

Summary
The lack of persistence of infused T cells is a principal limitation of adoptive immunotherapy in man. Interleukin (IL)‐15 can sustain memory T cell expansion when presented in complex with IL‐15Rα (15Rα/15). We developed a novel in‐vitro system for generation of stable 15Rα/15 complexes. Immunologically quantifiable amounts of IL‐15 were obtained when both IL‐15Rα and IL‐15 genes were co‐transduced in NIH 3T3 fibroblast‐based artificial antigen‐presenting cells expressing human leucocyte antigen (HLA) A:0201, β2 microglobulin, CD80, CD58 and CD54 [A2‐artificial antigen presenting cell (AAPC)] and a murine pro‐B cell line (Baf‐3) (A2‐AAPC15Rα/15and Baf‐315Rα/15). Transduction of cells with IL‐15 alone resulted in only transient expression of IL‐15, with minimal amounts of immunologically detectable IL‐15. In comparison, cells transduced with IL‐15Rα alone (A2‐AAPCRα) demonstrated stable expression of IL‐15Rα; however, when loaded with soluble IL‐15 (sIL‐15), these cells sequestered 15Rα/15 intracellularly and also demonstrated minimal amounts of IL‐15. Human T cells stimulated in vitro against a viral antigen (CMVpp65) in the presence of 15Rα/15 generated superior yields of high‐avidity CMVpp65 epitope‐specific T cells [cytomegalovirus‐cytotoxic T lymphocytes (CMV‐CTLs)] responding to ≤ 10− 13 M peptide concentrations, and lysing targets cells at lower effector : target ratios (1 : 10 and 1 : 100), where sIL‐15, sIL‐2 or sIL‐7 CMV‐CTLs demonstrated minimal or no activity. Both soluble and surface presented 15Rα/15, but not sIL‐15, sustained in‐vitro expansion of CD62L+ and CCR7+ central memory phenotype CMV‐CTLs (TCM). 15Rα/15 complexes represent a potent adjuvant for augmenting the efficacy of adoptive immunotherapy. Such cell‐bound or soluble 15Rα/15 complexes could be developed for use in combination immunotherapy approaches.
Keywords: adoptive immunotherapy, cancer immunotherapy, cytokine, immunomodulation, T cell memory, immune adjuvant
Introduction
The clinical success of adoptive immunotherapy has been hampered due to the limited persistence of infused self tumor antigen‐specific 1 or virus‐specific T cells 2 leading to recurrence of cancer or infection. TCM phenotype T cells expressing high levels of L‐selectin (CD62L), CCR7 and CD44 can home to and persist within lymphoid tissues, and therefore represent a desirable T cell population for adoptive immunotherapy that have the potential to provide durable protection from disease by virtue of their prolonged in‐vivo survival 3. In both animal models and humans, adoptively transferred TCM phenotype T cells directed against viral antigens such as cytomegalovirus (CMV) have demonstrated prolonged in‐vivo persistence and durable protection from infection 4, 5, 6. Common gamma‐chain cytokines, in particular interleukin (IL)−7 and IL‐15, can potentiate memory T cell survival and proliferation, respectively 7. Accordingly, cytokine cocktails incorporating IL‐7 and/or IL‐15 have been evaluated for their effect on supporting the in‐vitro expansion of memory phenotype antigen‐specific T cells for adoptive immunotherapy applications 8, 9.
Interleukin‐15 has been shown to be critical for the homeostatic proliferation of CD8+ memory T cells 10, 11, and it also functionally stimulates both memory T and natural killer (NK) cells 12, 13. Therefore, IL‐15 promises to be a valuable catalyst for augmenting the efficacy of adoptive immunotherapy. In animal models, IL‐15 treatment delivered by NK cells 14, intravenously 15, 16 or via transduced tumour cells 17, induced significant tumour regressions shown to be mediated by host‐derived or adoptively transferred CD8+ T cells and NK cells. Recent in‐vitro and animal model studies indicate that IL‐15 is most potent in stimulating CD8+ memory T cell and NK cell proliferation when it is bound exclusively with IL‐15Rα, forming an 15Rα/15 complex 18, 19. Such 15Rα/15 complexes, when infused into tumour‐bearing animals, have been shown to induce significant tumour regressions that are mediated by the sustained proliferation of memory CD8+ T cells 20, 21, 22, 23. These data suggest that 15Rα/15 would be a useful adjuvant for immunotherapy.
It is now recognized that both secreted and cell surface‐expressed forms of IL‐15 exist in complex with IL‐15Rα 24. These 15Rα/15 complexes can function in both cis and trans configurations and stimulate responding T and NK cells 25, 26. However, it remains unclear if the secreted 15Rα/15 differs from membrane bound 15Rα/15 in its functional effects on lymphocyte responses when exposed to antigen 27. To develop this agent appropriately for immunotherapy applications, we examined the soluble and membrane bound forms of 15Rα/15 in a series of in‐vitro experiments to determine the most functionally active form of 15Rα/15 that supports expansion of human antigen‐specific T cells. We developed and employed a novel cell based‐artificial antigen‐presenting cell (AAPC) system expressing human 15Rα/15, which permitted a controlled evaluation of soluble and membrane‐bound 15Rα/15 in comparison to soluble IL‐15 (sIL‐15). Genetically modified NIH 3T3‐based human leucocyte antigen (HLA) A2+ AAPC (A2‐AAPC) cell lines 28, as well as a third‐party murine pro‐B cell line Baf‐3 29, were transduced to co‐express either human IL‐15Rα alone or IL‐15Rα in complex with IL‐15 (A2‐AAPC15Rα, A2‐AAPC15Rα/15and Baf‐315Rα/15).
These studies established that co‐expression of IL‐15Rα and IL‐15 is essential for stable expression of 15Rα/15. Using cell lines transduced to co‐express IL‐15Rα and IL‐15, we examined the differential effects of soluble versus membrane‐bound 15Rα/15 in comparison to sIL‐15 in stimulating the in‐vitro expansion of memory phenotype epitope‐specific T cells in response to a viral antigen such as CMVpp65. We demonstrated that both soluble and secreted 15Rα/15 complexes can sustain the expansion of antigen‐specific TCM cells, more efficiently than soluble cytokine supplementation with IL‐15 or IL‐7. These data underscore the advantage of 15Rα/15 in stimulating the expansion of highly functional antigen‐specific TCM cells for adoptive immunotherapy applications. Such complexes could be harnessed for appropriate immunotherapy applications in conjunction with cell, vaccine or other immunomodulating agents.
Materials and methods
Donors
Blood was collected from six HLA A 02:01‐positive healthy, CMV‐seropositive, volunteer donors consenting to approved protocols by the Institutional Review Board at Memorial Sloan‐Kettering Cancer Center (MSKCC) after high‐resolution HLA typing (HLA Laboratory – MSKCC).
Generation of AAPC and Baf‐3 cells co‐expressing IL‐15Rα and IL‐15
Cloned plasmids encoding IL‐15 and IL‐15Rα genes and containing the CD8 leader sequence were inserted into SFG retroviral vectors at HindIII and BamHI sites and transduced sequentially into A2‐AAPC 55. The Kozak sequence (GCCGCCACC) inserted prior to the AUG initiator codon ensured enhanced expression of the transduced gene 56. IL‐15Rα transduced cells (A2‐AAPC15Rα) were isolated by fluorescence activated cell sorter (FACS) and stored [anti‐IL‐15Rα fluorescein isothiocyanate (FITC); BD Biosciences, San Jose, CA, USA]. Some aliquots of A2‐AAPC15Rα cells were then transduced with IL‐15, and cells expressing both IL‐15Rα and IL‐15 were cloned out by serial dilution. High‐expressing clones were isolated further by FACS [anti‐IL‐15 phycoerythrin (PE) and anti‐IL‐15Rα FITC; BD Biosciences], expanded in Dulbecco's modified Eagle's medium (DMEM; Invitrogen Inc., Carlsbad, CA, USA) + 10% heat‐inactivated defined calf serum (DCS; Hyclone, Logan, UT, USA) and stored in aliquots for T cell sensitization (A2‐AAPC15Rα/15). Similarly, the mouse pro‐B cell line Baf‐3 29, passaged in RPMI‐1640 with 10% fetal calf serum (FCS) (Life Technologies, Grand Island, NY, USA) was transduced sequentially with retroviral vectors containing the plasmid DNA for IL‐15Rα and IL‐15 genes (Baf‐315Rα/15), and irradiated aliquots were used in T cell cultures (Supporting information, Fig. S1).
Generation of CMV‐cytotoxic T lymphocytes (CMV‐CTLs)
T cells were enriched from Ficoll Hypaque separated peripheral blood mononuclear cells (PBMC) (Accurate Chemical and Scientific Corporation, Westbury, NY, USA) using immunomagnetic beads (Pan T cell Isolation Kit II; Miltenyi Biotec Inc., Auburn, CA, USA) 28. CMV‐CTLs were then generated as described previously 28 using A2‐AAPC at a stimulator to effector ratio of 1 : 10 in AIM‐V medium in eight different conditions: (1) A2‐AAPC + sIL‐2, (2) A2‐AAPC + sIL‐15, (3) A2‐AAPC + sIL‐2 + sIL‐15, (4) A2‐AAPC + sIL‐7 + sIL‐4, (5) A2‐AAPC15Rα + sIL‐2, (6) A2‐AAPC15Rα + sIL‐15, (7) A2‐AAPC15Rα/15 and (8) A2‐AAPC + Baf15Rα/15. T cells were restimulated every 10 days. T cells were supplemented with IL‐2 (20 U/ml) and or IL‐15 (10 ng/ml) or IL‐7 (10 ng/ml) + IL‐4 (1666 U/ml) (R&D Systems, Inc., Minneapolis, MN, USA) based on the assigned groups. Cytokines were first supplemented on day 8 and then three times per week. Group 8 received 1 × 106 irradiated Baf‐315Rα/15 cells at each restimulation, and group 7 was restimulated with A2‐AAPC15Rα/15 every 10 days without additional soluble cytokine supplementation.
