

Abstract
We demonstrate a strategy to trigger and finely control the assembly of supramolecular DNA nanostructures with pH. Control is achieved via a rationally designed strand displacement circuit that responds to pH and activates a downstream DNA tile self-assembly process. We observe that the DNA structures form under neutral/basic conditions, while the self-assembly process is suppressed under acidic conditions. The strategy presented here demonstrates a modular approach toward building systems capable of processing biochemical inputs and finely controlling the assembly of DNA-based nanostructures under isothermal conditions. In particular, the presented architecture is relevant for the development of complex DNA devices able to sense and respond to molecular markers associated with abnormal metabolism.
In Nature, several vital cellular tasks, such as the formation of the cell membrane or of stable host–guest complexes, rely on thermodynamically driven molecular assembly processes based on relatively weak interactions.1 The assembly of such complexes is usually finely controlled by a series of biological inputs and molecular cues.2 Inspired by this observation, researchers in the field of supramolecular chemistry have exploited non-covalent interactions to achieve controlled self-assembly of synthetic moieties and to build complex nanostructures of defined geometries.1,3 Because of its predictable base-pairing interactions and its low synthesis cost, DNA represents one of the best biomaterials to design and assemble complex structures with nanoscale features. Such structures have reached a level of complexity that would have been impossible to imagine 20 years ago: using synthetic DNA oligonucleotides, we can now build 2D and 3D nanoscale objects with virtually arbitrary shape.4,5 Input-responsive DNA assemblies, engineered to exhibit functional dynamic behaviors such as opening and closing or moving in response to biochemical inputs, have been also built.6
One of the current limitations of responsive DNA nanostructures is that responses are generally encoded in the structure itself (for instance, via input-responsive domains or aptamers) and can be modulated exclusively as a function of the input concentration or intensity. For example, DNA nanofabrication has been controlled directly using small synthetic ligands or light irradiation.7,8 Finer control of assembly is desirable in many applications, such as sensing, metabolic engineering,9 or nanomanufacturing. This limitation can be mitigated by using input-triggerable DNA strand displacement circuits to control assembly.10 Here, we demonstrate the viability of this approach with a pH-controlled DNA catalytic circuit to control the assembly of DNA-based nanostructures.
We focus on DNA structures self-assembling from DNA tiles, one of the best-characterized approaches to build scalable DNA architectures.11,12 In this strategy, DNA strands are designed to form rigid building blocks, called tiles (Supporting Information (SI), Figure SI1), that can self-assemble into lattices, ribbons, or tubular structures through single-stranded overhangs. DNA tile assembly can be triggered by an upstream DNA strand displacement circuit:10 inactive (protected) DNA tiles can be activated (deprotected) by the output of a DNA catalytic circuit,13 which is in turn triggered by a DNA catalyst molecule. This approach allows the isothermal assembly of DNA nanostructures, achieving control of the composition and timing of the process.14,15
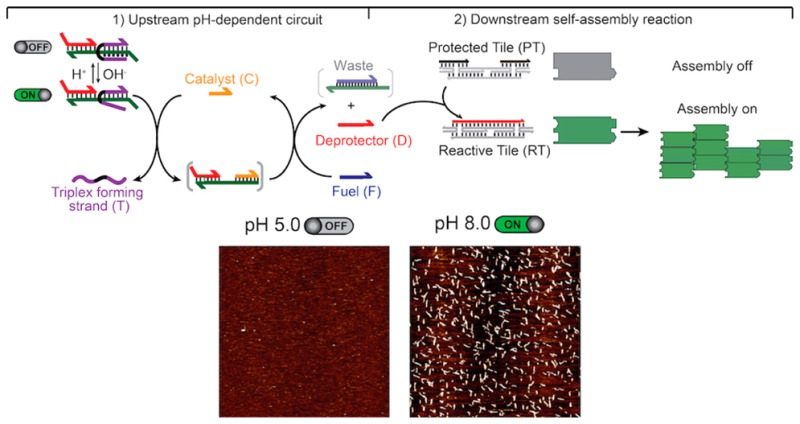
To achieve pH-controlled, circuit-tunable assembly of DNA nanostructures, we re-engineered a DNA strand displacement catalytic circuit (Figure 1) to be responsive to pH, and we used it to direct a well-known DNA tile self-assembly process based on double-crossover tiles (Figure SI1). pH control is achieved by taking advantage of the well-characterized pH sensitivity of triplex DNA, which requires the protonation of the N3 of cytosine in the third strand to form (average pKa of cytosines in triplex structure is ∼6.5).16 Specifically, we designed a pH-dependent substrate (Figure 1, purple strand) that prevents the strand displacement reaction with the catalyst strand (C) at acidic pH, due to the formation of Hoogsteen interactions in addition to the Watson–Crick base-pairings. Only at basic pHs (when Hoogsteen interactions are destabilized) is the accessibility of the DNA substrate, needed for enabling such strand displacement circuit, restored. In turn, tile assembly can occur efficiently upon catalyst addition (Figure 1, right) under these pH conditions.
