. Mol. Cell. Proteomics 8, 2080–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang C., Weerapana E., Blewett M. M., and Cravatt B. F. (2014) A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat. Methods 11, 79–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Papadopoulos K. P., Burris HA 3rd, Gordon M., Lee P., Sausville E. A., Rosen P. J., Patnaik A., Cutler R. E. Jr, Wang Z., Lee S., Jones S. F., and Infante J. R. (2013) A phase I/II study of carfilzomib 2–10-min infusion in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 72, 861–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Papadopoulos K. P., Siegel D. S., Vesole D. H., Lee P., Rosen S. T., Zojwalla N., Holahan J. R., Lee S., Wang Z., and Badros A. (2015) Phase I study of 30-minute infusion of carfilzomib as single agent or in combination with low-dose dexamethasone in patients with relapsed and/or refractory multiple myeloma. J. Clin. Oncol. 33, 732–739 [DOI] [PubMed] [Google Scholar]
- 34. Salamanca-Pinzon S. G., and Guengerich F. P. (2011) A tricistronic human adrenodoxin reductase-adrenodoxin-cytochrome P450 27A1 vector system for substrate hydroxylation in Escherichia coli. Protein Expr. Purif. 79, 231–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Palin M. F., Berthiaume L., Lehoux J. G., Waterman M. R., and Sygusch J. (1992) Direct expression of mature bovine adrenodoxin in Escherichia coli. Arch. Biochem. Biophys. 295, 126–131 [DOI] [PubMed] [Google Scholar]
- 36. Enright J. M., Toomey M. B., Sato S. Y., Temple S. E., Allen J. R., Fujiwara R., Kramlinger V. M., Nagy L. D., Johnson K. M., Xiao Y., How M. J., Johnson S. L., Roberts N. W., Kefalov V. J., Guengerich F. P., and Corbo J. C. (2015) Cyp27c1 Red-Shifts the Spectral Sensitivity of Photoreceptors by Converting Vitamin A1 into A2. Curr. Biol. 25, 3048–3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sagara Y., Wada A., Takata Y., Waterman M. R., Sekimizu K., and Horiuchi T. (1993) Direct expression of adrenodoxin reductase in Escherichia coli and the functional characterization. Biol. Pharm. Bull. 16, 627–630 [DOI] [PubMed] [Google Scholar]
- 38. Barnes H. J., Arlotto M. P., and Waterman M. R. (1991) Expression and enzymatic activity of recombinant cytochrome P450 17 alpha-hydroxylase in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 88, 5597–5601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stein R. L., Melandri F., and Dick L. (1996) Kinetic characterization of the chymotryptic activity of the 20S proteasome. Biochemistry 35, 3899–3908 [DOI] [PubMed] [Google Scholar]
- 40. Guengerich F. P. (2014) Analysis and characterization of enzymes and nucleic acids relevant to toxicology. Hayes' Principles Meth. Toxicol., 1905–1964 [Google Scholar]
- 41. Habig W. H., and Jakoby W. B. (1981) Assays for differentiation of glutathione S-transferases. Methods Enzymol. 77, 398–405 [DOI] [PubMed] [Google Scholar]
- 42. Kessner D., Chambers M., Burke R., Agus D., and Mallick P. (2008) ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tabb D. L., Fernando C. G., and Chambers M. C. (2007) MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 6, 654–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim S., and Pevzner P. A. (2014) MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 5, 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Elias J. E., and Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 46. Ma Z. Q., Dasari S., Chambers M. C., Litton M. D., Sobecki S. M., Zimmerman L. J., Halvey P. J., Schilling B., Drake P. M., Gibson B. W., and Tabb D. L. (2009) IDPicker 2.0: Improved protein assembly with high discrimination peptide identification filtering. J. Proteome Res. 8, 3872–3881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., and MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sherrod S. D., Myers M. V., Li M., Myers J. S., Carpenter K. L., Maclean B., Maccoss M. J., Liebler D. C., and Ham A. J. (2012) Label-free quantitation of protein modifications by pseudo selected reaction monitoring with internal reference peptides. J. Proteome Res. 11, 3467–3479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li M., Gray W., Zhang H., Chung C. H., Billheimer D., Yarbrough W. G., Liebler D. C., Shyr Y., and Slebos R. J. (2010) Comparative shotgun proteomics using spectral count data and quasi-likelihood modeling. J. Proteome Res. 9, 4295–4305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Speers A. E., Adam G. C., and Cravatt B. F. (2003) Activity-based protein profiling in vivo using a copper(i)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 125, 4686–4687 [DOI] [PubMed] [Google Scholar]
- 51. Prosser D. E., Guo Y., Jia Z., and Jones G. (2006) Structural motif-based homology modeling of CYP27A1 and site-directed mutational analyses affecting vitamin D hydroxylation. Biophys. J. 90, 3389–3409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Whitbread A. K., Masoumi A., Tetlow N., Schmuck E., Coggan M., and Board P. G. (2005) Characterization of the omega class of glutathione transferases. Methods Enzymol. 401, 78–99 [DOI] [PubMed] [Google Scholar]
- 53. Kaschani F., Verhelst S. H., van Swieten P. F., Verdoes M., Wong C. S., Wang Z., Kaiser M., Overkleeft H. S., Bogyo M., and van der Hoorn R. A. (2009) Minitags for small molecules: detecting targets of reactive small molecules in living plant tissues using ‘click chemistry’. Plant J. 57, 373–385 [DOI] [PubMed] [Google Scholar]
- 54. van der Linden W. A., Geurink P. P., Oskam C., van der Marel G. A., Florea B. I., and Overkleeft H. S. (2010) Proteasome selectivity towards Michael acceptor containing oligopeptide-based inhibitors. Org. Biomol. Chem. 8, 1885–1893 [DOI] [PubMed] [Google Scholar]
- 55. Cheng H., Nair S. K., Murray B. W., Almaden C., Bailey S., Baxi S., Behenna D., Cho-Schultz S., Dalvie D., Dinh D. M., Edwards M. P., Feng J. L., Ferre R. A., Gajiwala K. S., Hemkens M. D., Jackson-Fisher A., Jalaie M., Johnson T. O., Kania R. S., Kephart S., Lafontaine J., Lunney B., Liu K. K., Liu Z., Matthews J., Nagata A., Niessen S., Ornelas M. A., Orr S. T., Pairish M., Planken S., Ren S., Richter D., Ryan K., Sach N., Shen H., Smeal T., Solowiej J., Sutton S., Tran K., Tseng E., Vernier W., Walls M., Wang S., Weinrich S. L., Xin S., Xu H., Yin M. J., Zientek M., Zhou R., and Kath J. C. (2016) Discovery of 1-{(3R,4R)-3-[({5-Chloro-2-[(1-methyl-1H-pyrazol-4-yl)amino]-7H-pyrrolo[2,3-d]pyrimidin-4-yl}oxy)methyl]-4-methoxypyrrolidin-1-yl}prop-2-en-1-one (PF-06459988), a Potent, WT Sparing, Irreversible Inhibitor of T790M-Containing EGFR Mutants. J. Med. Chem. 59, 2005–2024 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.