Transwell T cell cultures
Parallel T cell co‐cultures were set up from three HLA‐A0201+ donors with irradiated A2‐AAPCs in Transwell tissue culture plates consisting of two chambers in each well separated by a 3 µm permeable membrane (Corning Costar #3414). The permeable membrane in each well allowed the passage of soluble cytokines as well as secreted soluble 15Rα/15, while separating the T cell co‐cultures from cell surface‐expressed 15Rα/15. In parallel co‐cultures, T cells stimulated with A2‐AAPCs were supplemented with (1) irradiated Baf‐315Rα/15 cells (106/ml), (2) irradiated A2‐AAPC15Rα/15 (106/ml), (3) sIL‐15 (10 ng/ml) or (4) sIL‐2 (20 units/ml). Soluble cytokines were added at day 8 and then thrice a week, and irradiated Baf‐315Rα/15 or A2AAPC15Rα/15 were replenished every 10 days.
Epstein–Barr virus (EBV)‐B lymphoblastoid cell lines (BLCLs)
Autologous EBV‐BLCLs were generated for each donor, as described previously 57. The cells were maintained in RPMI‐1640 + 10% FCS (Life Technologies, Grand Island, NY, USA).
CMV pp65 peptides
The HLA A 02:01 presented nonamer NLVPMVATV (NLV) within CMVpp65 was synthesized by the microchemistry and proteomics core facility at MSKCC, stored in small aliquots (2·4 µg/10 µl) and used to assess the responses in functional T cell assays.
Isolation and quantitation of IL‐15, IL‐15Rα and 15Rα/15 complexes
IL‐15 in all samples was quantitated by human IL‐15 Quantikine enzyme‐linked immunosorbent assay (ELISA) kit (R&D Systems, Inc., Minneapolis, MN, USA). Concentrated (3 kDa filtration units; Millipore Corp., Billerica, MA, USA) serum‐free cell supernatants (RPMI‐1640) were fractionated into 1 ml fractions running over a Superdex 200 10/30 column at 0·5 ml/min in 20 mM Tris, 50 mM NaCl, pH 8·0 buffer using a classic fast protein liquid chromatography (FPLC) system (GE Healthcare Bio‐Sciences Corp., Piscataway, NJ, USA). Bovine serum albumin (BSA) (66·4 kDa) and lysozyme (4·3 kDa) (1 mg/ml; Sigma‐Aldrich, St Louis, MO, USA), served as molecular weight (MW) markers (confirmed by Bradford protein assay and gel electrophoresis with Coomassie staining). FPLC fractions were analysed for IL‐15. Baf‐315Rα/15 supernatants were subjected to12·5% sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE) to distinguish free IL‐15 from 15Rα/15. Heat‐denatured, reduced and non‐reduced supernatants were then analysed by Western blot using anti‐human IL‐15 Rα and IL‐15 antibodies (R&D Systems, Inc.).
Phenotypical analysis of CMV‐CTLs
Quantitation of tetramer+ CD8+CMV‐CTLs. HLA A 02:01–NLV major histocompatibility complex (MHC)‐peptide tetramers (MSKCC tetramer core) were used to quantitate CMVpp65 NLV‐responsive T cells at days 0, 7, 14, 21 and 28 in culture, as described previously 28. HLA A 24:02‐QYDPVAALF and HLA B 07:02 TPRVTGGGAM peptide‐MHC tetramers (MSKCC tetramer core) were used as controls.
Memory phenotype of tetramer+ T cells. T cells were incubated with anti‐CD8 peridinin chlorophyll (PerCP), APC‐labelled tetrameric MHC–peptide complexes, anti‐CD62L FITC, anti‐CD45RA phycoerythin (PE) and anti‐CCR7 PE‐cyanin 7 (Cy7). CD8+ and Tet+ T cells were analysed to determine the proportion of CD45RA–CD62L+ or CCR7+ (TCM) or CD45RA–CD62L– or CCR7– (TEM). All antibodies for FACS analysis were purchased from BD Biosciences.
Cell proliferation and apoptosis
Carboxyfluorescein diacetate succinimidyl ester (CFSE) dilution assay. Day 14‐stimulated T cells were resuspended in phosphate‐buffered saline (PBS)/0·1% BSA at 107 cells/ml and incubated with a 5 mM dimethylsulphoxide (DMSO) stock solution of CFSE (Invitrogen) to achieve a final concentration of 10 μM CFSE for 10 min at 37°C. Labelled T cells were washed with 5 vol ice‐cold RPMI‐1640/10% FBS, incubated on ice for 5 min for quenching, then washed three times in T cell medium (AIM V + 5% DCS). Aliquots of 1–2 × 106/ml CFSE‐labelled T cells were then co‐cultured with irradiated A2 AAPC in separate six‐well plates supplemented with the same cytokines as previous stimulation: sIL‐2 (20 U/ml), sIL‐15 (10 ng/ml), sIL‐7 (10 ng/ml) + sIL‐4 (1666 U/ml), 1 × 106 irradiated Baf‐315Rα/15 or with irradiated A2‐AAPC15Rα/15. Primary T cells stimulated with CD3‐CD28 beads at a 1 : 1 ratio + 50 U/ml sIL‐2 served as positive control. CFSE‐labelled T cells were then stained with CD3, CD8 and A2‐NLV tetramer and analysed by FACS at 2 and 7 days in culture after CFSE labelling.
Apoptosis assay
Non‐viable T cells in the different culture conditions were assessed by FACS using the dead cell stain 7‐aminoactinomycin D (7AAD). Epitope specific A2‐NLV tetramer + T cells labelled with 7AAD were quantitated.
Functional analysis of CMV‐CTLs
T helper type 1 (Th1) cytokine generation. T cell responses to the nonamer peptide (NLV) were evaluated by quantitating interferon (IFN)‐γ+ CD8+ T cells upon secondary stimulation with peptide‐loaded autologous APCs (PBMC or BLCL), as described previously 57, 58. Autologous APCs loaded with serial dilutions of NLV peptide (10 nM–0·1 pM) were also used to elicit differential T cell responses.
Intracellular granzyme B. NLV peptide‐loaded autologous BLCL were co‐incubated with CMV‐CTLs for 4–6 h at a 5 : 1 responder to stimulator ratio in the presence of brefeldin A. Fluorescent antibody‐labelled T cells (anti‐CD3, CD4, CD8; BD Biosciences) were fixed, then permeabilized (BD Biosciences fix and perm kit) and labelled with anti‐human granzyme B antibody (GB11, eBiosciences, San Diego, CA, USA) and analysed by FACS.
In‐vitro cytotoxicity. T cell cytotoxic activity was evaluated in a standard in‐vitro 51Cr release assay 57. T cell targets included: autologous EBV‐BLCLs (1) loaded with titrated concentrations of the NLV peptide (2·4 µg–2·4 ng/106 EBV‐BLCLs), (2) loaded with 2·4 µg/106 EBV‐BLCLs at progressively diminishing effector : target (E : T) ratios, (3) NLV peptide‐loaded HLA mismatched EBV‐BLCL and (4) BLCL lines without peptide. Groups 3 and 4 served as controls.
Statistics
Wilcoxon's rank sum test was used to compare groups.
Results
Soluble IL‐15 augments expansion of CMV‐CTLs in vitro and prevents T cell apoptosis
Our goal has been to develop strategies for robust in‐vitro expansion of antigen‐specific T cells. We initially compared the effects of the prosurvival cytokine IL‐15 in comparison to IL‐2 on the enrichment and overall expansion of CMVpp65‐specific T cells in our AAPC model system. This panel of HLA class‐I‐expressing AAPCs is designed specifically for the expansion of CD8+ CMV‐CTLs responding to HLA class‐I‐presented epitopes 28. To generate CMV‐CTLs, T cells from six healthy CMV‐seropositive HLA A02:01+ donors were stimulated using A2‐AAPC and supplemented with either sIL‐2 (20 U/ml) or sIL‐15 (10 ng/ml). Using this approach, CTLs supplemented with sIL‐15 demonstrated a steady enrichment through 28 days of epitope‐specific T cells responding to the HLA A02:01‐presented NLV epitope in MHC‐peptide tetramer binding assays. Strikingly, sIL‐15 supplementation maintained a high proportion of Tet+ T cells even beyond 21 days of continuous antigenic stimulation (Fig. 1a shows one representative example). In comparison, the enrichment of Tet+ T cells in sIL‐2‐supplemented CMV‐CTLs peaked at 21 days, after which Tet+ T cells underwent an attrition in both proportion and numbers between 21 and 28 days (Fig. 1b). As a result, sIL‐15 generated a significantly higher overall yield of Tet+ T cells with a median of 1·8 × 107 compared to 3·4 × 106 Tet+ T cells in sIL‐2 CTLs (P < 0·01) (Fig. 1b,c), providing a median fold expansion of 900 versus 375 (Table 1). This also correlated with proportionately lower numbers of 7AAD+ apoptotic T cells observed in sIL‐15 CTLs compared to sIL‐2 CTLs (3–5% and 24–32%, respectively) (P < 0·001) (Fig. 1d). We examined simultaneously combinations of γ‐chain cytokines for their effect on overall yields of Tet+ T cells. When sIL‐15 was supplemented together with sIL‐2, an augmented yield of Tet+ T cells was achieved at 28 days in comparison to sIL‐2 CTLs, but the yield remained below that obtained with sIL‐15 alone (median = 1 × 107 and 1·8 × 107, respectively or 550‐ versus 900‐fold expansion with sIL‐15 alone) (P < 0·01) (Fig. 1b,c). We then also examined sIL‐7 + sIL‐4 in three separate T cell donors based on previously reported T cell expansion in short‐term in‐vitro cultures 8. As shown in Fig. 1a, although this combination led to an excellent overall T cell expansion CTLs expanded in the presence of sIL‐7 and sIL‐4 contained a sizable proportion of CD4+ T cells (38–51%). Importantly, enrichment of Tet+ T cells was achieved in these cultures within the first 15–21 days that then reached a plateau between 21 and 28 days. This resulted in an overall higher yield of Tet+ T cells with sIL‐7 + sIL‐4 than in sIL‐2‐supplemented CTLs, but also remained lower than in sIL‐15‐only CTLs in our AAPC system, which fosters expansion of CD8+ T cells (Fig. 1b,c). The proportion of apoptotic T cells in sIL‐7 + sIL‐4 cultures was low, as with sIL‐15‐supplemented CTLs (Fig. 1d,e). Overall, in this in‐vitro system, supplementation with sIL‐15 demonstrated the most robust CTL expansion.
