

Key Clinical Message
A focused genetic workup is useful in determining the cause of familial microcephaly, especially in the setting of mildly different phenotypes. As illustrated by this case from an impoverished international urban location, one must not assume the etiology for the apparent familial microcephaly is the same for all affected members.
Keywords: 10p15.3 deletion, familial microcephaly, neurodevelopmental delays
Introduction
Microcephaly is defined as an occipitofrontal head circumference measuring >2 standard deviations below the mean for a specific age and gender, often associated with neurodevelopmental delays. Microcephaly can be primary, in which an individual is microcephalic at birth, or acquired, in which the individual is within two standard deviations of the mean at birth, but falls to >2 standard deviations below the mean over time. A head circumference measurement at birth is especially important, as knowing whether microcephaly is primary or acquired can assist in the complex evaluation of determining its etiology 1.
Because growth of the cranial vault occurs in part due to growth of the brain, microcephaly is significant as it also indicates micrencephaly 1. Microcephaly can be isolated, or it can be syndromic when associated with other congenital anomalies. In certain cases, microcephaly may be the first clue to the presence of a syndromic phenotype. As such, determining the cause for microcephaly may aid in earlier recognition of disease processes and better management of these individuals and their families.
Clinical Reports
Hispanic family presented here consists of a mother and her two daughters from an impoverished urban area of Honduras (Fig. 1). The 26‐year‐old mother exhibits microcephaly with an occipitofrontal circumference of 50.5 cm (<3rd percentile). She also exhibits some mild facial dysmorphism including micrognathia (Fig. 1) but has no history of seizures. On neuropsychological testing, she was found to have a functional IQ between 70 and 80 and was described by the examiner as having difficulties with problem‐solving and decision‐making, as well as displaying some depression‐like symptoms. She completed school through the 7th grade. She reports that multiple family members have subjectively small heads, but they were not available to be examined.
Figure 1.

Mother and two daughters with microcephaly. Mother, top left; older daughter, bottom right; younger daughter, bottom left.
The early neurodevelopment of the older 7‐year‐old daughter was within the broad limits of normal. She walked at 13 months and was able to speak three words at 7 months. She was also microcephalic, with an occipitofrontal circumference of 47.5 cm at age 7 years (<3rd percentile, 50th percentile for a 2‐year‐old). Her height and weight are also less than the 3rd percentile for her age. She has no facial dysmorphism and no known history of seizures. Neuropsychological testing revealed a functional IQ between 70 and 80. She was described as “impulsive, stressed, and anxious” during testing by the neuropsychological examiner.
The younger 4‐year‐old daughter has a different father from her older sister. She was born at 30 weeks of gestation complicated by placenta previa with a birthweight of 1.45 kg. She was discharged home from the hospital at 6 days of age. She went on to display myoclonic jerks at 8 months of age and developed tonic–clonic/myoclonic epilepsy at 21 months. Her seizures were initially difficult to control with antiepileptic medications. Currently, she has not had a seizure in several months and has been successfully weaned off her antiepileptic medications. She currently exhibits severe microcephaly, with an occipitofrontal circumference of 40.5 cm at 48 months of age (≪3rd percentile, 50th percentile for a 3.5‐month‐old). Her earliest recorded occipitofrontal circumference was at 11 months of age and was 37 cm (≪3rd percentile, 50th percentile for a 1‐month‐old). Her height and weight are also below the 3rd percentile. She displays facial dysmorphism (Fig. 2) including micrognathia. Her sleep is irregular despite trials of sleep medications.
Figure 2.
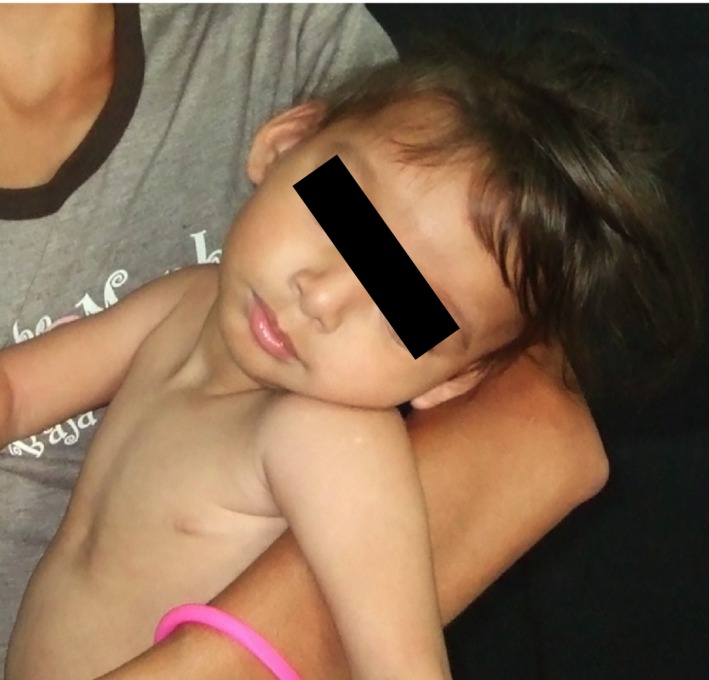
Younger daughter with visible hypotonia including a head tilt, micrognathia, and mild left strabismus in this image.
