

Abstract
Outgrowth of metastases expressing ERα mutations Y537S and D538G is common after endocrine therapy for estrogen receptor α (ERα) positive breast cancer. The effect of replacing wild type ERα in breast cancer cells with these mutations was unclear. We used the CRISPR-Cas9 genome editing system and homology directed repair to isolate and characterize 14 T47D cell lines in which ERαY537S or ERαD538G replace one or both wild-type ERα genes. In 2-dimensional, and in quantitative anchorage-independent 3-dimensional cell culture, ERαY537S and ERαD538G cells exhibited estrogen-independent growth. A progestin further increased their already substantial proliferation in micromolar 4-hydroxytamoxifen and fulvestrant/ICI 182,780 (ICI). Our recently described ERα biomodulator, BHPI, which hyperactivates the unfolded protein response (UPR), completely blocked proliferation. In ERαY537S and ERαD538G cells, estrogen-ERα target genes were constitutively active and partially antiestrogen resistant. The UPR marker sp-XBP1 was constitutively activated in ERαY537S cells and further induced by progesterone in both cell lines. UPR-regulated genes associated with tamoxifen resistance, including the oncogenic chaperone BiP/GRP78, were upregulated. ICI displayed a greater than 2 fold reduction in its ability to induce ERαY537S and ERαD538G degradation. Progestins, UPR activation and perhaps reduced ICI-stimulated ERα degradation likely contribute to antiestrogen resistance seen in ERαY537S and ERαD538G cells.
Endocrine therapy for estrogen receptor α (ERα) positive breast cancers employs aromatase inhibitors to block estrogen production and tamoxifen and fulvestrant/Faslodex/ICI 182,780 (ICI) that compete with estrogens for binding to ERα. For advanced metastatic breast cancer, selection and outgrowth of tumors resistant to endocrine therapy and expressing ERα mutations ERαY537S and ERαD538G is common1,2,3,4. There is compelling evidence these mutations are resistant to aromatase inhibitors1,2,3,4,5,6,7. While most evidence suggests they are also clinically resistant to tamoxifen and fulvestrant/ICI4,8,9, recent studies demonstrated increased prevalence of ERα mutations in breast cancers of patients treated with aromatase inhibitors, but not in patients treated with fulvestrant5, or tamoxifen6. These researchers question the association of ERα mutations with clinical resistance to fulvestrant and tamoxifen. In studies mostly using transfected ERα negative cells, the mutants were reported to be resistant to tamoxifen and ICI4, resistant to tamoxifen but sensitive to ICI3 and sensitive to antiestrogen inhibition2,10. Previously described systems for analyzing the ERαY537S and ERαD538G mutations were not ideal. Cell lines derived from circulating tumor cells exhibit multiple genetic changes and lack a control cell line. Transfected ERα negative cell lines do not exhibit estrogen-ERα regulated proliferation and display a different ERα-regulated gene expression pattern than ERα positive breast cancer cells11. A better experimental model would compare cells expressing the ERα mutations and wild type ERα in a defined genetic background in an ERα positive breast cancer cell whose proliferation is stimulated by estrogen. We therefore used the CRISPR-Cas9 gene editing system to produce multiple cell lines in which one or both copies of the wild type ERα gene was replaced by ERαY537S or ERαD538G.
Although the most common application of the CRISPR-Cas9 system is targeted gene inactivation by non homologous end joining (NHEJ) to repair the Cas9 generated DNA break, when a homologous repair donor is present, a homology-directed repair process (HDR) can precisely insert a sequence containing the desired modification into the gene of interest. Because the frequency of HDR is usually extremely low12,13,14,15,16, the CRISPR-Cas9 system has rarely been used to successfully repair or insert specific mutations in both copies of endogenous genes in a cancer cell line. We used the CRISPR-Cas9 gene editing system to generate 50 clonal cell lines with one or both copies of endogenous wild-type ERα replaced with ERαY537S or ERαD538G.
Although progesterone reportedly plays a role in breast cancer progression17,18, a recent study concluded that when E2 is present, progesterone enhances tamoxifen’s effectiveness as an antiestrogen19. The effect of progestins in cells expressing ERα mutations had not been explored.
We showed that the estrogen, 17β-estradiol (E2), acts through ERα to elicit extremely rapid and functionally important anticipatory activation of the endoplasmic reticulum stress sensor, the unfolded protein response (UPR)20. Moreover, activation of a UPR gene index at diagnosis is a powerful prognostic indicator, tightly correlated with subsequent resistance to tamoxifen therapy20. This ERα-regulated UPR pathway is targeted by BHPI, our recently described noncompetitive ERα biomodulator. BHPI hyperactivates the UPR, converting it from cytoprotective to cytotoxic21,22. While BHPI is effective in tamoxifen-resistant breast cancer cells expressing wild type ERα, its effectiveness in cells expressing ERα mutations associated with metastases was unknown.
Here we describe the effects of OHT, ICI and BHPI on proliferation of the ERαY537S and ERαD538G cells in anchorage dependent and anchorage independent culture with and without a progestin, analyze gene expression, and evaluate estrogen-independent and progestin-stimulated UPR activation and reduced ERα degradation as potential contributors to antiestrogen resistance.
Results
Using CRISPR-Cas9 to replace wild-type ERα with ERαY537S or ERαD538G
Our strategy is illustrated in Supplementary Fig. S1. To increase the frequency of HDR, we used 2 guide sequences. Additionally, to abolish repeated cutting of homology-replaced sequences by Cas9, we changed one nucleotide in both PAM sequences of the HDR template without altering the ERα amino acid sequence. To facilitate identification of cells in which the mutant ERα fragment replaced the wild-type ERα fragment, we inserted AclI and SpeI sites into the HDR template.
We chose T47D cells because they contain 2 copies of the ERα gene and exhibit E2 dependent growth23. Previous transfection studies suggested ERαY537S and ERαD538G cells grow without E21,2,3. We exploited this phenotype during outgrowth and selection of colonies of ERαY537S and ERαD538G cells.