Figure 1.
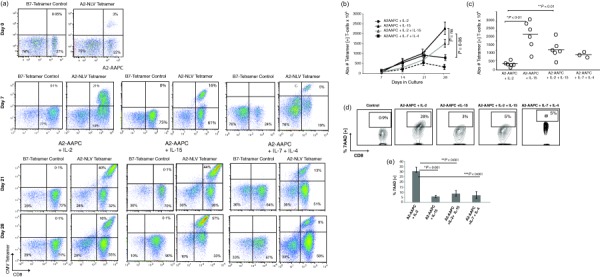
Soluble interleukin (IL)−15 augments expansion of cytomegalovirus‐specific cytotoxic T lymphocytes (CMV‐CTLs) in vitro and prevents T cell apoptosis. T cells from parallel co‐cultures of A2‐artificial antigen‐presenting cells (AAPCs) supplemented with either soluble interleukin (sIL)‐2, sIL‐15 or sIL‐7 + sIL‐4 were incubated with anti‐CD3 fluorescein isothiocyanate (FITC), anti‐CD8 peridinin chlorophyll (PerCP) (BD Biosciences) and antigen‐presenting cell (APC)‐conjugated major histocompatibility complex (MHC)–peptide tetrameric complex (20 min at 4ºC). Data were acquired by fluorescence activated cell sorter (FACS) (LSR‐II flow cytometer; BD Biosciences) and analyzed using FlowJo software (Tree Star Inc.). CD3+, CD8+ gated T cells were analysed for percentage of CD8+ Tet+ T cells binding the A2‐NLVPMVATV (NLV) tetramer in each culture. (a) CD8+ Tet+ T cells at day 7 (upper panel), 21 and 28 (lower panel). (b) The total yield of Tet+ T cells was calculated from the percentages of CD8+ Tet+ T cells within the total CD3+ T cells. The number of Tet+ T cells present at 7, 14, 21 and 28 days is plotted. (c) The total yield of Tet+ T cells at day 28 is plotted for each donor in each cytokine condition to determine differences in total yields of Tet+ T cells between sIL‐2 and sIL‐15 (P < 0·01). (d, e). The proportion of apoptotic T cells within A2‐AAPC‐sensitized T cells supplemented with either sIL‐2, sIL‐15, sIL‐2 + sIL‐15 or sIL‐7 + sIL‐4 were analysed using FACS after labelling with 7‐aminoactinomycin D (7AAD). Analysis was performed 3 days after each A2‐AAPC restimulation, and 2 days after cytokine supplementation to avoid including cell death resulting from depletion of alloreactive cells after restimulation or from activation‐induced cell death (AICD). (d) CD8+ 7AAD + T cells are shown in a representative donor and (e) among all donors tested.
Table 1.
Summary of in‐vitro analysis of T cells cultured under different cytokine conditions
| Culture condition | Fold expansion Tet+ CD8+ | Fold expansion Tet[+] CD62L[+] CD8+ | IFN‐γ+CD8[106] NLV | % in vitro cytotoxicity | ||
|---|---|---|---|---|---|---|
| nM | 0.1pM | E : T = 1 : 1 | E : T = 1 : 10 | |||
| A2‐AAPC+ IL‐2 | 200–600 | 0 | 1–2 | 0 | 12–21 | 0 |
| A2‐AAPC+ IL‐15 | 300–1300 | 3–5 | 2–4 | < 1–2 | 15–23 | 0 |
| A2‐AAPC+ IL‐2+ IL‐15 | 250–750 | 0 | 1–3 | < 1 | 11–19 | 0 |
| A2‐AAPC+ IL‐7+ IL‐4 | 330–675 | 7–11 | 1–4 | 1–2 | 17 −21 | 3 |
| A2‐AAPCIL‐15Rα+ IL‐2 | 25–100 | 0 | < 1–2 | 0 | 8–14 | 0 |
| A2‐AAPCIL‐15Rα+ IL‐15 | 100–300 | 7–10 | 1–4 | 1–2 | 13–24 | 3–5 |
| A2‐AAPCIL‐15Rα/IL‐15 | 1200–2300 | 600–1000 | 10–16 | 7–12 | 52–73 | 12–20 |
| A2‐AAPC + Baf‐3IL‐15Rα/IL‐15 | 1100–1600 | 550–700 | 10–14 | 7–10 | 40–60 | 16–25 |
A2‐AAPC = A2‐artificial antigen‐presenting cells; IFN = interferon; IL = interleukin.
Generation of an AAPC system providing IL‐15Rα/IL‐15 complex for robust expansion of antigen‐specific T cells requires both IL‐15 and IL‐15Rα genes
Previous studies have shown that the stimulatory effect of IL‐15 on CD8+ memory T cell expansion is mediated through the 15Rα/15 complex, which is also expressed on DCs. We therefore sought to develop an off‐the‐shelf APC system providing molecules for both in‐vitro expansion of antigen‐specific T cells as a strategy to provide potentially superior and more physiological T cell stimulation. The requisite in‐vitro conditions for the formation and cell surface expression of 15Rα/15 were examined initially. We transduced A2AAPC as well as Baf‐3 cells with the IL‐15 gene alone and evaluated the expression and secretion of IL‐15. In several independent experiments, IL‐15 transduced cells lost expression after a few in‐vitro passages, and minimal amounts of IL‐15 (64–145 pg/ml) were detected in the supernatants of these cells by ELISA (Fig. 2a). This suggested that the IL‐15 gene is unstable when transduced alone, and requires IL‐15Rα to form a stable complex. Thereafter, A2 AAPC transduced with IL‐15Rα alone (A2‐AAPC15Rα) were generated, which demonstrated stable expression of IL‐15Rα. These cells were then loaded with saturating doses of sIL‐15 (10–50 ng/ml) to evaluate the expression of 15Rα/15 and secretion of IL‐15. Surprisingly, sIL‐15‐loaded A2‐AAPC15Rα cells also demonstrated a markedly lower level of immunologically detectable IL‐15 (94–270 pg/ml IL‐15) in comparison to A2‐AAPC supernatants supplemented with the same concentrations of sIL‐15 (6000–10 000 pg/ml) (Fig. 2b), and did not express 15Rα/15 on the cell surface. In T cell co‐cultures, sIL‐15‐loaded A2‐AAPC15Rα elicited a lower yield of epitope‐specific Tet+ T cell numbers compared to sIL‐15 supplemented A2‐AAPC (Fig. 2c). To elucidate reasons for lower IL‐15 concentrations detected in A2‐AAPC15Rα cells, we performed time–sequence studies quantitating cell surface‐expressed IL‐15 and observed that all detectable IL‐15 was intracellular, suggesting that A2‐AAPC15Rα cells bound and rapidly internalized the supplemented sIL‐15 from the cell medium, without recycling for surface presentation (data not shown). The inferior T cell expansion in A2‐AAPC15Rα co‐cultures was therefore ascribed to the non‐ availability of IL‐15 due to intracellular sequestration within these AAPCs. Although, in other systems, IL‐15Rα‐expressing cells loaded with sIL‐15 have demonstrated surface expression of 15Rα/15 complexes 30, these data suggested that, in this system, both IL‐15 and IL‐15Rα genes would be required within the same cell for secretion of IL‐15 and stable expression of 15Rα/15 complexes. Accordingly, we generated AAPC transduced to express both IL‐15 and IL‐15Rα genes (A2‐AAPC15Rα/15). These cells demonstrated high expression levels of 15Rα/15 complex on the cell surface (Supporting information, Fig. S1) and also secreted detectable quantities of IL‐15 by ELISA (3000–6000 pg/ml of IL‐15) (Fig. 2a).
Figure 2.
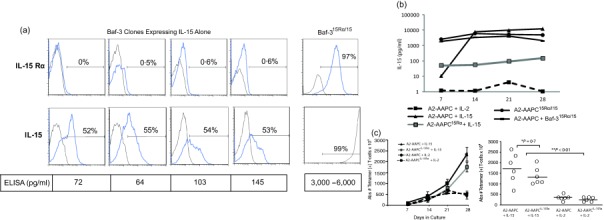
Artificial antigen‐presenting cells (AAPCs) genetically modified to co‐express interleukin (IL)−15Rα and IL‐15 secrete IL‐15 and are potent stimulators of antigen‐specific T cell expansion. (a) Baf 3 cells not expressing IL‐15Rα (top panel) were sorted and then transduced with the IL‐15 gene alone. IL‐15 expressing Baf‐3 cells were cloned by limiting dilution, and individual clones were then analysed for intracellular expression of IL‐15 protein by fluorescence activated cell sorter (FACS) after 2, 5 and 7 passages (lower panel). The IL‐15 expression within Baf‐3 cells expressing IL‐15 alone was compared to Baf‐3 cells co‐expressing IL‐15Rα and IL‐15. (b) The cell culture supernatants from A2‐AAPCIL‐15Rα and A2‐AAPC co‐incubated with sIL‐15 (10–50 ng/ml) were analysed for IL‐15 in an enzyme‐linked immunosorbent assay (ELISA) 10–30 min after IL‐15 supplementation. Parallel analysis was performed for A2‐AAPC15Rα/15and Baf‐315Rα/15containing 106 cells/ml. (c) Parallel in‐vitro T cell cultures stimulated with A2‐AAPC and A2‐AAPCIL‐15Rα supplemented with either soluble IL‐2 or IL‐15 were established. Total yield of Tet+ T cells (analysed by FACS) at 7, 14, 21 and 28 days (left) is shown (error bars = standard error of the mean). The scattergraph (right) shows the overall yields of Tet+ T cells at 28 days after culture initiation for each of the six donors tested. The horizontal line = median. sIL‐15 supplemented T cells stimulated with A2‐AAPC or A2‐AAPCIL‐15Rα generated similar yields of Tet+ T cells (P = 0·7), while soluble IL (sIL)‐2‐supplemented cytotoxic T lymphocytes (CTLs) elicited significantly lower yields of Tet+ T cells (P < 0·01).