At 3 years of age, significant neurodevelopmental delays included being nonambulatory and exhibiting significant hypotonia in all extremities. Her gross and fine motor development included some scooting, but no sitting, crawling, walking, or normally grasping objects. Her language development at 3 years of age included only vowel sounds.
At 4 years of age, she can move by scooting. She has not had any neurodevelopmental regression. She still does not roll, crawl, sit, or stand. She uses a small number of single words and appears to understand some simple concepts. She is able to communicate her wants primarily using visual gaze estimated at the 18‐ to 24‐month‐old level. On physical examination, her deep tendon reflexes are 2+ and symmetric in her upper extremities, and 3+ and symmetric in her lower extremities with up‐going great toes bilaterally.
A head CT scan was completed on the younger sister at age 29 months. This revealed a brain that fills the microcephalic cranial vault and hydrocephalus ex vacuo. A recent brain MRI at 4 years of age revealed significant abnormal findings in nearly all areas of the brain (Fig. 3). These findings included diffusely thickened cortex, polymicrogyria, ventriculomegaly, smooth‐appearing frontal and parietal cortical surfaces, patchy increased T2 signal in the white matter, and some cobblestone‐like cortex in the temporal lobes.
Figure 3.
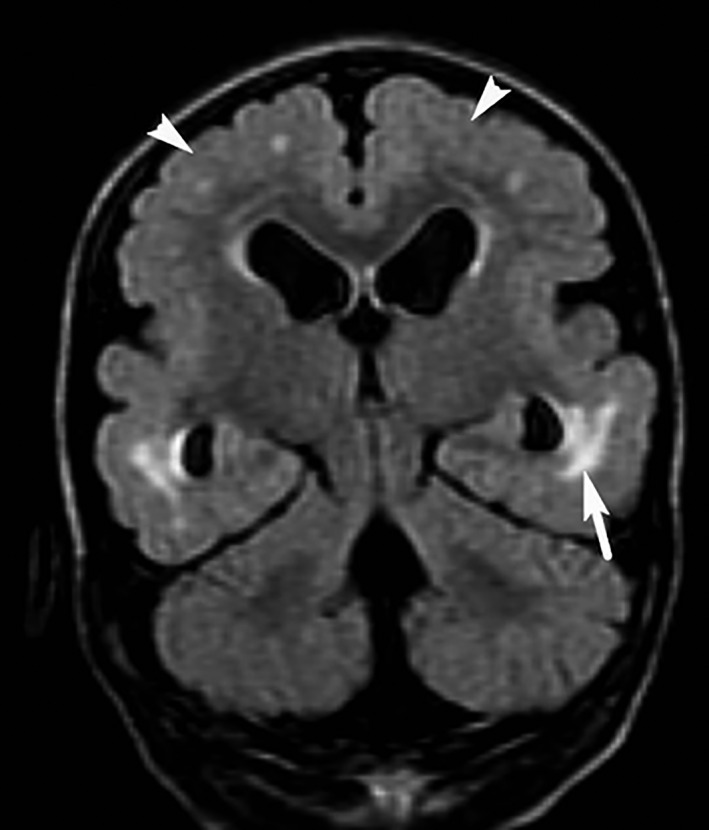
Brain MRI of younger daughter – FLAIR coronal view, clearly illustrating patches of increased T2 signal (arrow), as well as polymicrogyria and thickened cerebral cortex (arrow heads) with poorly defined boundaries between gray and white matter.
Initial genetic testing was completed for the mother and her two daughters via chromosomal microarray analysis (CytoScan HD Microarray; Affymetrix, Inc., USA). The mother and older daughter were shown via microarray to have a gain at 14q11.2 [chr14:20,511,672‐20,903,963; hg19]. This variant is not known to have any specific phenotypic effect and was not able to be directly correlated with the microcephaly. The younger daughter did not have a gain at 14q11.2, but did exhibit a deletion at 10p15.3 [chr10:219,859‐344,821; hg19]. As this had not been seen in the mother, a microarray analysis was performed on the father. His microarray was normal. This confirmed that the loss at 10p15.3 was de novo and likely clinically significant. Deletions in this region have been reported to be a rare cause of cognitive deficits, speech disorders, motor delay, and hypotonia 2, 3. However, based on previous reports of 10p15.3 deletions, it is unlikely that the deletion fully explains the significant cerebral dysgenesis seen on the brain MRI. To further investigate the cerebral dysgenesis and associated epilepsy history, next‐generation sequencing was performed on a 103‐gene panel known for its association with epilepsy. The epilepsy gene panel revealed heterozygous variants in the CHRNB2, MCPH1, and CLN3 genes which were not felt to be clinically significant. No significant genetic findings were found to explain the variability of the phenotypes seen in the affected family members other than the 10p15.3 deletion in the younger child.