Individual colonies were pulled, grown out and genotyped. Genomic DNA spanning the HDR template was amplified and analyzed by digestion with AclI or SpeI. Replacement of wild-type ERα results in two bands due to AclI or SpeI cutting (Supplementary Fig. S1). 50 of 65 clonal cell lines contained single or double gene replacements (Supplementary Fig. S2). Expression of the ERα mutations in mRNA was confirmed by synthesizing and digesting cDNA. The presence of the mutated ERα sequences in the cell lines was verified by DNA sequencing (Supplementary Fig. S1). We used ERαY537S-4 because its properties were typical of the ERαY537S cell lines and because mapping and sequencing confirmed 2 distinct and independent gene replacement events (Supplementary Fig. S1 legend); ERαD538G-1 was chosen as typical of the ERαD538G cell lines. Sequencing and Western blotting with N-terminal ERα antibody showed that NHEJ introduced diverse indels in the non-replaced copy of ERα (Supplementary Figs 1 and 2). We therefore focused primarily on the double replacement cell lines.
Western blotting to compare ERα levels in parental T47D cells to levels of ERαY537S and ERαD538G in single and double replacement cell lines showed some heterogeneity in expression level of ERαY537S and ERαD538G. The ERα mutations were expressed at levels similar to or lower than wild-type ERα (Supplementary Fig. S2).
ERαY537S and ERαD538G cells exhibit E2-independent growth and partial resistance to OHT and ICI
Although the clonal cell lines were isolated and maintained without E2, it was unknown whether E2 further stimulates their growth. As expected, the parental T47D cells exhibited little growth without E2 and dose-dependent increases in growth with E2 (Fig. 1a)21. The ERαY537S-4 cells and the other ERαY537S cell lines were completely E2-independent for growth. E2 further stimulated the more modest E2-independent proliferation of ERαD538G-1 and other ERαD538G cells (Fig. 1a and Supplementary Fig. 3).
Figure 1. In standard anchorage-dependent 2D culture, ERαY537S-4 and ERαD538G-1 cells exhibit E2-independent proliferation and partial resistance to OHT and ICI.

(a) Dose-response study comparing the effects of increasing concentrations of E2 on the proliferation of T47D, ERαY537S-4 and ERαD538G-1 cells. (b,c) Dose-response studies comparing the effects of increasing concentrations of OHT (b) and ICI (c) on the proliferation of T47D, ERαY537S-4 and ERαD538G-1 cells. Data is mean ± SEM (n = 8).
We next performed dose response studies evaluating the effect of OHT and ICI on proliferation of the T47D, ERαY537S and ERαD538G cell lines. To facilitate comparisons, T47D and mutant cells were maintained in medium containing E2. In T47D cells, OHT and ICI nearly abolished proliferation at a 50–1,000 fold molar excess over E2 (Fig. 1b,c). In contrast, the mutant cell lines, especially ERαY537S-4, exhibited some resistance (Fig. 1b,c; blue bars). Additionally, while T47D cells displayed negligible growth in ICI, all seven ERαY537S and ERαD538G double replacement cell lines displayed at least some growth in ICI (Supplementary Fig. S3). In longer 8-day cultures, compared to T47D cells, the mutant cells displayed increased proliferation in OHT and ICI (Supplementary Fig. S4).
BHPI blocks growth of ERαY537S and ERαD538G cells
Since the ERαY537S and ERαD538G cell lines displayed partial resistance to OHT and ICI, we evaluated the ability of our recently described noncompetitive ERα biomodulator, BHPI, to inhibit proliferation of ERαY537S and ERαD538G cells21. At 25 nM, BHPI completely inhibited proliferation of T47D, ERαY537S-4 and ERαD538G-1 cells and the 14 characterized ERα mutant cell lines (Fig. 2a and Supplementary Fig. S5). In longer 8-day cultures, BHPI continued to completely block proliferation of T47D, ERαY537S-4 and ERαD538G-1 cells (Supplementary Fig. S4).
Figure 2. ERαY537S-4 and ERαD538G-1 cells show antiestrogen resistant growth in 2D and 3D culture, but are killed by BHPI.

(a) Dose-response study of the effect of BHPI on the proliferation of T47D, ERαY537S-4 and ERαD538G-1 cells in standard anchorage-dependent 2D culture. (b) BHPI inhibits protein synthesis >90% in T47D, ERαY537S-4 and ERαD538G-1 cells. Cells were treated with vehicle, 1 nM E2 or 1 nM P4 with or without 100 nM BHPI for 3 hr. Data is mean ± SEM (n = 5). (c) In 2D culture, R5020 reduces the ability of OHT and ICI to inhibit proliferation of ERαY537S-4 and ERαD538G-1 cells. (d) In 3D culture, ERαY537S-4 and ERαD538G-1 cells are highly antiestrogen resistant and resistance is robustly increased by R5020, but the cells are killed by BHPI. Conditions (a,c,d) as noted, with 100 pM E2 unless otherwise stated. Data (a,c,d) is mean ± SEM (n = 8). For (b) letters indicate a significant difference among groups (p < 0.05) using one-way ANOVA followed by Tukey’s post hoc test within each cell line. -E2 in each cell line set to 100%. For (c,d) *p < 0.05, **p < 0.01, ***p < 0.001 (by Student’s T test) comparing treatment to treatment + R5020.
BHPI potently inhibits protein synthesis in ERαY537S and ERαD538G cells
BHPI acts through ERα to induce toxic hyperactivation of all three arms of the unfolded protein response (UPR)21,22,24. By hyperactivating the protein synthesis inhibiting PERK arm of the UPR, BHPI induces near quantitative inhibition of protein synthesis21. Potent long-term inhibition of protein synthesis is central to BHPI’s ability to block growth and kill ERα positive breast cancer cells21. While the hormones E2 and progesterone (P4) had minimal effects on protein synthesis, BHPI (100 nM) rapidly inhibited protein synthesis by >90% in T47D, ERαY537S-4 and ERαD538G-1 cells (Fig. 2b). Thus, the ERαY537S and ERαD538G mutations have no effect on BHPI’s ability to block cell proliferation and strongly inhibit protein synthesis by hyperactivating the UPR.