IL‐15 detected in the supernatants of A2‐AAPC15Rα/15, Baf‐315Rα/15 and A2‐AAPC15 Rα is bound predominantly to IL‐15Rα
Studies in various mouse and in‐vitro models have suggested that IL‐15 exists preferentially as a complex bound to IL‐15Rα. We examined if this was true for the genetically modified cells expressing both human IL‐15 and IL‐15Rα genes (A2‐AAPC15Rα/15 and Baf‐315Rα/15) and compared this to sIL‐15‐loaded A2‐AAPC15Rα. In Western blot analysis, performed on concentrated cell supernatants that had retained all detectable IL‐15 (see Methods), both IL‐15 and IL‐15Rα proteins were detected as a high molecular weight (HMW) band under non‐reducing conditions in Baf‐315Rα/15, A2‐AAPC15Rα/15 and A2‐AAPC15Rα cultures (Fig. 3a). Upon fractionation of the concentrated supernatants and FPLC analysis, we confirmed that the immunologically detectable IL‐15 was present exclusively in the HMW fractions (Fig. 3b). Nevertheless, in sIL‐15 supplemented supernatants of A2‐AAPC, IL‐15 was detected only in low molecular weight (LMW) fractions (Fig. 3c). Based on these data, we inferred that IL‐15 existed as a complex with IL‐15Rα in both Baf‐315Rα/15 and A2‐AAPC15Rα/15 (Fig. 3b,c).
Figure 3.
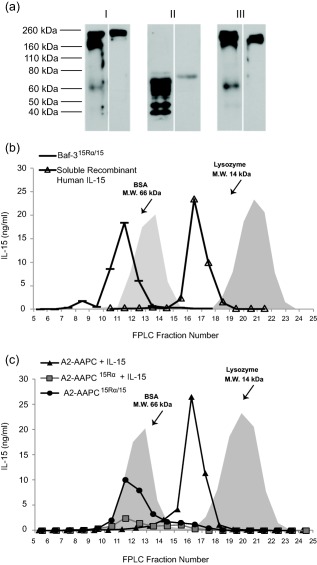
Interleukin (IL)‐15 detected in the supernatants of A2‐artificial antigen‐presenting cells (AAPCs)15Rα/15, Baf‐315Rα/15 and A2‐AAPCIL‐15 Rα is bound predominantly to IL‐15Rα. Concentrated supernatant samples were analysed by Western blot under non‐reducing non‐heat‐denaturing [no dithiothreitol (DTT), 100' at room temperature]; reducing, heat‐denaturing conditions (50 m M DTT, 10' at 95ºC or 98ºC); non‐reducing, heat‐denaturing conditions (no DTT, 10' at 95ºC). (a) Representative Western blots of Baf‐315Rα/15 supernatants are shown. Baf‐315Rα/15 cells were first incubated in serum‐free RPMI for 48 h, then 20 µl of concentrated supernatant was subjected to 12·5% sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE) under: (i) non‐reducing, non‐heat‐denaturing conditions; (ii), reducing, heat‐denaturing; and (iii), non‐reducing, heat‐denaturing conditions. 15Rα/15 complex and IL‐15Rα were detected using antibody against IL‐15Rα (left panels) and against IL‐15 (right panels). (b) Baf‐315Rα/15 cells were incubated in serum‐free RPMI for 24 h, filtered and concentrated. Serum free (RPMI‐1640; Life Technologies) cell supernatants were concentrated 14–20‐fold using 3 kDa filtration units (Millipore Corporation). One‐ml fractions of the supernatants were obtained a classic fast protein liquid chromatography (FPLC) system. Recombinant human soluble IL‐15 (10 ng/ml) (R&D Systems) in RPMI was prepared in parallel. Conditioned media (Baf‐315Rα/15 supernatants and sIL‐15 10 ng/ml) was run through the FPLC system using bovine serum albumin (BSA) [molecular weight (MW) 66 kDa] and lysozyme (MW 14 kDa) as MW markers. IL‐15 was detected in each fraction by enzyme‐linked immunosorbent assay (ELISA). FPLC fractions (volumes 8–30 ml, ranging from retention volumes below BSA and above lysozyme) were analysed for IL‐15. As shown, all IL‐15 activity in Baf‐315Rα/15supernatants was detected in fractions containing molecules greater than 66 kDa MW (BSA). Medium containing recombinant human sIL‐15 was detected in fractions comparable to MW of lysozyme (14 kDa). (c) Concentrated supernatants from A2‐AAPC15Rα/15and A2‐AAPCIL‐15Rα or sIL‐15 (10 ng/ml)‐loaded A2‐AAPC were run in parallel through the FPLC system using BSA and lysozyme as MW markers, and fractions analysed for IL‐15 by ELISA. In both A2‐AAPC15Rα/15and sIL‐15‐loaded A2‐AAPCIL‐15Rα, IL‐15 was detected exclusively in the high MW fractions > 66 kDa (BSA). In contrast, IL‐15 detected in sIL‐15‐loaded A2‐AAPC was exclusively in the low MW fractions ∼ 16 kDa, similar to the peak for recombinant human IL‐15.
AAPC co‐expressing IL‐15Rα and IL‐15 support continuous enrichment of antigen‐specific CD8+ T cells during prolonged in‐vitro expansion
We next wished to compare the enrichment of antigen‐specific T cells when stimulated in the presence of 15Rα/15 complexes versus sIL‐15 or sIL‐2. CMV‐CTLs from six seropositive donors were expanded in vitro in parallel co‐cultures with A2 AAPC15Rα/15 with A2 AAPC supplemented with sIL‐2 or sIL‐15. As shown in a representative example in Figs 4a and 1a, in the first 7 days after culture initiation we observed a lower proportion of Tet+ T cells within A2‐AAPC15Rα/15‐stimulated T cells (5·8%) compared to sIL‐15 or sIL‐2 supplemented A2‐AAPC T cell cultures (21 and 16%, respectively, Fig. 1a). However, after the initial week, A2‐AAPC15Rα/15 sensitized T cells demonstrated robust enrichment of NLV epitope‐specific Tet+ T cells from 5·8 to 92% at 28 days, thus achieving the highest enrichment within all conditions. This enhanced enrichment of Tet+ CMV‐CTLs with A2‐AAPC15Ra/15 was confirmed in triplicate analyses of CTLs from each donor (P < 0·01). In T cell proliferation assays measuring CFSE dilution, Tet+ T cells within T cells stimulated with A2‐AAPC15Ra/15 or with A2‐AAPC + Baf‐315Ra/15 demonstrated a higher proliferative rate compared to sIL‐15, sIL‐7 or sIL‐2 supplemented T cells. A higher proliferation of TetNeg T cells was also observed within A2‐AAPC + Baf‐315Ra/15 and IL‐7+ IL‐4‐stimulated T cells. However, for A2‐AAPC+ Baf‐315Ra/15, the proliferation of Tet+ T cells remained higher than the TetNeg T cells (Fig. 4c). The delayed enrichment of Tet+ T cells with A2‐AAPC15Rα/15 could therefore be attributed to early non‐specific expansion of T cells mediated by the 15Rα/15 complexes. Expansion of non‐specific T cells within sIL‐7+ sIL‐4‐stimulated T cells would also explain the lower enrichment of Tet+ T cells compared to IL‐15‐stimulated T cells.
Figure 4.
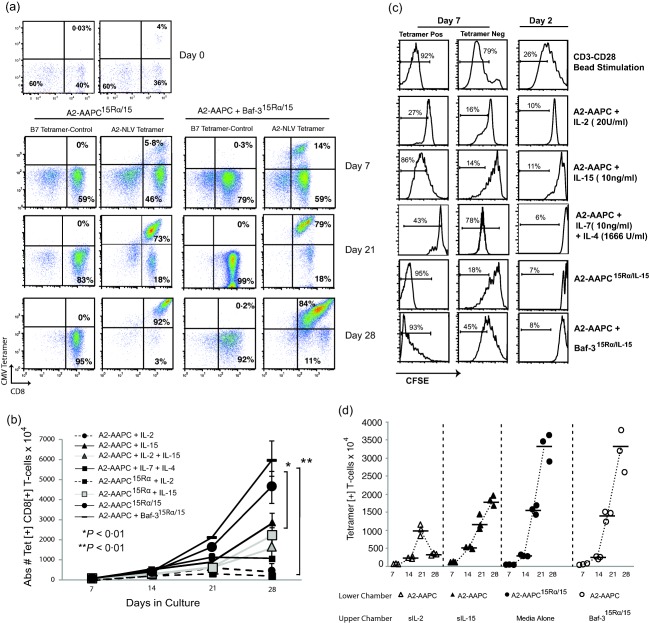
Artificial antigen‐presenting cells (AAPCs) co‐expressing interleukin (IL)−15Rα and IL‐15 support continuous enrichment of antigen‐specific CD8+ T cells during prolonged in‐vitro expansion. T cells from human leucocyte antigen (HLA) A 02:01+ and cytomegalovirus (CMV)‐seropositive donors were sensitized in parallel using (a) A2‐AAPC15Rα/15and A2‐AAPC+ Baf‐315Rα/15with no exogenously supplemented cytokines. Tet+ T cells were quantitated by fluorescence activated cell sorter (FACS) analysis at 7, 21 and 28 days after incubation with anti‐CD3, anti‐CD8 and A2‐NLV tetrameric complexes at 4ºC for 20 min. (b) The mean total yield of Tet+ T cells calculated after FACS analysis is plotted for each time‐point (error bars = standard error of the mean). For cultures sensitized with either A2‐AAPC15Rα/15 or A2‐AAPC+ Baf‐315Rα/IL‐15, the yield of Tet+ T cells was 5–6 × 107 compared to 1·8–2·3 × 107 for T cells sensitized with A2‐AAPC or A2‐AAPCIL‐15Rα and supplemented with soluble IL (sIL)−15 (P < 0·01). (c) T cells stimulated for 14 days with A2‐AAPC15Rα/15, A2‐AAPC+ Baf‐315Rα/IL‐15, sIL‐2, sIL‐15 or sIL‐7 + sIL‐4‐loaded A2‐AAPC were labelled with carboxyfluorescein succinimidyl ester (CFSE), and then further stimulated for 5 days in the same condition: i.e. with A2‐AAPC15Rα/15, A2‐AAPC+ Baf‐315Rα/IL‐15 or sIL‐2, sIL‐15 or sIL‐7 + sIL‐4‐loaded A2‐AAPC. sIL‐2‐loaded A2‐AAPC T cells stimulated with CD3/CD28 beads (1 : 1) were used as a positive control. T cells in each condition were then stained with CD3 fluorescein isothiocyanate (FITC), CD8 phycoerythrin (PE) and A2‐NLVPMVATV (NLV) antigen‐presenting cell (APC) tetrameric complexes and analysed by FACS. CFSE dilution was analysed within A2‐NLV Tet+ T cells as well as TetNeg CD8+ T cells to compare the proliferative potential of antigen‐specific and non‐specific CD8+ T cells in each condition. (d) T cells from three HLA A2+ donors were co‐cultured in six Transwell plates containing a 3‐µm permeable membrane with (i) A2‐AAPC supplemented with either sIL‐2 or sIL‐15 or Baf315Rα/15 or A2‐AAPC15Rα/15 separated from T cell co‐cultures by the permeable membrane and (ii) A2‐AAPC15Rα/15co‐cultured with T cells in direct contact. The proportion of antigen‐specific T cells in each culture condition were quantitated at 7, 14, 21 and 28 days by tetramer analysis and the total yield of tetramer+ T cells, calculated based on the proportion within the total CD3+ T cells is shown.