Figure 1.

pH-controlled self-assembly of DNA tiles. We have achieved pH-controlled DNA tile assembly by coupling an upstream re-engineered pH-controlled circuit with a downstream DNA tile self-assembly process. (1) In the pH-dependent upstream circuit, a catalyst (C) binds to a pH-dependent substrate, leading to the release of a deprotector strand (D). (2) The D strand, in turn, activates a downstream self-assembly reaction by irreversibly associating with a protected tile (PT). This leads to reactive double-crossover tiles (RT) self-assembling into lattices and nanotubes (right). The pH-dependent substrate is implemented with a clamp-like triplex-forming DNA strand (T) that, under acidic pHs, can form a triplex complex, inhibiting the strand displacement reaction with the catalyst.
The designed upstream pH-dependent circuit can be finely controlled with pH. To demonstrate this, we have characterized in isolation the pH-dependent strand displacement circuit by using an external, optically labeled reporter (R) that stoichiometrically reacts with the liberated deprotector strand (D) (Figure 2a). We first characterized the pH dependency of the substrate complex by employing a triplex-forming strand (T) (responsible for the substrate formation) labeled with a fluorophore and a quencher (Figure 2b, left). This allowed us to monitor the folding and unfolding of the triplex structure at different pHs (Figure 2b, right). As expected, under acidic pHs (favoring triplex formation) a low fluorescence signal is observed, suggesting folding of the triplex complex. Upon increasing the solution’s pH, the fluorescence signal increases, consistent with the destabilization of Hoogsteen interactions (Figure 2b, right). The pH of semiprotonation (the average pKa due to several interacting protonation sites) for this triplex complex is 7.5, which is in agreement with previous observations of similar triplex-forming sequences.16,17
Figure 2.
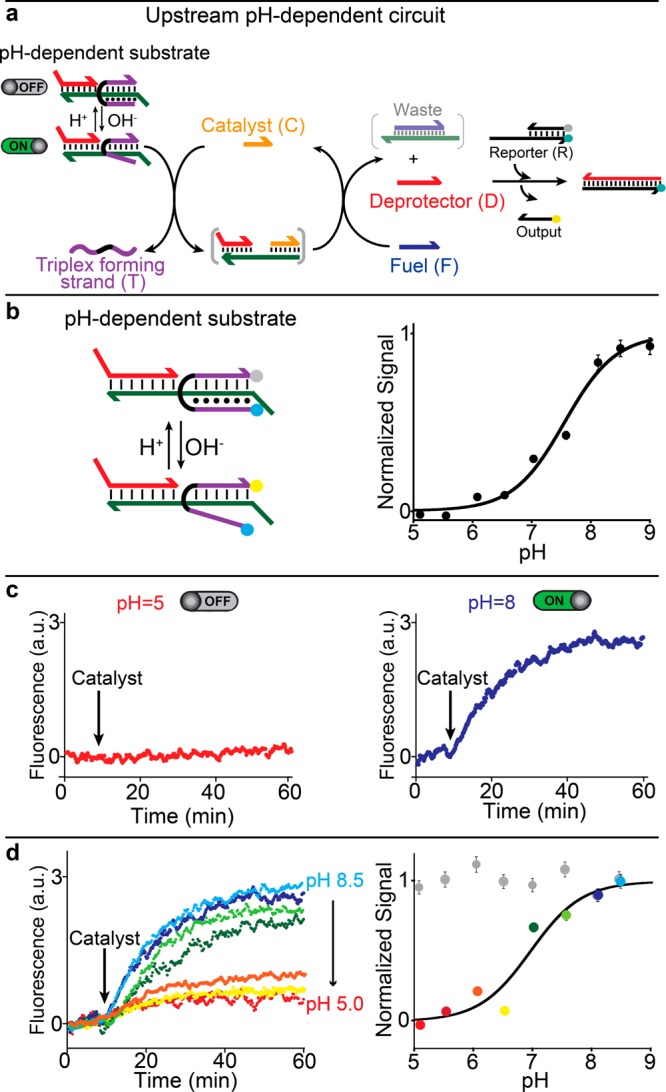
(a) Upstream pH-dependent DNA circuit. (b) Triplex formation in the pH-dependent substrate complex, studied by incorporating a pH-insensitive FRET pair at the ends of the clamp-like triplex-forming strand (left) and measuring the fluorescence signal at different pHs (right). Stable triplex formation is observed only at pHs below 7.0. (c) The pH-dependent triplex complex in the substrate inhibits strand displacement reaction upon C addition (pH 5.0, left). At basic pHs the destabilization of Hoogsteen interactions leads to substrate activation, which allows strand displacement reaction in the presence of catalyst strand (C) (pH 8.0, right). (d) The pH dependence of the catalyst/substrate reaction can be finely controlled at different pHs. Here, strand displacement reaction is followed by fluorescence measurements in a solution containing the pH-dependent substrate (10 nM), the fuel strand (F) (20 nM), and an external, optically labeled reporter (30 nM) (see experimental details in the SI) that stoichiometrically reacts with the released D to give a fluorescence signal. The catalyst was added at a high concentration (30 nM) to better highlight the pH dependence of the circuit. All experiments were performed in a TAE 1x buffer + 15 mM MgCl2 at 25 °C with the pH adjusted using small aliquots of HCl (1 M) and NaOH (1 M). Error bars here and in the following figures represent the average and standard deviations (average RSD = 6%) of three independent measurements.