ERαY537S and ERαD538G cells exhibit progesterone-stimulated antiestrogen-resistant, BHPI-sensitive, growth in anchorage-independent culture
Although anchorage-independent growth in 3-dimensional (3D) culture is a hallmark of cancer, it has been difficult to study quantitatively. Modifying earlier methods25, we developed a protocol for quantitative assessment of cell proliferation in a more biologically relevant anchorage-independent 3D culture in soft agar.
We compared the effects of OHT (1 μM; 10,000 fold excess over E2), ICI (1 and 5 μM), BHPI (100 nM) and the synthetic progestin R5020 (10 nM), on proliferation of ERαY537S-4 and ERαD538G-1 cells in anchorage-dependent 2D and anchorage-independent 3D culture. 5 μM OHT was not used because it exhibits nonspecific toxicity26,27,28. In standard 2D culture, the ERαY537S-4 cells exhibited reduced, but significant proliferation in OHT and ICI, which was increased by R5020. The ERαD538G-1 cells exhibited little proliferation in OHT and ICI, which increased slightly in R5020 (Fig. 2c, compare open bars: no R5020, hatched bars: +R5020).
In quantitative 3D culture, ICI was more effective than OHT in blocking growth of T47D cells. Impressively, proliferation of the mutants in 1 μM OHT and ICI was >50% of control, a dramatic increase compared to 2D culture (Fig. 2d). At 1 μM, ICI was no more effective than OHT in inhibiting proliferation of the mutants. Notably, in 3D culture, ERαY537S-4 and ERαD538G-1 cells grown in R5020 were completely resistant to growth inhibition by 1 μM OHT and ICI (Fig. 2d).
In both 2D and 3D culture, BHPI stopped growth and killed the ERαY537S-4 and ERαD538G-1 cells grown with or without R5020 (Fig. 2a,c,d). Neither growth in a progestin, nor growth in 3D culture, impaired BHPI’s impressive ability to block growth and kill ERαY537S-4 and ERαD538G-1 cells.
E2-ERα regulated genes are constitutively expressed and antiestrogen resistant in ERαY537S and ERαD538G cells
To explore factors that might contribute to antiestrogen resistance in the ERαY537S and ERαD538G mutants, we evaluated the effects of E2, OHT and ICI on ERα-regulated transcription. Our endogenous test genes were progesterone receptor (PgR), the oncogenic co-regulator, GREB1, and the down-regulated gene, IL1-R1. In parental T47D cells, E2 induction of PgR and GREB1 mRNAs and down regulation of IL1-R1 mRNA was abolished by OHT and ICI (Fig. 3a–c). Consistent with cell proliferation data (Fig. 1a), induction and repression of gene expression by ERαY537S was estrogen independent, but was only partially constitutive for ERαD538G (Fig. 3a–c). Induction of PgR and GREB1 mRNAs was partially OHT and ICI resistant in both ERαY537S-4 and ERαD538G-1 cells (Fig. 3a,b). In contrast, down-regulation of IL1-R1 was highly resistant to OHT and ICI in ERαY537S-4 cells (Fig. 3c). Perhaps because IL1-R1 down-regulation was mostly estrogen dependent in ERαD538G cells, it was highly sensitive to inhibition by OHT and ICI (Fig. 3c).
Figure 3. ERαY537S-4 and ERαD538G-1 cells exhibit constitutive, E2 independent, gene expression with significant resistance to inhibition by OHT and ICI.
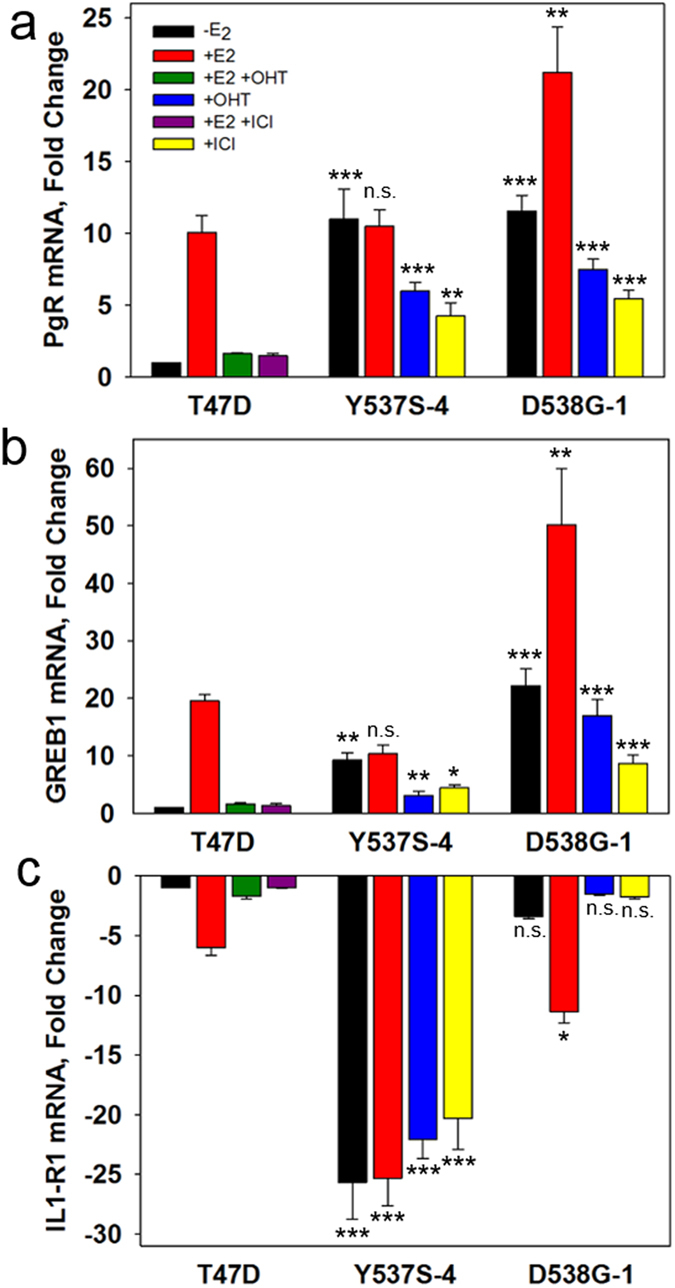
(a) Levels of PgR mRNA or (b) GREB1 mRNA after 4 hr treatment and (c) IL1-R1 mRNA after 24 hr treatment in T47D, ERαY537S-4 and ERαD538G-1 cells. *p < 0.05, **p < 0.01, ***p < 0.001, for each treatment, comparing mutant cell lines to wild type T47D using one-way ANOVA followed by Dunnett’s post hoc test. E2 1 nM; OHT 1 μM; ICI 1 μM; Data is mean ± SEM (n = 3); qRT-PCR; -E2 in parental T47D cells set to 1 in (a,b) and −1 in (c).