Soluble and membrane‐bound 15Rα/15 complexes are equally efficient in stimulating high proportions of antigen‐specific T cell expansion
Previous work has demonstrated that 15Rα/15 complexes, either expressed on cells or secreted, can engage responsive CD8+ T cells 18, 31. Thus far, we could demonstrate secretion of significant quantities of IL‐15, existing predominantly as a stable 15Rα/15 complex, in cell supernatants of A2‐AAPC15Rα/15 and Baf‐315Rα/15. We next examined whether 15Rα/15 complexes presented on neighbouring non‐APC cells or soluble/secreted complexes could mediate the same effects as APC‐expressed 15Rα/15. We established parallel T cell co‐cultures with A2‐AAPC in Transwell culture plates where the supplemented cytokines were separated from the T cell co‐cultures by a 3 μm permeable membrane that would permit the diffusion of soluble cytokines (sIL‐15, sIL‐2) and secreted 15Rα/15 complexes from Baf‐315Rα/15 or A2‐AAPC15Rα/15, but would not enable cellular contact with the membrane‐bound 15Rα/15 complexes. Within T cells stimulated by A2‐AAPCs in the presence of soluble 15Rα/15 permeating through the Transwell membrane, we observed a significantly higher enrichment of Tet+ T cells compared to sIL‐2 or sIL‐15 supplemented T cells (P < 0·01). These yields were similar to the overall yields of Tet+ T cells obtained with CMV‐CTLs generated by direct co‐culture with A2‐AAPC15Rα/15 (Fig. 4d).
15Rα/15 stimulation supports the expansion of central memory phenotype antigen‐specific T cells
The above data demonstrated clearly that 15Rα/15 supported superior enrichment of antigen‐specific T cells. For adoptive immunotherapy applications, we asked if 15Rα/15 could potentiate the enrichment antigen‐specific T cells bearing a central memory phenotype that would have longer in‐vivo persistence after infusion. We examined the expression of CD62L and CCR7 within A2‐NLV Tet+ T cells expanded in vitro under different cytokine conditions. As with sIL‐2, within the first 14 days all sIL‐15 CMV‐CTLs also had minimally detectable proportions of CD62L+ and CCR7+ T cells (Fig. 5a), but because of the overall T cell stimulatory effects of sIL‐15, these residual CD62L+/CCR7+ Tet+ T cells (TCM) expanded three‐ to fivefold between 14 and 21 days in culture (Table 1), at which time no Tet+ TCM cells could be detected in sIL‐2 CTLs. In contrast, A2‐AAPC15Rα/15 stimulated CMV‐CTLs demonstrated a sustained expansion of Tet+ TCM through 28 days (Fig. 5a), resulting in a 600–1000 fold expansion (Table 1), and a total yield at 21 days of 2–3 × 106 and approximately 5 × 106 by 28 days. These yields of Tet+ TCM were significantly higher than in sIL‐15 CTLs, which generated only 0·5–1 × 106 at 21 days and 1·5 × 106 Tet+ TCM at 28 days (P < 0·01). In a representative example shown (Fig. 5b), 15Rα/15‐stimulated T cells (A2‐AAPC15Rα/15 or A2‐AAPC + Baf‐315Rα/15) maintained a sizable proportion of Tet+ CD62L+ T cells even at later time‐points between 21 and 28 days after initial stimulation, ranging from 16 to 36%, suggesting a role for 15Rα/15 complexes in sustaining TCM expansion during continuous antigenic stimulation. Of note, we also observed expansion of CD62L+ and CCR7+ Tet+ TCM cells within with sIL‐7 + sIL‐4 CTLs, which was intermediate between sIL‐15‐ and 15Rα/15‐stimulated T cells (Fig. 5a). The Tet+ T cells expanded in the presence of sIL‐7+ sIL‐4 demonstrated a higher proportion of CD62L+ at day 21 that was comparable to 15Rα/15 and much higher than sIL‐15 and sIL‐2 stimulated T cells. However, by day 28, the highest proportion of CD62L+ Tet+ TCM cells were elicited within 15Rα/15 stimulated T cells, as shown in a representative example (Fig. 5b).
Figure 5.
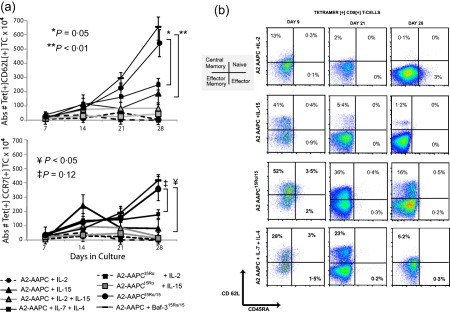
15Rα/15 stimulation endorses the expansion of central memory phenotype antigen‐specific T cells. T cell memory phenotype was evaluated after 7, 14, 21 and 28 days in culture for each culture condition using CCR7 and CD62L as markers of central memory phenotype (TCM). T cells sensitized for 21–28 days under the different culture conditions were labelled with immunofluorescent antibodies: anti‐CD3 phycoerythrin (PE), anti‐CD8 peridinin chlorophyll (PerCP), anti‐CD62L fluorescein isothiocyanate (FITC) and anti‐CCR7 PE‐cyanin‐7 (Cy7) and antigen‐presenting cell (APC)‐labelled A2‐NLVPMVATV (NLV) tetrameric complexes for 20 min at 4ºC and analysed by fluorescence activated cell sorter (FACS). CD8+ Tet+ T cells were gated to determine the proportion of antigen‐specific T cells expressing CD62L and CCR7. T cells labelled with HLA B 07:02‐TPR tetramers and unstained tubes served as controls for CD62L and CCR7. The total yield of CD62L+/CCR7+ Tet+ T cells was calculated based on the proportion of each population within CD3+ T cells. (a) CD62L+/CCR7+ Tet+ T cells at 7, 14, 21 and 28 days is shown for each donor in each culture condition (error bars = standard error of the mean). (b) A representative example demonstrating the proportion of CD62L+/CD45RA– Tet+ T cells detected at 21 days (left panel) and 28 days (right panel) of culture initiation for each culture condition is shown.
15Rα/15 complexes support the generation of high‐avidity antigen‐specific T cells
We next evaluated the effect of 15 Rα/15 complexes on the functional capacity of CMV‐CTLs in comparison to sIL‐15. T cell cytokine secretion was examined initially 21 days after stimulation in response to secondary stimulation with 10 nM NLV‐loaded autologous APCs. As shown in one representative donor (Fig. 6a), 15Rα/15‐stimulated T cells (A2‐AAPC15Rα/15 and Baf‐315Rα/15) elicited a markedly higher proportion of IFN‐γ+ CD8+ T cells (43·4 and 32·4%) compared to sIL‐15‐stimulated T cells with either A2‐AAPC or A2‐AAPC15Rα (19·2 and 22·6%). sIL‐2‐supplemented CMV‐CTLs elicited lower proportions of NLV‐responsive IFN‐γ+ CD8+ T cells with either A2‐AAPC or A2‐AAPC15Rα stimulation (9·7 and 3·7%), which could be augmented with additional sIL‐15, but the yields were still lower than those achieved within sIL‐15 alone supplemented T cells (15·5 versus 19·2%) (Fig. 6a). Overall, 15Rα/15‐stimulated T cells (A2‐AAPC15Rα/15 and Baf‐315Rα/15) produced the highest yield of NLV‐responsive IFN‐γ+ CD8+ T cell numbers, generating a median of 1 × 107 and 8·3 × 106 epitope‐specific T cells, respectively, compared to a median of 1–3 × 106 IFN‐γ+ CD8+ T cells in other conditions (P < 0·001) (Fig. 6b and Table 1). T cells stimulated with soluble, secreted 15Rα/15 complexes delivered via a permeable membrane also demonstrated similarly high proportions of IFN‐γ+ CD8+ T cells in response to NLV peptide (Fig. 6c) to those observed in T cells stimulated by direct co‐culture with A2‐AAPC15Rα/15 and Baf‐315Rα/15.
Figure 6.