Discussion
On initial presentation, these three individuals were presumed to have a familial syndrome that consisted of microcephaly and cognitive delay along a spectrum. The younger female child was presumed to have a worse phenotype than her other family members due to a genetic phenomenon such as anticipation or variable expressivity. However, upon genetic analysis, she was shown to have a de novo 10p15.3 deletion which has likely played an important role in her significant neurodevelopmental delays, epilepsy, and hypotonia.
Deletions at 10p15.3 have only been reported in 24 cases. Only two of these 24 cases were microcephalic 2, 4. The other individuals with this deletion have been normocephalic or macrocephalic, indicating that this deletion is probably not the sole cause of the younger daughter's microcephaly. However, individuals with a deletion at 10p15.3 are noted to be at increased risk of intellectual disability, speech and motor delay, hypotonia, brain anomalies, and seizures. There are two genes with OMIM entries contained within the deleted region of 10p15.3, ZMYND11 (exons 2‐15; CCDS7052.2), and DIP2C (exons 33‐37; CCDS7054.1). None of the cases reported severe brain abnormalities or significant intellectual disability to the extent seen in the younger daughter in this report. The variants detected on the next‐generation sequencing panel were not associated with any reports of known significance.
Genetic testing in this family provided clues as to the cause of the younger daughter's presentation and revealed information that is useful in providing reproductive counseling to her parents. Chromosomal microarray is currently indicated as the most cost‐effective genetic test of choice in such instances, as it is superior to G‐banded karyotype with regard to diagnostic yield in individuals with multiple congenital anomalies, neurodevelopmental delays, intellectual disability, and autism, especially in more severely impacted individuals, and is useful in providing diagnostic information for individuals with epilepsy 5, 6, 7.
The syndromic presentation seen in our youngest patient may not be entirely caused by the 10p15.3 deletion, as it appears to be more extreme and includes more complex brain malformations than others who have been reported with this deletion. However, the variants of unknown significance found on the 103‐gene epilepsy panel may have had some interaction with the 10p15.3 deletion, worsening the phenotype. Another possibility is that the genetic anomaly that is the true etiology of this patient's presentation has not been found yet using only chromosomal microarray and the 103‐gene epilepsy panel. If further investigation is indicated in the future, whole‐exome sequencing would likely be the diagnostic test of choice.
This family provides evidence for the utility of a genetic workup for familial microcephaly, even though a cause of the microcephaly in this family has not been determined. Without further workup, genetic counseling to her family would have been unable to predict the likelihood of a similar phenotype in future offspring of the mother and older child. Genetic testing and imaging, instead, did provide clear evidence of familial microcephaly of unknown origin at this time, but with a de novo cause of the remainder of the younger daughter's extensive neurodevelopmental delays and epilepsy.
Conflict of Interest
None declared.
Acknowledgments
We would like to thank the family for their participation and the Greenwood Genetic Center's Diagnostic Laboratories for performing the extensive genetic testing studies.
References
- 1. Holden, K. R. , and Lyons M. J.. 2009. Microcephaly‐Acquired Pp. 421–428 in Maria B. L., ed. Current management in child neurology, 4th ed. BC Decker Inc, PMPH‐USA, Shelton, Connecticut. [Google Scholar]
- 2. DeScipio, C. , Conlin L., Rosenfeld J., Tepperberg J., Pasion R., Patel A., et al. 2012. Subtelomeric deletion of chromosome 10p15.3: clinical findings and molecular cytogenetic characterization. Am. J. Med. Genet. A 158A:2152–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vargiami, E. , Ververi A., Kyriazi M., Papathanasiou E., Gioula G., Gerou S., et al. 2013. Severe clinical presentation in monozygotic twins with 10p15.3 microdeletion syndrome. Am. J. Med. Genet. A 164:764–768. [DOI] [PubMed] [Google Scholar]
- 4. Cobben, J. , Weiss M., van Dijk F., De Reuver R., de Kruiff C., Pondaag W., et al. 2014. A de novo mutation in ZMYND11, a candidate gene for 10p15.3 deletion syndrome, is associated with syndromic intellectual disability. Eur. J. Med. Genet. 57:636–638. [DOI] [PubMed] [Google Scholar]
- 5. Miller, D. , Adam M., Aradhya S., et al. 2010. Consensus statement: chromosomal microarray is a first‐tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 86:749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bartnik, M. , Nowakowska B., Derwińska K., et al. 2013. Application of array comparative genomic hybridization in 256 patients with developmental delay or intellectual disability. J. Appl. Genet. 55:125–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ezugha, H. , Anderson C., Marks H., Khurana D., Legido A., and Valencia I.. 2010. Microarray analysis in children with developmental disorder or epilepsy. Pediatr. Neurol. 43:391–394. [DOI] [PubMed] [Google Scholar]