In the ERαY537S-5 and ERαD538G-4 cell lines, in which one copy of the ERα gene was replaced with a mutant ERα and the other contained an indel resulting in reading frame shift and truncated ERα, induction of PgR and GREB1 and down-regulation of IL1-R1 was estrogen independent and was much reduced compared to the double replacement cell lines. The modest induction and repression of gene expression in ERαY537S-5 and ERαD538G-4 cells was highly resistant to inhibition by OHT and ICI (Supplementary Fig. 6).
ICI 182,780 exhibits a reduced ability to induce degradation of ERαY537S and ERαD538G
ICI induces ERα degradation. Since the ERαY537S-4 and ERαD538G-1 cells were ICI resistant in 3D culture, we examined the effect of ICI and other ligands on levels of ERαY537S and ERαD538G. In the presence of E2, wild-type ERα exhibited modest down-regulation, which was less evident in the mutants. OHT is a weak agonist and stabilizes ERα. OHT increased levels of ERαY537S but had little effect on the level of ERαD538G. There was a highly significant >2 fold reduction in the ability of ICI to induce degradation of both the double and single replacement ERαY537S and ERαD538G mutants (Fig. 4a,b and Supplementary Fig. S7).
Figure 4. ERαY537S and ERαD538G exhibit altered ligand-dependent degradation.

(a) Western blot comparing the effects of E2, OHT and ICI on levels of ERα in T47D, ERαY537S-4 and ERαD538G-1 cells. Cells were treated with vehicle (C) or 10 nM E2 (18 hr), or with 1 μM OHT or 1 μM ICI (24 hr). (b) Quantitation of densitometry of ERα levels in ligand-treated T47D, ERαY537S and ERαD538G double replacement cell lines, within each cell line treatments were compared to vehicle control. Data is mean ± SEM (ERαY537S: n = 4 cell lines; ERαD538G: n = 3 cell lines; T47D n = 3 experiments); (b) p < 0.01 (by Student’s T test), for each treatment, comparing mutant to wild-type ERα in T47D cells.
Markers of UPR activation are elevated in ERαY537S and ERαD538G cells
E2 elicits a weak anticipatory activation of the UPR that is a strong prognostic marker correlated with tamoxifen resistance20,24,29. The effect of P4 on the UPR had not been explored. We therefore investigated the effect of estrogen and progesterone on UPR markers in the tamoxifen-resistant ERαY537S-4 and ERαD538G-1 cells. Activation of IRE1α, resulting in cleavage of XBP1 mRNA to spliced XBP1 (sp-XBP1), is a widely used marker of UPR activation. Activation of the UPR’s ATF6α and IRE1α arms induces the strongly oncogenic chaperone BiP/GRP78/HSPA5 by 1.5-3 fold30,31. Activation of these UPR arms also induces the anti-apoptotic protein p58IPK, the angiogenesis-related protein ERO1a, and the oncogenic UPR-related protein SERP132,33,34. In T47D cells, E2 modestly induced sp-XBP1 mRNA (Fig. 5a)20. In the ERαY537S-4 cells sp-XBP1 levels were constitutively elevated and were not further increased by E2. In the ERαD538G-1 cells sp-XBP1 levels were minimally higher than basal levels and were not increased by E2 (Fig. 5a). UPR-induced BiP protein and ERO1a and SERP1 mRNAs were all constitutively elevated in the ERαY537S-4 cells and BiP and SERP1 were elevated in the ERαD538G-1 cells (Fig. 5c,d and Supplementary Fig. S8). Pooling data from all 14 mutant cell lines, BiP was significantly elevated in ERαY537S cells and p58IPK was elevated in both ERαY537S and ERαD538G cells (Fig. 5c).
Figure 5. The UPR is activated in ERαY537S-4 and ERαD538G-1 cells and is further activated by progesterone.

(a) sp-XBP1 mRNA is upregulated in ERαY537S-4 cells and P4 causes further upregulation in both cell lines. Letters indicate a significant difference among groups (p < 0.05) using one-way ANOVA followed by Tukey’s post hoc test. Cells were treated with vehicle, 1 nM E2 and/or 1 nM P4 for 4 hr. Data is mean ± SEM (n = 3); qRT-PCR; -E2 in T47D cells set to 1. (b) Progesterone receptor is highly upregulated in ERαY537S-4 and ERαD538G-1 cells. (c) Western blots (left) and quantitation (right) showing BiP and p58IPK induction in all ERαY537S and ERαD538G cell lines. (d) Western blot showing induction of BiP and p58IPK in ERαY537S-4 and ERαD538G-1 cells. (e) BiP is strongly induced and p58IPK is induced by P4 in T47D cells, p58IPK but not BiP is induced by P4 in ERαY537S-4 cells and neither is significantly induced by P4 in ERαD538G-1 cells. For (b–d) T47D cells were treated with vehicle or 10 nM E2 for 24 hr, mutant cell lines were treated with vehicle. In (e) T47D cells were treated with vehicle for 48 hr or with E2 for 24 hr to induce PR then P4 for 24 hr; ERαY537S-4 and ERαD538G-1 cells were treated with vehicle or P4 for 24 hr. Data is mean ± SEM. In (c) ERαY537S: n = 8 cell lines; ERαD538G: n = 6 cell lines; T47D n = 3 experiments. In (d,e) quantitation shown below is from the average of three identical blots. For (c,d,e) *p < 0.05, **p < 0.01, ***p < 0.001 (by Student’s T test), for each treatment, comparing each mutant to wild-type vehicle control (c,d) or comparing vehicle treated to P4 within each cell line (e).