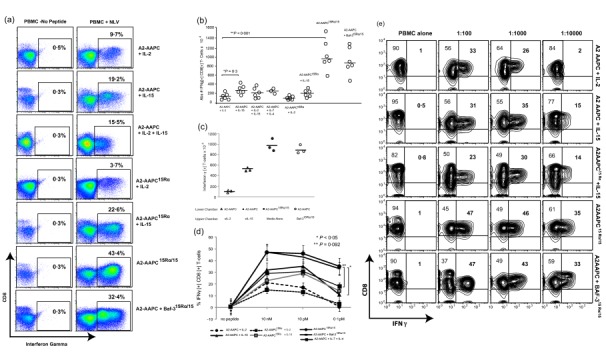
15Rα/15 complexes support the generation of high‐avidity antigen‐specific T cells. The proportion of CD8+ interferon (IFN)‐γ+ T cells responding to the cytomegalovirus (CMV)pp65 epitope NLVPMVATV (NLV) presented by human leucocyte antigen (HLA) A 02:01 were quantitated on day 21 for each parallel culture condition. (i) A2‐artificial antigen‐presenting cells (AAPCs)+ soluble interleukin (sIL)−2 (20U/ml) or sIL‐15 (10 ng/ml), sIL‐2 + sIL‐15 or sIL‐7 (10 ng/ml) + sIL‐4 (1666 U/ml); (ii) A2‐AAPCIL‐15Rα + sIL‐2 or sIL‐15; and (iii) A2‐AAPC15Rα/15 or A2‐AAPC + Baf‐3IL‐15Rα/IL‐15, with no exogenous cytokines. Aliquots of autologous peripheral blood mononuclear cells (PBMC) were loaded (37ºC × 3 h) with serial dilutions of NLV peptide (10 nM, 10 pM, 0·1pM), and co‐incubated with T cells at a responder : target ratio of 5 : 1 × 12 h in the presence of brefeldin A (BFA). T cells were labelled with immunofluorescent antibodies against CD3, CD4, CD8, fixed and then permeabilized (fix and perm kit; Invitrogen) and then incubated with anti‐human IFN‐γ fluorescein isothiocyanate (FITC). Data were acquired on a BD LSRII flow cytometer and analysed using FlowJo software. (a) One representative example demonstrating the proportion of IFN‐γ+ CD8+ T cells in response to 10 nM peptide‐loaded targets within CD3+ T cells is shown. (b) The total yield of IFN‐γ+ CD8+ T cells generated in response to 10 nM peptide was calculated from the percentage of IFN‐γ+ CD8+ T cells and plotted for each donor in each culture condition. (c) T cells from three separate HLA A2 + donors that were sensitized in six‐well Transwell plates according to cytokine conditions providing sIL‐2, sIL‐15 or 15Rα/15 complexes via the permeable transmembrane. Antigen‐specific T cells generating functional cytokines in response to 10 nM NLV peptide were evaluated on day 21 to quantitate the proportion of NLV‐specific CD8+ IFN‐γ+ T cells. (d) After 21 days of stimulation, the proportion of IFN‐γ+ CD8+ T cells elicited upon secondary stimulation with autologous targets loaded with serial dilutions of NLV peptide is shown for each donor in each culture condition (error bars = standard error of the mean), and (e) in one representative donor, IFN‐γ+ CD8+ T cells elicited in response targets loaded with serial peptide dilutions is shown. The proportion IFN‐γ+ CD8+ T cells in 15Rα/15‐stimulated T cells was significantly greater than sIL‐2 or sIL‐15 cultures at all peptide dilutions (P = 0·001). There was a significant reduction in the proportion of IFN‐γ+ CD8+ T cells at 10 pM versus 0·1 pM peptide concentrations for sIL‐15 cultures (P < 0·05).
To delineate further the most functionally avid T cells, we examined T cell cytokine secretion in response to titrated doses of the NLV peptide (10 nM, 10 pM, 0·1 pM). In these studies, significant differences in T cell responses could be discerned only at peptide concentrations of ≤ 10−13 M (0·1 pM) within T cells expanded under different cytokine conditions (Fig. 6d). At higher peptide concentrations, there were minimal differences in T cell responses in any of the cytokine conditions. In a representative example (Fig. 6e), at 10−13 M peptide, no responses were elicited in sIL‐2‐supplemented T cells, while 15Rα/15‐stimulated T cells (A2‐AAPC15Rα/15 or A2‐AAPC + Baf‐315Rα/15) elicited robust IFN‐γ + CD8+ T cell responses. At 10−13 M peptide, diminishing responses were elicited in T cells supplemented with sIL‐15 as well as sIL‐7 + sIL‐4, with 10 and 14% IFN‐γ+ CD8+ T cells compared to 23 and 32% enumerated in response to 10−12 M peptide in sIL‐15 CTLs (P < 0·05) (Fig. 6d,e).
15Rα/15‐stimulated antigen‐specific T cells lyse targets efficiently at lower E : T ratios
We then evaluated the T cell cytotoxic activity of CMV‐CTLs as another differentiating parameter of functional activity. Lysis of autologous targets loaded with titrated doses of the NLV peptide and at graded E : T ratios was examined. At concentrations ≥ 0·1 nM, all IL‐15‐supplemented CTLs lysed equally the peptide‐loaded autologous targets without exhibiting any explicit cytotoxicity hierarchy. T cells supplemented with sIL‐7 + sIL‐4 also demonstrated similar cytotoxic activity at graded peptide concentrations. In comparison, sIL‐2‐supplemented CTLs exhibited inferior cytotoxicity at all peptide concentrations (P < 0·05) (Fig. 7a). Peptide concentrations lower than 0·1 nM did not elicit CTL toxicity in any condition.
Figure 7.
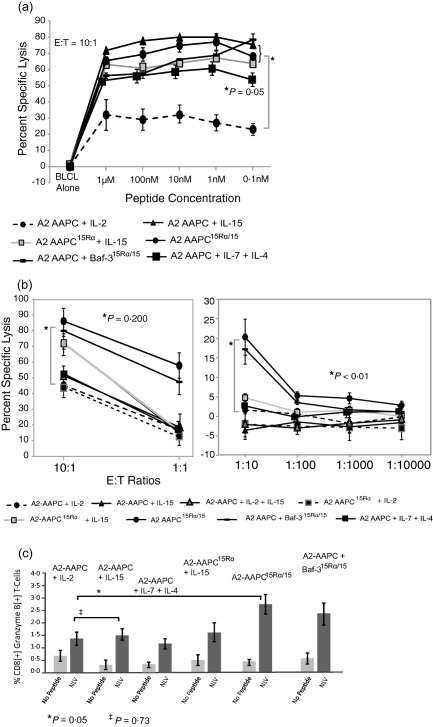
15Rα/15‐stimulated antigen‐specific T cells efficiently lyse targets at lower effector : target (E : T) ratios. T cell cytotoxic capacity was measured in a standard 51Cr release assay, performed at 21–28 days after culture initiation using peptide‐loaded autologous BLCL as targets. BLCL not loaded with peptide were usedas control. (a) A fixed E : T ratio of 10 T cells to one target cell was used and the cytotoxic activity of T cells sensitized in all culture conditions was tested against targets loaded with serial dilutions of the NLVPMVATV (NLV) peptide (10 nM, 1 nM, 0·1nM, 10 pM and 0·1pM at 37ºC × 3 h in serum free medium). (b) The cytotoxic activity of T cells was evaluated at decreasing E : T ratios against targets loaded with a fixed concentration (10 nM) of peptide. (c) T cells in all culture conditions were evaluated for expression of intracellular granzyme B upon secondary restimulation with NLV peptide‐loaded autologous peripheral blood mononuclear cells (PBMC) 21–28 days after culture initiation. T cells co‐incubated with peptide‐loaded autologous PBMC were labelled with fluorescently labelled anti‐CD3, anti‐CD8 and anti‐CD4, followed by incubation with anti‐human granzyme B after cell permeabilization and analysed by fluorescence activated cell sorter (FACS). The proportion of granzyme B‐positive T cells CD8+ T cells was evaluated. T cells sensitized in the presence of 15Rα/15 complexes generated significantly higher proportions of granzyme B+ T cells compared to sensitization in the presence of soluble IL‐2 (P = 0·05).
We then evaluated the cytotoxic activity of CMV‐CTLs at graded E : T ratios. This permitted the recognition of differential cytotoxic activity for 15Rα/15‐stimulated CMV‐CTLs compared to other conditions. At E : T ratios lower than 10 : 1, only 15Rα/15‐stimulated CMV‐CTLs demonstrated sufficient cytotoxic activity, which was diminished markedly at this E : T ratio in all other cytokine conditions (P < 0·01) (Fig. 7b). A higher proportion of granzyme B‐generating CD8+ T cells was also observed within 15Rα/15‐stimulated CMV‐CTLs in comparison to sIL‐2, sIL‐15 or sIL‐7 + sIL‐4‐supplemented T cells (P < 0·05) (Fig. 7c). Taken together, this analysis permitted a functional distinction between CTLs stimulated in different cytokine conditions, and 15Rα/15 stimulation emerged as a means to generate high‐avidity CD8+ antigen‐specific T cells for adoptive immunotherapy applications.
Discussion
Adoptive therapy with antigen‐specific transplant donor‐derived T cells is now established as a viable and effective approach for the treatment of life‐threatening viral infections complicating allogeneic haematopoietic cell or organ transplants 32, 33, 34, 35. Induction of cancer remission has also been achieved in a proportion of chemotherapy refractory patients after infusion of in‐vitro expanded autologous tumour‐infiltrating lymphocytes 36 as well as tumour‐antigen‐specific T cells 37, 38, 39. However, sustained responses have been achieved only in patients with detectable in‐vivo expansion of the adoptively transferred T cells 40. Since then, studies in animal tumour models have shown that infusion of highly differentiated tumour‐antigen‐specific T cells are less effective in eradicating tumours compared to naive and early effector T cells 41. These observations have placed a major emphasis on the development of methodologies that not only enhance the yields of antigen‐specific T cells for adoptive therapy, but also the selective expansion of less differentiated long‐lived memory T cells capable of inducing durable responses 42. Techniques for augmenting the efficacy of adoptively transferred T cells are equally desirable to attain higher rates of remission.