pH-dependent triplex formation in the substrate of the downstream catalytic network allows rational control of the output concentration by simply changing the solution’s pH. At pH 5.0, which is acidic enough for clamp-like strand to form an inactive triplex complex, the addition of the catalyst strand (C) results in no significant fluorescence change (Figure 2c, left). This suggests that the circuit is fully suppressed. At pH 8.0, which inhibits triplex formation, strand displacement successfully proceeds with fast kinetics upon C addition (Figure 2c, right). The activation level of the circuit can be modulated by changing the pH of the solution (Figures 2d and SI2) or the C concentration (Figure SI3). However, probably due to the presence of the triplex-forming sequence, the circuit catalytic efficiency is poorer than previously reported (Figure SI4).10 At high C concentration, the complete circuit converts the gradual pH dependency shown in Figure 2b into a digital-like response; in contrast, at low C concentration, the circuit response remains gradual. A control experiment involving a pH-independent substrate where the triplex-forming portion has been substituted with a random sequence unable to form a triplex structure shows no effect of pH over the entire pH range explored, and over a wide range of C concentrations (Figure 2d, right, gray dots; Figures SI5, SI6, and SI7).
The pH-controlled DNA catalytic circuit can be used to direct the assembly of DNA nanostructures using pH. To do this we have interconnected the above-characterized pH-dependent circuit with a DNA tile self-assembly process.10,11a,11b,12 Fluorescence microscopy images (Figure 3, top) and AFM images (Figure 3, center) confirm that tiles assemble only at neutral/basic pHs, while no assemblies are observed over the same reaction time at acidic pHs (pH 5.0 and 6.0). Tile assembly largely yields tubular structures according to fluorescence microscopy and AFM images. As a control experiment we used a pH-independent substrate and observed assembly of DNA tiles in the entire pH range investigated (Figure 3, bottom). Moreover, statistical analysis shows that the length and yield of nanotubes formed with the pH-dependent substrate under basic conditions (average length = 0.91 μm and yield = 23 ± 10%, at pH 8.0) is comparable to those of the control pH-independent substrate at both pH 8.0 (length = 0.75 μm and yield = 12 ± 6%) and 5.0 (length = 0.79 μm and yield = 19 ± 12%) (see SI for experimental details and Figure SI8). As a further demonstration that pH does not affect the downstream tile assembly reaction, we have exogenously added D to a solution containing protected tiles and observed pH-independent tile assembly (Figure SI9). Conversely, the absence of C leads to no formation of nanotubes (Figure SI10).
Figure 3.