In T47D cells, E2-induced sp-XBP1 levels were further increased by P4. In the ERαY537S-4 and ERαD538G-1 cells, P4 elicited a strong E2-independent induction of sp-XBP1 mRNA (Fig. 5a). Consistent with the larger effect of progesterone on sp-XBP1 levels in the ERαY537S-4 and ERαD538G-1 cells (Fig. 5a), and with the constitutive induction of PgR mRNA (Fig. 3a), progesterone receptor (PR) was constitutively elevated to levels higher than in E2-treated T47D cells (Fig. 5b). P4 significantly induced levels of BiP and p58IPK in T47D cells and further increased p58IPK in ERαY537S-4 cells, but neither BiP nor p58IPK were significantly further induced in ERαD538G-1 cells (Fig. 5e). Thus, progesterone further increases expression of some, but not all, UPR markers.
Discussion
While the CRISPR-Cas9 genome editing system has been widely used to inactivate genes35,36,37,38,39,40, gene replacement studies often use a few cell lines exhibiting unusually high HDR frequency14,15,16,41. While our use of 2 guide sequences to increase the frequency of HDR may increase the possibility of off-target events42,43,44, our ability to confirm key observations in 4 ERαY537S and 3 ERαD538G clonal cell lines strongly suggests that the observed properties are due to ERα gene replacement. Because indirect evidence suggests long templates might increase HDR45, we used double-stranded templates with 1.2 kb arms, not the short ~50 bp single stranded arms usually used in HDR46.
Consistent with the frequency of error-prone NHEJ being much higher than the frequency of HDR12,13,14,15,16, all sequenced single replacement clones contained indels in the non-HDR ERα copy. Since ERα in these clones may exist in heterodimers, in which the indel-containing ERα monomer is non-functional, we focused on the double replacement cell lines. Notably, ~30% of cell lines replaced both copies of ERα. This suggests each wild-type ERα copy may undergo multiple NHEJ events before an indel destroys the guide-matched DNA region or a rare HDR event inactivates the PAM sequence.
This work exploited our ability to select for multiple clones expressing ERα mutants that grew out in the absence of E2. Based on calculations from our large sample size, it seems feasible, but very challenging, to replace genes in typical breast cancer cell lines without a selection. Recent studies, largely in HEK293 cells, describe methods for increasing the probability of HDR using inhibitors of NHEJ or silencing key molecules involved in NHEJ and optimized HDR templates that target the asymmetric mechanism of Cas914,15,16.
Our data indicates that the ERαY537S and ERαD538G mutations are sufficient to convert an ERα positive cancer cell, exhibiting E2-dependent proliferation and growth inhibition by antiestrogens, to one exhibiting E2-independent proliferation and resistance to high concentrations of OHT and ICI. Importantly, since these experiments were the initial exposure of the ERαY537S and ERαD538G cells to antiestrogens, there was no prior selection for other changes favoring antiestrogen resistance. Consistent with earlier work, E2-independent proliferation will make tumors expressing these mutations resistant to aromatase inhibitors.
In more biologically relevant anchorage-independent 3D culture, the ERαY537S and ERαD538G cell lines exhibited robust proliferation in the presence of 1 μM z-OHT and 1 μM fulvestrant/faslodex/ICI 182,780 (Fig. 2d). Notably, the mutant cell lines continued to proliferate in 5 μM ICI. Since serum ICI in patients is 20–30 nM47,48, it is unlikely tumors can concentrate ICI to levels that exceed the 1 and 5 μM in which the mutant cell lines proliferate in 3D culture.
It is widely accepted that ICI is a more nearly pure antagonist than OHT. Since OHT stabilizes and ICI induces degradation of ERα, the ~9 fold lower level of ERα in ICI-treated compared to OHT-treated cells likely makes an important contribution to the increased effectiveness of ICI in T47D cells. However, in the ERαY537S and ERαD538G cell lines, ICI was no more effective in blocking cell growth than OHT, and ICI exhibited a greater than 2 fold reduced ability to induce degradation of the mutant ERαs. Instead of the ~9 fold higher level of ERα in OHT-treated versus ICI-treated T47D cells, the difference in mutant ERα levels was reduced to 2–3 fold. The diminished ability of ICI to induce degradation of the ERα mutants likely contributes to ICI resistance of ERαY537S and ERαD538G cells. Notably, the ERαY537N mutation is also resistant to ICI-induced degradation49. However, in a very recent report, ERαD538G in one patient’s circulating tumor cells was not resistant to degradation50. This suggests that ICI-induced ERα degradation may also depend on subtle genetic differences in tumors. Of note, using double replacement cell lines may facilitate degradation studies; using current antibodies it is not possible to distinguish between wild type ERα and the mutant ERαs containing these single amino acid changes.
OHT is an antagonist because it induces a different conformational change in ERα than E2. Very recently, using structural, computational and biophysical approaches Greene and coworkers suggested that the ERαY537S and ERαD538G mutations confer partial antiestrogen resistance because they exhibit reduced affinity for OHT and because they induce a constitutively active conformation that resists deformation by OHT. Moreover, they suggested that ICI, which disorders helix 12 of ERα, might be a therapeutically effective antagonist, blocking activity of ERαY537S and ERαD538G51. They also report that ERαY537S is in a conformation that exhibits higher constitutive binding of coactivators than ERαD538G51. Our experimental data on differences in proliferation and gene expression between the ERαY537S and ERαD538G cell lines is consistent with their proposed differences in coactivator interaction. Supporting the view that ERαY537S and ERαD538G are in a constitutively active conformation that resists antiestrogens, both gene expression in standard cell culture and proliferation in 3D culture are constitutive and largely resistant to saturating OHT and ICI. Elevated coactivator expression and a shift in coactivators, often including down-regulation of GREB1, have been linked to antiestrogen resistance52,53,54,55,56,57. In contrast, GREB1 is constitutively upregulated in the mutant cell lines, suggesting antiestrogen resistance is not caused by a shift away from GREB1.