IL‐15 has been shown to play a central role in the stimulation and maintenance of antigen‐specific CD8+ memory T cells when presented in complex with its high‐affinity receptor IL‐15Rα to responding T cells 10, 26, 43, 44, 45, and therefore represents a potentially valuable immunomodulating agent for augmenting the efficacy of adoptive immunotherapy. IL‐15 signalling through the PI3K/AKT pathway has been shown to even revive the exhausted proliferative function of effector memory phenotype T cells specific for infectious agents or tumours in a T cell receptor (TCR)‐independent manner 46. In a study evaluating acute graft rejection in renal transplant recipients, IL‐15 was shown to induce proliferation of CD8+ memory T cells that was independent of B7‐CD28 co‐stimulation 47. These data suggest that in tumours that express HLA or tumour antigens poorly, or lack expression of co‐stimulatory molecules, IL‐15 would be able to endow the host T cells or adoptively transferred T cells with the necessary signals to proliferate and lyse tumour cell targets. As the expression of IL‐15Rα is not optimal in vivo, IL‐15 monotherapy would not be as effective without IL‐15Rα.
The studies herein define the conditions required for the stable expression and generation of 15Rα/15 in vitro, and then compare the effects of soluble IL‐15 and other gamma‐chain cytokines with 15Rα/15 in their capacity to stimulate antigen‐specific T cell expansion. We describe a novel cell‐based APC system that can present and secrete stable 15Rα/15 that has been generated using genetically modified cells either transduced with IL‐15Rα alone or with both IL‐15 and IL‐15Rα genes (A2‐AAPC15Rα/15 and Baf‐315Rα/15). These studies established that both IL‐15 and IL‐15Rα genes are required to be expressed in the same cell to form stable 15Rα/15 complexes, and that the IL‐15 gene was not expressed when transduced without IL‐15Rα. Such an obligate requirement for binding with the alpha chain receptor for stabilization and effect has not been described for other gamma chain cytokines, including IL‐7 or IL‐2. We examined the effects of 15Rα/15 complexes generated by these cells on the in‐vitro enrichment, memory phenotype and functional capacity of antigen‐specific T cells. The data demonstrate that 15Rα/15 complexes can augment not only the yields of antigen‐specific T cells, but also specifically enrich TCM phenotype cells that have the potential to induce durable remissions after adoptive transfer. Importantly, cells generating 15Rα/15 complexes supported the steady expansion of antigen‐specific CD62L+ Tet+ TCM cells, which was not observed in sIL‐15‐supplemented cultures. Furthermore, in cultures where 15Rα/15‐expressing cells were separated from T cells by semipermeable membranes, the secreted and soluble 15Rα/15, potentially presenting IL‐15 in a cis configuration, permeated the membrane and stimulated responding CD8+ T cells efficiently without cell‐to‐cell contact. In fact, 15Rα/15 complexes can not only signal to responding neighbouring lymphocytes when bound to cell membranes but also, as soluble complexes, they can become internalized into responding lymphocytes and lead to sustained stimulation 24, 30, 48. However, in the elegant study by Mortier et al. 30, cell‐to‐cell contact was shown to be essential for the internalization of 15Rα/15 released upon cleavage from the cell surface to stimulate responding CD8 and NK cells effectively. This study also demonstrated stable surface expression of 15Rα/15 using IL‐15Rα‐expressing cells loaded with sIL‐15, which was not observed in our study using sIL‐15‐loaded A2‐AAPC15Rα. In our system, we cannot exclude the possibility that the soluble 15Rα/15 complexes could have bound to AAPCs not expressing either IL‐15Rα or IL‐15, or to activated T cells themselves, and thereby cross‐stimulate adjacent T cells through direct cell contact 45. In order to develop this agent for clinical use, these issues are being explored.
Importantly, the data herein illustrate that 15Rα/15 not only stimulate the expansion of TCM phenotype CTLs, but the T cells generated exhibit high functional activity, as evidenced by high IFN‐γ and granzyme B secretion, and response to minute concentrations of NLV 49 (Fig. 5 and Table 1). Such high‐affinity pMHC/TCR interactions can override the requirement for CD8 engagement for cytotoxic activity 50, suggesting that, by promoting the expansion of high‐avidity T cells, 15Rα/15 could be an invaluable reagent for the expansion of antigen‐specific T cells responding to less immunogenic antigens such as self‐tumour antigens. Indeed, 15Rα/15 complexes expressed on langerhans cells have been shown to overcome tolerance and stimulate the expansion of Wilms' tumour (WT)−1‐specific T cells when electroporated with WT‐1 mRNA 51. Such 15Rα/15 complexes could also be tremendously valuable for the expansion of T cells responding to subdominant epitopes that presumably have lower TCR avidities. Furthermore, infusion of these complexes may also enhance the function of tumour‐resident, low‐avidity T cells. In a recent study using the transgenic adenocarcinoma of the mouse prostate (TRAMP)‐C2 murine tumour model, treatment with agonistic anti‐CD40 in combination with sIL‐15 resulted in tumour regressions in 70–100% of treated animals in comparison to 0–30% treated with antibody alone 52. Here, treatment with anti‐CD40 augmented the IL‐15 Rα expression on host DC resulting in the formation of 15Rα/15 complexes upon exposure to sIL‐15, which then supported the expansion and cytotoxic activity of host tumour‐specific CD8+ T cells and enhanced anti‐tumour activity. Synergistic anti‐tumour activity has also been demonstrated using a combination of IL‐15 and immune check‐point inhibitors 53, 54.
In conclusion, 15Rα/15 complexes are required for optimal IL‐15 activity. These 15Rα/15 complexes represent a potent biological reagent for in‐vitro expansion of highly functional long‐lived antigen‐ specific TCM suitable for adoptive immunotherapy and may also prove useful as therapeutic agents for augmentation of anti‐tumour activity when used in conjunction with other immunotherapies.
Disclosure
AN Hasan and RJ O'Reilly receive Royalties from Atara Biotherapeutics.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Expression of transduced IL‐15Rα and IL‐15 genes. A2‐artificial antigen‐presenting cells (AAPCs) transduced to express IL‐15Rα alone and A2‐AAPC or Baf‐3 cells transduced to co‐express IL‐15Rα and IL‐15, were evaluated for the protein level expression of the transduced genes by FACS. As shown (L –R) high expression of the transduced genes was observed in all cell lines.
Acknowledgements
This research was supported by NIH CA59350, CA 23766, the Aubrey Foundation, the Major Family Fund for Cancer Research, Sportsmen for Charity, the NY Yankees Universe Fund, the Claire Tow Endowment, the Max Cure Fund for Pediatric Cancer Research, the Baker Street Foundation and the Hyundai Hope Grant. The authors have no conflicting financial interests. We thank Eric Pamer and Ingrid Leiner (MSKCC Tetramer Core Facility) for providing the MHC‐peptide tetramers, Dana Bakalar and Christy Kuo for technical support and Joseph Olechnowicz for editing assistance.
References
- 1. Huang J, Khong HT, Dudley ME et al Survival, persistence, and progressive differentiation of adoptively transferred tumor‐reactive T cells associated with tumor regression. J Immunother 2005; 28:258–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Walter EA, Greenberg PD, Gilbert MJ et al Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T cell clones from the donor. N Engl J Med 1995; 333:1038–44. [DOI] [PubMed] [Google Scholar]
- 3. Wherry EJ, Teichgraber V, Becker TC et al Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol 2003; 4:225–34. [DOI] [PubMed] [Google Scholar]
- 4. Quinn M, Turula H, Tandon M et al Memory T cells specific for murine cytomegalovirus re‐emerge after multiple challenges and recapitulate immunity in various adoptive transfer scenarios. J Immunol 2015; 194:1726–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berger C, Jensen MC, Lansdorp PM et al Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest 2008; 118:294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stemberger C, Graef P, Odendahl M et al Lowest numbers of primary CD8(+) T cells can reconstitute protective immunity upon adoptive immunotherapy. Blood 2014; 124:628–37. [DOI] [PubMed] [Google Scholar]
- 7. Schluns KS, Lefrancois L. Cytokine control of memory T cell development and survival. Nat Rev Immunol 2003; 3:269–79. [DOI] [PubMed] [Google Scholar]
- 8. Gerdemann U, Keirnan JM, Katari UL et al Rapidly generated multivirus‐specific cytotoxic T lymphocytes for the prophylaxis and treatment of viral infections. Mol Ther 2012; 20:1622–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wolfl M, Merker K, Morbach H et al Primed tumor‐reactive multifunctional CD62L+ human CD8+ T cells for immunotherapy. Cancer Immunol Immunother 2011; 60:173–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang X, Sun S, Hwang I et al Potent and selective stimulation of memory‐phenotype CD8+ T cells in vivo by IL‐15. Immunity 1998; 8:591–9. [DOI] [PubMed] [Google Scholar]
- 11. Sprent J, Surh CD. Generation and maintenance of memory T cells. Curr Opin Immunol 2001; 13:248–54. [DOI] [PubMed] [Google Scholar]
- 12. Kennedy MK, Glaccum M, Brown SN et al Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15‐deficient mice. J Exp Med 2000; 191:771–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schluns KS, Williams K, Ma A et al Cutting edge: requirement for IL‐15 in the generation of primary and memory antigen‐specific CD8 T cells. J Immunol 2002; 168:4827–31. [DOI] [PubMed] [Google Scholar]
- 14. Imamura M, Shook D, Kamiya T et al Autonomous growth and increased cytotoxicity of natural killer cells expressing membrane‐bound interleukin‐15. Blood 2014; 124:1081–8. [DOI] [PubMed] [Google Scholar]
- 15. Roychowdhury S, May KF Jr, Tzou KS et al Failed adoptive immunotherapy with tumor‐specific T cells: reversal with low‐dose interleukin 15 but not low‐dose interleukin 2. Cancer Res 2004; 64:8062–7. [DOI] [PubMed] [Google Scholar]