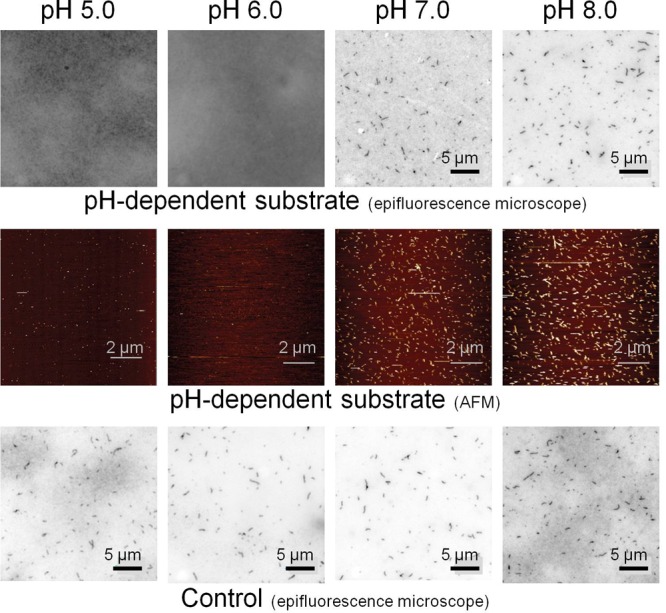
pH-dependent self-assembly of DNA tiles. (Top) The upstream, pH-dependent DNA circuit coupled with a downstream tile self-assembly process (Figure 1) allows to control DNA tile self-assembly with pH. At acidic pHs (pH 5.0 and 6.0) no formation of assemblies is observed with optical fluorescence microscopy. By increasing the pH of the solution (pH 7.0 and 8.0) we achieve evident formation of DNA lattices. (Center) pH-dependent lattices were also imaged with atomic force microscopy (AFM). (Bottom) A control experiment using a pH-independent substrate (unable to form a pH-dependent triplex structure (Figure SI5)) leads to pH-independent assembly of DNA tiles. All the experiments shown here and in Figure 4 were performed using the following concentrations of reagents: protected tile (PT), 200 nM; fuel (F), 440 nM; pH-dependent substrate or control substrate, 220 nM; and catalyst (C), 20 nM. The assembly was achieved in TAE 1x buffer + 15 mM MgCl2, at 25 °C with the pH adjusted using small aliquots of HCl (1 M) and NaOH (1 M). For all the fluorescence microscopy experiments, a cy3-labeled tile central strand (t4, see SI) was used to detect nanotubes formation. AFM images of the pre-adsorbed nanostructures on freshly cleaved mica (see SI) were obtained with AC mode in TAE 1x buffer + 15 mM MgCl2 buffer, with 1 Hz scan rate, 256 pixel × 256 pixel image definition and processed with second-rder flattening.
The kinetics of DNA tile assembly was studied with AFM. Images derived from the reaction samples (upstream pH-dependent circuit + protected tiles) at different intervals after the addition of the catalyst strand were obtained. No tiles assembly at pH 5.0 (Figure 4, top) and pH 6.0 (Figure SI11) was observed during the entire time frame investigated. At pH 8.0 a substantial amount of assembled tiles can be observed within 30 min after catalyst addition (Figure 4, bottom). We note that assembly of small lattices at pH 5.0 was observed under AFM after 4 days since the start of the reaction, presumably due to leak of the deprotector from the substrate (Figure SI12).10
Figure 4.

Kinetics of pH-dependent self-assembly of DNA tiles. The self-assembly of DNA tiles was followed by AFM images at different time intervals. No tile assembly is observed within the time frame of the experiment at pH 5.0. In contrast, at basic pH (pH 8.0), DNA lattices can be clearly observed just 15 min after catalyst addition. See Figure 3 caption for experimental details.
In this work we have demonstrated a modular architecture to regulate the self-assembly of DNA nanostructures with the sole change of pH. We did so by integrating an upstream pH-dependent strand displacement circuit into an already-characterized, downstream DNA tile self-assembly process. Our architecture is potentially relevant in biomedical applications of DNA nanotechnology. For example, pH disregulation is a hallmark of several diseases,18 including cancer. The availability of pH-triggerable DNA nanostructures will have intriguing potential applications in drug delivery research.19 Moreover, because we have recently demonstrated that the pH dependence of triplex DNA could be finely regulated by simply changing the relative content of TAT vs CGC triplets in the triplex-forming sequence,16b we anticipate the possibility of programming the assembly of different DNA nanostructures at different pH thresholds. Finally, many enzymes are known that catalyze either proton-producing or proton-consuming reactions.20 Therefore, different enzymes and enzymatic substrates could be used as molecular, functional inputs to control the isothermal assembly of DNA nanostructures.