In breast cancer cells expressing wild type ERα and PR, progesterone increases stemness and markers associated with therapy resistance17. Moreover, progesterone can be mitogenic and mediate cell survival in 3D cultures17. However, recent xenograft and explant studies show that when estrogen is present, progestins enhance sensitivity of breast cancers expressing wild type ERα to tamoxifen19. Consistent with an earlier study58, PR was constitutively expressed to extremely high levels in the ERαY537S and ERαD538G cells, suggesting it might play an important role. We report that a progestin decreases sensitivity of ERαY537S and ERαD538G cells to growth inhibition by OHT and ICI in both 2D and 3D culture. In 3D cultures treated with a progestin, 1 μM OHT and ICI have lost the ability to inhibit proliferation of ERαY537S and ERαD538G cells. Since progestin had little ability to reverse OHT and ICI inhibition of T47D cell growth, the progestin effect is likely related to the ERαY537S and ERαD538G mutations. Consistent with the possibility that serum progestin might influence antiestrogen resistance in women whose breast cancers express the ERαY537S and ERαD538G mutations, progesterone levels in post-menopausal women are 0.6–3 nM59,60,61. Moreover, breast cancer cells expressing both ERα and PR, or overexpressing PR alone, may be sensitive to very low concentrations of hormone17,18. Since the ERαY537S and ERαD538G cells constitutively express very high levels of PR, the low serum concentrations of progestins in postmenopausal women together with high PR could well have functional effects on antiestrogen resistance in metastatic breast cancers expressing ERα mutations.
Weak activation of the UPR is protective in cancer and is associated with a poor prognosis, while extensive and sustained UPR activation by BHPI is toxic21,22,24,29. Since we cannot currently analyze protein and mRNA levels in cells from 3D cultures in soft agar, the UPR studies were carried out in standard 2D cell culture. The ERαY537S cells exhibited constitutive activation of the IRE1α arm of the UPR, as indicated by estrogen-independent expression of sp-XBP1 and induction of the oncogenic chaperone BiP. In contrast, the ERαD538G cells exhibited low expression of sp-XBP1 and a much smaller induction of BiP. This is consistent with the ERαD538G mutant showing a lower level of constitutive growth and antiestrogen resistance in cell proliferation and in E2-ERα regulated gene expression. Since our 2D cell culture data shows that ERαY537S cells exhibit significant resistance to OHT and ICI and the ERαD538G cells exhibited very little resistance (Fig. 2c), this data is consistent with a link between constitutive UPR activation and antiestrogen resistance. Moreover, progesterone increased sp-XBP1 expression to a higher level in the ERαY537S cells (Fig. 5a) than in the ERαD538G cells. Compared to the ERαD538G cells, the ERαY537S cells exhibited increased progesterone-stimulated resistance to OHT and ICI (Fig. 2c). Some oncogenic UPR markers, notably SERP1 were elevated in the mutant cell lines, but others, such as p58IPK and ERO1a behaved differently in different cell lines. Although progesterone strongly induced sp-XBP1, it did not significantly further increase the strongly elevated level of BiP in the mutant cell lines and only further induced p58IPK in the ERαY537S cells. Thus, while constitutive UPR activation likely contributes to the antiestrogen resistant phenotype seen in the ERαY537S and ERαD538G cell lines, the extent to which it does is unclear, and effects are likely complex.
In 2D culture R5020 increased proliferation of the ERαY537S and ERαD538G cells in OHT and ICI; in 3D culture, R5020 abolished growth inhibition by OHT and ICI. This effect of progestin-PR may not only be mediated through further activation of the UPR but also through modulation of ERα action and PR’s own transcriptomic program19. Although further study is needed, progestins seem likely to enhance antiestrogen effectiveness in cancer cells expressing the ERαY537S and ERαD538G mutations.
Although the mutations exhibit an altered ERα conformation5, and are close to the proposed BHPI binding site21, BHPI was as effective in blocking proliferation of all 14 of the ERαY537S and ERαD538G cell lines as it was in T47D cells. Moreover, in 3D culture where the ERαY537S-4 and ERαD538G-1 cells exhibited robust proliferation in 1 μM OHT and ICI, 100 nM BHPI completely blocked proliferation and killed the cells. In cancer cells expressing wild-type ERα, BHPI strongly activates the PERK arm of the UPR, resulting in rapid, near-quantitative inhibition of protein synthesis21. In the parental T47D cells and in the ERαY537S-4 and ERαD538G-1 cells, 100 nM BHPI elicited >90% inhibition of protein synthesis. This supports BHPI acting in the ERα mutant cell lines by the same pathway we described in cancer cells expressing wild-type ERα21.
We used the CRISPR-Cas9 genome editing system to generate precise replacements of both copies of a steroid receptor gene. Breast cancer cell lines expressing these ERα mutations associated with metastatic breast cancer are highly resistant to endocrine therapy. Taken together, our data and the recent structural and biophysical studies suggest that a constitutively active ERα conformation, altered ICI-induced degradation of ERα, progestin and UPR activation may all contribute to the antiestrogen resistant phenotype. Notably, the preclinical drug candidate BHPI retains its effectiveness against these resistant breast cancer cells.
Methods
Cell culture, reagents and western blotting
Cell culture medium and conditions were as previously described62. T47D cells were from ATCC (VA). z-OHT was from Sigma-Aldrich (MO) and ICI 182,780 was from Tocris Biosciences (Bristol, UK). Additional details are in Supplementary Methods.
Generation of ERαY537S and ERαD538G cell lines
Our protocol was loosely based on previous work46. Two guide sequences (Supplementary Fig. S1) were cloned into pSpCas9(BB)-2A-Puro(PX459) (Addgene, MA). An HDR template with mutations was cloned into pUC18, and linearized before transfection. Two plasmids with guide sequences and the HDR template carrying either the ERαY537S or ERαD538G mutation were co-transfected into T47D cells. After 3-day selection in 2.5 μg/ml puromycin, followed by 3 weeks of E2-free growth, colonies were transferred to a 24-well plate. Clones were genotyped at the DNA and mRNA levels using PCR and restriction enzyme digestion. Sequencing confirmed selected clones. Oligonucleotide sequences and additional details are in Supplementary Methods.