- 16. Perna SK, De Angelis B, Pagliara D et al Interleukin 15 provides relief to CTLs from regulatory T cell‐mediated inhibition: implications for adoptive T cell‐based therapies for lymphoma. Clin Cancer Res 2013; 19:106–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu RB, Engels B, Schreiber K et al IL‐15 in tumor microenvironment causes rejection of large established tumors by T cells in a noncognate T cell receptor‐dependent manner. Proc Natl Acad Sci USA 2013; 110:8158–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dubois S, Mariner J, Waldmann TA et al IL‐15Ralpha recycles and presents IL‐15 In trans to neighboring cells. Immunity 2002; 17:537–47. [DOI] [PubMed] [Google Scholar]
- 19. Chertova E, Bergamaschi C, Chertov O et al Characterization and favorable in vivo properties of heterodimeric soluble IL‐15.IL‐15Ralpha cytokine compared to IL‐15 monomer. J Biol Chem 2013; 288:18093–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sato N, Patel HJ, Waldmann TA et al The IL‐15/IL‐15Ralpha on cell surfaces enables sustained IL‐15 activity and contributes to the long survival of CD8 memory T cells. Proc Natl Acad Sci USA 2007; 104:588–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stoklasek TA, Schluns KS, Lefrancois L. Combined IL‐15/IL‐15Ralpha immunotherapy maximizes IL‐15 activity in vivo . J Immunol 2006; 177:6072–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu W, Jones M, Liu B et al Efficacy and mechanism‐of‐action of a novel superagonist interleukin‐15: interleukin‐15 receptor alphaSu/Fc fusion complex in syngeneic murine models of multiple myeloma. Cancer Res 2013; 73:3075–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Epardaud M, Elpek KG, Rubinstein MP et al Interleukin‐15/interleukin‐15R alpha complexes promote destruction of established tumors by reviving tumor‐resident CD8+ T cells. Cancer Res 2008; 68:2972–83. [DOI] [PubMed] [Google Scholar]
- 24. Bergamaschi C, Rosati M, Jalah R et al Intracellular interaction of interleukin‐15 with its receptor alpha during production leads to mutual stabilization and increased bioactivity. J Biol Chem 2008; 283:4189–99. [DOI] [PubMed] [Google Scholar]
- 25. Rowley J, Monie A, Hung CF et al Expression of IL‐15RA or an IL‐15/IL‐15RA fusion on CD8+ T cells modifies adoptively transferred T cell function in cis. Eur J Immunol 2009; 39:491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Burkett PR, Koka R, Chien M et al Coordinate expression and trans presentation of interleukin (IL)−15Ralpha and IL‐15 supports natural killer cell and memory CD8+ T cell homeostasis. J Exp Med 2004; 200:825–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mortier E, Woo T, Advincula R et al IL‐15Ralpha chaperones IL‐15 to stable dendritic cell membrane complexes that activate NK cells via trans presentation. J Exp Med 2008; 205:1213–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hasan AN, Kollen WJ, Trivedi D et al A panel of artificial APCs expressing prevalent HLA alleles permits generation of cytotoxic T cells specific for both dominant and subdominant viral epitopes for adoptive therapy. J Immunol 2009; 183:2837–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rodriguez‐Tarduchy G, Collins M, Lopez‐Rivas A. Regulation of apoptosis in interleukin‐3‐dependent hemopoietic cells by interleukin‐3 and calcium ionophores. EMBO J 1990; 9:2997–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tamzalit F, Barbieux I, Plet A et al IL‐15.IL‐15Ralpha complex shedding following trans‐presentation is essential for the survival of IL‐15 responding NK and T cells. Proc Natl Acad Sci USA 2014; 111:8565–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kokaji AI, Hockley DL, Kane KP. IL‐15 transpresentation augments CD8+ T cell activation and is required for optimal recall responses by central memory CD8+ T cells. J Immunol 2008; 180:4391–401. [DOI] [PubMed] [Google Scholar]
- 32. Doubrovina E, Oflaz‐Sozmen B, Prockop SE et al Adoptive immunotherapy with unselected or EBV‐specific T cells for biopsy‐proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood 2012; 119:2644–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heslop HE, Ng CY, Li C et al Long‐term restoration of immunity against Epstein‐Barr virus infection by adoptive transfer of gene‐modified virus‐specific T lymphocytes. Nat Med 1996; 2:551. [DOI] [PubMed] [Google Scholar]
- 34. Koehne G, Hasan A, Doubrovina E et al Immunotherapy with donor T cells sensitized with overlapping pentadecapeptides for treatment of persistent cytomegalovirus infection or viremia. Biol Blood Marrow Transplant 2015; 21:1663–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Feuchtinger T, Opherk K, Bethge WA et al Adoptive transfer of pp65‐specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood 2010; 116:4360–7. [DOI] [PubMed] [Google Scholar]
- 36. Rosenberg SA, Packard BS, Aebersold PM et al Use of tumor‐infiltrating lymphocytes and interleukin‐2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med 1988; 319:1676–80. [DOI] [PubMed] [Google Scholar]
- 37. Rosenberg SA, Dudley ME. Cancer regression in patients with metastatic melanoma after the transfer of autologous antitumor lymphocytes. Proc Natl Acad Sci USA 2004; 101 Suppl 2:14639–45.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hunder NN, Wallen H, Cao J et al Treatment of metastatic melanoma with autologous CD4+ T cells against NY‐ESO‐1. N Engl J Med 2008; 358:2698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Morgan RA, Dudley ME, Wunderlich JR et al Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006; 314:126–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Klebanoff CA, Gattinoni L, Torabi‐Parizi P et al Central memory self/tumor‐reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci USA 2005; 102:9571–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gattinoni L, Klebanoff CA, Palmer DC et al Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest 2005; 115:1616–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ahmed R, Gray D. Immunological memory and protective immunity: understanding their relation. Science 1996; 272:54–60. [DOI] [PubMed] [Google Scholar]
- 43. Waldmann TA, Dubois S, Tagaya Y. Contrasting roles of IL‐2 and IL‐15 in the life and death of lymphocytes: implications for immunotherapy. Immunity 2001; 14:105–10. [PubMed] [Google Scholar]
- 44. Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity 2008; 29:848–62. [DOI] [PubMed] [Google Scholar]
- 45. Schluns KS, Klonowski KD, Lefrancois L. Transregulation of memory CD8 T cell proliferation by IL‐15Ralpha+ bone marrow‐derived cells. Blood 2004; 103:988–94. [DOI] [PubMed] [Google Scholar]
- 46. Kim HR, Hwang KA, Kang I. Dual roles of IL‐15 in maintaining IL‐7R alpha low CCR7– memory CD8+ T cells in humans via recovering the phosphatidylinositol 3‐kinase/AKT pathway. J Immunol 2007; 179:6734–40. [DOI] [PubMed] [Google Scholar]
- 47. Traitanon O, Gorbachev A, Bechtel JJ et al IL‐15 induces alloreactive CD28(–) memory CD8 T cell proliferation and CTLA4‐Ig resistant memory CD8 T cell activation. Am J Transplant 2014; 14:1277–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mortier E, Advincula R, Kim L et al Macrophage‐ and dendritic‐cell‐derived interleukin‐15 receptor alpha supports homeostasis of distinct CD8+ T cell subsets. Immunity 2009; 31:811–22. [DOI] [PubMed] [Google Scholar]
- 49. Oh S, Hodge JW, Ahlers JD et al Selective induction of high avidity CTL by altering the balance of signals from APC. J Immunol 2003; 170:2523–30. [DOI] [PubMed] [Google Scholar]
- 50. Kerry SE, Buslepp J, Cramer LA et al Interplay between TCR affinity and necessity of coreceptor ligation: high‐affinity peptide‐MHC/TCR interaction overcomes lack of CD8 engagement. J Immunol 2003; 171:4493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Romano E, Cotari JW, Barreira da Silva R et al Human Langerhans cells use an IL‐15R‐alpha/IL‐15/pSTAT5‐dependent mechanism to break T cell tolerance against the self‐differentiation tumor antigen WT1. Blood 2012; 119:5182–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang M, Ju W, Yao Z et al Augmented IL‐15Ralpha expression by CD40 activation is critical in synergistic CD8 T cell‐mediated antitumor activity of anti‐CD40 antibody with IL‐15 in TRAMP‐C2 tumors in mice. J Immunol 2012; 188:6156–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yu P, Steel JC, Zhang M et al Simultaneous inhibition of two regulatory T cell subsets enhanced Interleukin‐15 efficacy in a prostate tumor model. Proc Natl Acad Sci USA 2012; 109:6187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yu P, Steel JC, Zhang M et al Simultaneous blockade of multiple immune system inhibitory checkpoints enhances antitumor activity mediated by interleukin‐15 in a murine metastatic colon carcinoma model. Clin Cancer Res 2010; 16:6019–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Latouche JB, Sadelain M. Induction of human cytotoxic T lymphocytes by artificial antigen‐presenting cells. Nat Biotechnol 2000; 18:405–9. [DOI] [PubMed] [Google Scholar]
- 56. Kozak M. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J Mol Biol 1987; 196:947–50. [DOI] [PubMed] [Google Scholar]
- 57. Koehne G, Smith KM, Ferguson TL et al Quantitation, selection, and functional characterization of Epstein–Barr virus‐specific and alloreactive T cells detected by intracellular interferon‐gamma production and growth of cytotoxic precursors. Blood 2002; 99:1730–40. [DOI] [PubMed] [Google Scholar]
- 58. Waldrop SL, Pitcher CJ, Peterson DM et al Determination of antigen‐specific memory/effector CD4+ T cell frequencies by flow cytometry: evidence for a novel, antigen‐specific homeostatic mechanism in HIV‐associated immunodeficiency. J Clin Invest 1997; 99:1739–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Expression of transduced IL‐15Rα and IL‐15 genes. A2‐artificial antigen‐presenting cells (AAPCs) transduced to express IL‐15Rα alone and A2‐AAPC or Baf‐3 cells transduced to co‐express IL‐15Rα and IL‐15, were evaluated for the protein level expression of the transduced genes by FACS. As shown (L –R) high expression of the transduced genes was observed in all cell lines.