The approach described here could also be expanded to consider a wider range of molecular inputs controlling the self-assembly of supramolecular structures. In fact, apart from pH, it is in principle possible to engineer upstream DNA strand displacement circuits activated by the presence of proteins, antibodies, or other relevant biomolecular inputs so that the downstream self-assembly process is input-specific. In principle, logic or dynamic circuits could replace the catalytic network we considered, providing the system with expandable signal-processing capacity. Finally, because strand displacement circuits can be designed to be highly specific and to respond orthogonally to a specific input, it would be possible to control the simultaneous assembly of different structures using multiple inputs in a highly programmable fashion, thus leading to a better temporal and spatial regulation of the assembly processes.
Acknowledgments
This work was supported by Associazione Italiana per la Ricerca sul Cancro, AIRC (project no. 14420 to F.R.), by the European Research Council, ERC (project no. 336493 to F.R.; project no. 269051-Monalisa’s Quidproquo to M.C.), by the Italian Ministry of Education, University and Research (PRIN, to F.R.), and by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences (Award No. DE-SC0010595 to E.F.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b07676.
Supporting methods and Figures SI1–SI12 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Mattia E.; Otto S. Nat. Nanotechnol. 2015, 10, 111. 10.1038/nnano.2014.337. [DOI] [PubMed] [Google Scholar]
- a Desai A.; Mitchison T. J. Annu. Rev. Cell Dev. Biol. 1997, 13, 83. 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]; b Lloyd C.; Chan J. Nat. Rev. Mol. Cell Biol. 2004, 5, 13. 10.1038/nrm1277. [DOI] [PubMed] [Google Scholar]; c Li R.; Gundersen G. G. Nat. Rev. Mol. Cell Biol. 2008, 9, 860. 10.1038/nrm2522. [DOI] [PubMed] [Google Scholar]
- a Li J.; Nowak P.; Otto S. J. Am. Chem. Soc. 2013, 135, 9222. 10.1021/ja402586c. [DOI] [PubMed] [Google Scholar]; b Subhabrata M.; Fortuna I.; Ferrante C.; Scrimin P.; Prins J. L. Nat. Chem. 2016, 8, 725. 10.1038/nchem.2511. [DOI] [PubMed] [Google Scholar]
- a He Y.; Tian Y.; Ribbe A. E.; Mao C. D. J. Am. Chem. Soc. 2006, 128, 15978. 10.1021/ja0665141. [DOI] [PubMed] [Google Scholar]; b Ke Y.; Ong L. L.; Shih W. M.; Yin P. Science 2012, 338, 1177. 10.1126/science.1227268. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wei B.; Dai M.; Yin P. Nature 2012, 485, 623. 10.1038/nature11075. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Zhao Z.; Liu Y.; Yan H. Nano Lett. 2011, 11, 2997. 10.1021/nl201603a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fu J.; Liu M.; Liu Y.; Woodbury N. W.; Yan H. J. Am. Chem. Soc. 2012, 134, 5516. 10.1021/ja300897h. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fu J.; Yang Y. R.; Johnson-Buck A.; Liu M.; Liu Y.; Walter N. G.; Woodbury N. W.; Yan H. Nat. Nanotechnol. 2014, 9, 531. 10.1038/nnano.2014.100. [DOI] [PubMed] [Google Scholar]
- a Douglas S. M.; Bachelet I.; Church G. M. Science 2012, 335, 831. 10.1126/science.1214081. [DOI] [PubMed] [Google Scholar]; b Gerling T.; Wagenbauer K. F.; Neuner A. M.; Dietz H. Science 2015, 347, 1446. 10.1126/science.aaa5372. [DOI] [PubMed] [Google Scholar]