MTS cell proliferation assay
Cell proliferation assays in 2D culture were performed as described21. Briefly, 2,000 cells/well were incubated with treatment for 4 days with one medium change after 2 days. After 4 days, 20 μl of CellTiter 96 Aqueous One Solution (Promega, WI) was added to each well and absorbance measured.
Quantitative proliferation assays in 3D culture
Quantitative assays of colonies in soft agar were as in ref. 25 with minor modifications. Briefly, 2,000 cells in 60 μl 0.4% agar were plated in 96-well plates above 50 μl 0.6% agar. Cells were grown for 5 days with one medium change, treated with AlamarBlue (Fisher, NH) and fluorescence measured.
Quantitative reverse transcriptase-PCR (qRT-PCR)
qRT-PCR was as described63.
Protein synthesis
Protein synthesis was quantitated by measuring incorporation of 35S-Methionine into newly synthesized protein21.
Additional Information
How to cite this article: Mao, C. et al. Antiestrogen Resistant Cell Lines Expressing Estrogen Receptor α Mutations Upregulate the Unfolded Protein Response and are Killed by BHPI. Sci. Rep. 6, 34753; doi: 10.1038/srep34753 (2016).
Supplementary Material
Acknowledgments
We thank Drs Han Xiao and Arindam Chakraborty for advice on CRISPR. This work was supported by grants NIH RO1 DK 071909 and DOD BCRP W81XWH-13 (to DS) and by the NSF Graduate Research Fellowship Program under grant DGE-1144245 (to ML).
Footnotes
A patent application on BHPI has been filed.
Author Contributions C.M., M.L., J.E.K. and D.J.S. conceived and designed experiments. C.M., M.L. and J.E.K. performed experiments. C.M., M.L., J.E.K. and D.J.S. wrote the manuscript.
References
- Merenbakh-Lamin K. et al. D538G Mutation in Estrogen Receptor-α: A Novel Mechanism for Acquired Endocrine Resistance in Breast Cancer. Cancer Res. 73, 6856–6864 (2013). [DOI] [PubMed] [Google Scholar]
- Robinson D. R. et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 45, 1446–1451 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toy W. et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 45, 1439–1445 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q. X., Borg A., Wolf D. M., Oesterreich S. & Fuqua S. A. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res. 57, 1244–1249 (1997). [PubMed] [Google Scholar]
- Spoerke J. M. et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat. Commun. 7, 11579 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fribbens C. et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor-Positive Advanced Breast Cancer. J. Clin. Oncol., doi: 10.1200/JCO.2016.67.3061 (2016). [DOI] [PubMed] [Google Scholar]
- Schiavon G. et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci. Transl. Med. 7, 313ra182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M. et al. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 345, 216–220 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti C. et al. Heterogeneous estrogen receptor expression in circulating tumor cells suggests diverse mechanisms of fulvestrant resistance. Mol. Oncol. 10, 1078–1085 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis K. E., Ekena K., Thomas J. A., Lazennec G. & Katzenellenbogen B. S. Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol. Endocrinol. 10, 1388–1398 (1996). [DOI] [PubMed] [Google Scholar]
- Stender J. D. et al. Estrogen-regulated gene networks in human breast cancer cells: involvement of E2F1 in the regulation of cell proliferation. Mol. Endocrinol. 21, 2112–2123 (2007). [DOI] [PubMed] [Google Scholar]
- Doudna J. A. & Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096 (2014). [DOI] [PubMed] [Google Scholar]
- Smith C. et al. Efficient and allele-specific genome editing of disease loci in human iPSCs. Mol. Ther. 23, 570–577 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T. et al. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 33, 538–542 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu V. T. et al. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 33, 543–548 (2015). [DOI] [PubMed] [Google Scholar]
- Richardson C. D., Ray G. J., DeWitt M. A., Curie G. L. & Corn J. E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 34, 339–344 (2016). [DOI] [PubMed] [Google Scholar]
- Diep C. H., Daniel A. R., Mauro L. J., Knutson T. P. & Lange C. A. Progesterone action in breast, uterine, and ovarian cancers. J. Mol. Endocrinol. 54, R31–R53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obr A. E. & Edwards D. P. The biology of progesterone receptor in the normal mammary gland and in breast cancer. Mol. Cell. Endocrinol. 357, 4–17 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed H. et al. Progesterone receptor modulates ERα action in breast cancer. Nature 523, 313–317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andruska N., Zheng X., Yang X., Helferich W. G. & Shapiro D. J. Anticipatory estrogen activation of the unfolded protein response is linked to cell proliferation and poor survival in estrogen receptor α-positive breast cancer. Oncogene 34, 3760–3769 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andruska N. D. et al. Estrogen receptor α inhibitor activates the unfolded protein response, blocks protein synthesis, and induces tumor regression. Proc. Natl. Acad. Sci. 112, 4737–4742 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X. et al. Targeting multidrug-resistant ovarian cancer through estrogen receptor α dependent ATP depletion caused by hyperactivation of the unfolded protein response. Oncotarget, doi: 10.18632/oncotarget.10819 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kytölä S. et al. Chromosomal alterations in 15 breast cancer cell lines by comparative genomic hybridization and spectral karyotyping. Genes. Chromosomes Cancer 28, 308–317 (2000). [DOI] [PubMed] [Google Scholar]
- Shapiro D. J., Livezey M., Yu L., Zheng X. & Andruska N. Anticipatory UPR Activation: A Protective Pathway and Target in Cancer. Trends Endocrinol. Metab, doi: 10.1016/j.tem.2016.06.002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke N. et al. One-week 96-well soft agar growth assay for cancer target validation. BioTechniques 36, 826–828, 830, 832–833 (2004). [DOI] [PubMed] [Google Scholar]
- Mao C. et al. A New Small Molecule Inhibitor of Estrogen Receptor α Binding to Estrogen Response Elements Blocks Estrogen-dependent Growth of Cancer Cells. J. Biol. Chem. 283, 12819–12830 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrero M., Yu D. V. & Shapiro D. J. Estrogen Receptor-dependent and Estrogen Receptor-independent Pathways for Tamoxifen and 4-Hydroxytamoxifen-induced Programmed Cell Death. J. Biol. Chem. 277, 45695–45703 (2002). [DOI] [PubMed] [Google Scholar]
- Zhou J.-H., Yu D. V., Cheng J. & Shapiro D. J. Delayed and persistent ERK1/2 activation is required for 4-hydroxytamoxifen-induced cell death. Steroids 72, 765–777 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X. et al. Interplay between steroid hormone activation of the unfolded protein response and nuclear receptor action. Steroids, doi: 10.1016/j.steroids.2016.03.014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A. S. GRP78 Induction in Cancer: Therapeutic and Prognostic Implications. Cancer Res. 67, 3496–3499 (2007). [DOI] [PubMed] [Google Scholar]
- Zhang J. et al. Association of elevated GRP78 expression with increased lymph node metastasis and poor prognosis in patients with gastric cancer. Clin. Exp. Metastasis 23, 401–410 (2006). [DOI] [PubMed] [Google Scholar]
- Gao D. et al. ERp29 induces breast cancer cell growth arrest and survival through modulation of activation of p38 and upregulation of ER stress protein p58IPK. Lab. Investig. J. Tech. Methods Pathol. 92, 200–213 (2012). [DOI] [PubMed] [Google Scholar]
- May D. et al. Ero1-L alpha plays a key role in a HIF-1-mediated pathway to improve disulfide bond formation and VEGF secretion under hypoxia: implication for cancer. Oncogene 24, 1011–1020 (2005). [DOI] [PubMed] [Google Scholar]
- Jiang Y. et al. Gene expression profiling in a renal cell carcinoma cell line: dissecting VHL and hypoxia-dependent pathways. Mol. Cancer Res. 1, 453–462 (2003). [PubMed] [Google Scholar]
- Hsu P. D., Lander E. S. & Zhang F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 157, 1262–1278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. et al. Correction of a Genetic Disease in Mouse via Use of CRISPR-Cas9. Cell Stem Cell 13, 659–662 (2013). [DOI] [PubMed] [Google Scholar]
- Schwank G. et al. Functional Repair of CFTR by CRISPR/Cas9 in Intestinal Stem Cell Organoids of Cystic Fibrosis Patients. Cell Stem Cell 13, 653–658 (2013). [DOI] [PubMed] [Google Scholar]
- Wang H. et al. One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Cell 153, 910–918 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu Y. et al. Generation of Gene-Modified Cynomolgus Monkey via Cas9/RNA-Mediated Gene Targeting in One-Cell Embryos. Cell 156, 836–843 (2014). [DOI] [PubMed] [Google Scholar]
- Liang P. et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell 6, 363–372 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q. et al. Precise gene deletion and replacement using the CRISPR/Cas9 system in human cells. BioTechniques 57, 115–124 (2014). [DOI] [PubMed] [Google Scholar]
- Cradick T. J., Fine E. J., Antico C. J. & Bao G. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 41, 9584–9592 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S. W. et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24, 132–141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y. et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 31, 822–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasty P., Rivera-Pérez J. & Bradley A. The length of homology required for gene targeting in embryonic stem cells. Mol. Cell. Biol. 11, 5586–5591 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack P. & Sapunar F. Pharmacokinetic profile of the fulvestrant loading dose regimen in postmenopausal women with hormone receptor-positive advanced breast cancer. Clin. Breast Cancer 8, 347–351 (2008). [DOI] [PubMed] [Google Scholar]
- Croxtall J. D. & McKeage K. Fulvestrant: a review of its use in the management of hormone receptor-positive metastatic breast cancer in postmenopausal women. Drugs 71, 363–380 (2011). [DOI] [PubMed] [Google Scholar]
- Jeselsohn R. et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res. 20, 1757–1767 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladd B. et al. Effective combination therapies in preclinical endocrine resistant breast cancer models harboring ER mutations. Oncotarget, doi: 10.18632/oncotarget.10852 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanning S. W. et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y. & Brown M. Molecular determinants for the tissue specificity of SERMs. Science 295, 2465–2468 (2002). [DOI] [PubMed] [Google Scholar]
- Fowler A. M., Solodin N. M., Valley C. C. & Alarid E. T. Altered Target Gene Regulation Controlled by Estrogen Receptor-α Concentration. Mol. Endocrinol. 20, 291–301 (2006). [DOI] [PubMed] [Google Scholar]
- Mohammed H. et al. Endogenous Purification Reveals GREB1 as a Key Estrogen Receptor Regulatory Factor. Cell Reports 3, 342–349 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naughton C. et al. Progressive loss of estrogen receptor alpha cofactor recruitment in endocrine resistance. Mol. Endocrinol. 21, 2615–2626 (2007). [DOI] [PubMed] [Google Scholar]
- McCune K. et al. Prognosis of hormone-dependent breast cancers: implications of the presence of dysfunctional transcriptional networks activated by insulin via the immune transcription factor T-bet. Cancer Res. 70, 685–696 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C. et al. A modulated empirical Bayes model for identifying topological and temporal estrogen receptor α regulatory networks in breast cancer. Bmc Syst. Biol. 5, 67 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep 4, 1116–1130 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunar F., Gormus Z. I., Baltaci A. K. & Mogulkoc R. The effect of low dose zinc supplementation to serum estrogen and progesterone levels in post-menopausal women. Biol. Trace Elem. Res. 126 Suppl 1, S11–S14 (2008). [DOI] [PubMed] [Google Scholar]
- Lucisano A. et al. Ovarian and peripheral plasma levels of progestogens, androgens and oestrogens in post-menopausal women. Maturitas 6, 45–53 (1984). [DOI] [PubMed] [Google Scholar]
- Yoo K. Y. et al. Female sex hormones and body mass in adolescent and postmenopausal Korean women. J. Korean Med. Sci. 13, 241–246 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andruska N., Mao C., Cherian M., Zhang C. & Shapiro D. J. Evaluation of a luciferase-based reporter assay as a screen for inhibitors of estrogen-ERα-induced proliferation of breast cancer cells. J. Biomol. Screen. 17, 921–932 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherian M. T., Wilson E. M. & Shapiro D. J. A Competitive Inhibitor That Reduces Recruitment of Androgen Receptor to Androgen-responsive Genes. J. Biol. Chem. 287, 23368–23380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.