- a Liu Z.; Li Y.; Tian C.; Mao C. Biomacromolecules 2013, 14, 1711. 10.1021/bm400426f. [DOI] [PubMed] [Google Scholar]; b McLaughlin C. K.; Hamblin G. D.; Sleiman H. F. Chem. Soc. Rev. 2011, 40, 5647. 10.1039/c1cs15253j. [DOI] [PubMed] [Google Scholar]; c Yang Y.; Endo M.; Hidaka K.; Sugiyama H. J. Am. Chem. Soc. 2012, 134, 20645. 10.1021/ja307785r. [DOI] [PubMed] [Google Scholar]
- a Dohno C.; Atsumi H.; Nakatani K. Chem. Commun. 2011, 47, 3499. 10.1039/c0cc05473a. [DOI] [PubMed] [Google Scholar]; b Han D.; Huang J.; Zhu Z.; Yuan Q.; You M.; Chen Y.; Tan W. Chem. Commun. 2011, 47, 4670. 10.1039/c1cc10893j. [DOI] [PubMed] [Google Scholar]; c Yang Y.; Endo M.; Hidaka K.; Sugiyama H. J. Am. Chem. Soc. 2012, 134, 20645. 10.1021/ja307785r. [DOI] [PubMed] [Google Scholar]
- Delebecque C. J.; Lindner A. B.; Silver P. A.; Aldaye F. A. Science 2011, 333, 470. 10.1126/science.1206938. [DOI] [PubMed] [Google Scholar]
- Zhang D. Y.; Hariadi R. F.; Choi H. M. T.; Winfree E. Nat. Commun. 2013, 4, 1965. 10.1038/ncomms2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mao C.; LaBean T. H.; Reif J. H.; Seeman N. C. Nature 2000, 407, 493. 10.1038/35035038. [DOI] [PubMed] [Google Scholar]; b Winfree E.; Liu F.; Wenzler L. A.; Seeman N. C. Nature 1998, 394, 539. 10.1038/28998. [DOI] [PubMed] [Google Scholar]; c Zhang S.; Fu T. J.; Seeman N. C. Biochemistry 1993, 32, 8062. 10.1021/bi00083a002. [DOI] [PubMed] [Google Scholar]
- a Chen H.; Weng T.-W.; Riccitelli M. M.; Cui Y.; Irudayaraj J.; Choi J. H. J. Am. Chem. Soc. 2014, 136, 6995. 10.1021/ja500612d. [DOI] [PubMed] [Google Scholar]; b Rothemund P. W. K.; Ekani-Nkodo A.; Papadakis N.; Fygenson D. K.; Winfree E. J. Am. Chem. Soc. 2004, 126, 16344. 10.1021/ja044319l. [DOI] [PubMed] [Google Scholar]
- a Zhang D. Y.; Seelig G. Nat. Chem. 2011, 3, 103. 10.1038/nchem.957. [DOI] [PubMed] [Google Scholar]; b Zhang D. Y.; Turberfield A. J.; Yurke B.; Winfree E. Science 2007, 318, 1121. 10.1126/science.1148532. [DOI] [PubMed] [Google Scholar]
- a Bath J.; Turberfield A. J. Nat. Nanotechnol. 2007, 2, 275. 10.1038/nnano.2007.104. [DOI] [PubMed] [Google Scholar]; b Krishnan Y.; Simmel F. C. Angew. Chem., Int. Ed. 2011, 50, 3124. 10.1002/anie.200907223. [DOI] [PubMed] [Google Scholar]
- a Seelig G.; Soloveichik D.; Zhang D. Y.; Winfree E. Science 2006, 314, 1585. 10.1126/science.1132493. [DOI] [PubMed] [Google Scholar]; b Turberfield A. J.; Mitchell J. C.; Yurke B.; Mills A. P. Jr.; Blakey M. I.; Simmel F. C. Phys. Rev. Lett. 2003, 90, 118102. 10.1103/PhysRevLett.90.118102. [DOI] [PubMed] [Google Scholar]; c Wang F.; Lu C.-H.; Willner I. Chem. Rev. 2014, 114, 2881. 10.1021/cr400354z. [DOI] [PubMed] [Google Scholar]
- a Amodio A.; Zhao B.; Porchetta A.; Idili A.; Castronovo M.; Fan C.; Ricci F. J. Am. Chem. Soc. 2014, 136, 16469. 10.1021/ja508213d. [DOI] [PubMed] [Google Scholar]; b Idili A.; Vallée-Bélisle A.; Ricci F. J. Am. Chem. Soc. 2014, 136, 5836. 10.1021/ja500619w. [DOI] [PubMed] [Google Scholar]
- Porchetta A.; Idili A.; Vallée-Bélisle A.; Ricci F. Nano Lett. 2015, 15, 4467. 10.1021/acs.nanolett.5b00852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb B. A.; Chimenti M.; Jacobson M. P.; Barber D. L. Nat. Rev. Cancer 2011, 11, 671. 10.1038/nrc3110. [DOI] [PubMed] [Google Scholar]
- a Sellner S.; Kocabey S.; Nekolla K.; Krombach F.; Liedl T.; Rehberg M. Biomaterials 2015, 53, 453. 10.1016/j.biomaterials.2015.02.099. [DOI] [PubMed] [Google Scholar]
- Del Grosso E.; Dallaire A. M.; Vallée-Bélisle A.; Ricci F. Nano Lett. 2015, 15, 8407. 10.1021/acs.nanolett.5b04566. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.